Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes

 , , , , ,
, , , , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
2.1. Clinical Evaluation
2.2. Genetic Studies
2.3. Cardiac Differentiation
3. Results
3.1. Clinical Characterization and In Silico Modeling
3.2. Reprogramming of Patient’s Skin Fibroblasts and Characterization of CPVT-hiPSCs
3.3. CPVT hiPSC Differentiate into Functional Cardiomyocytes
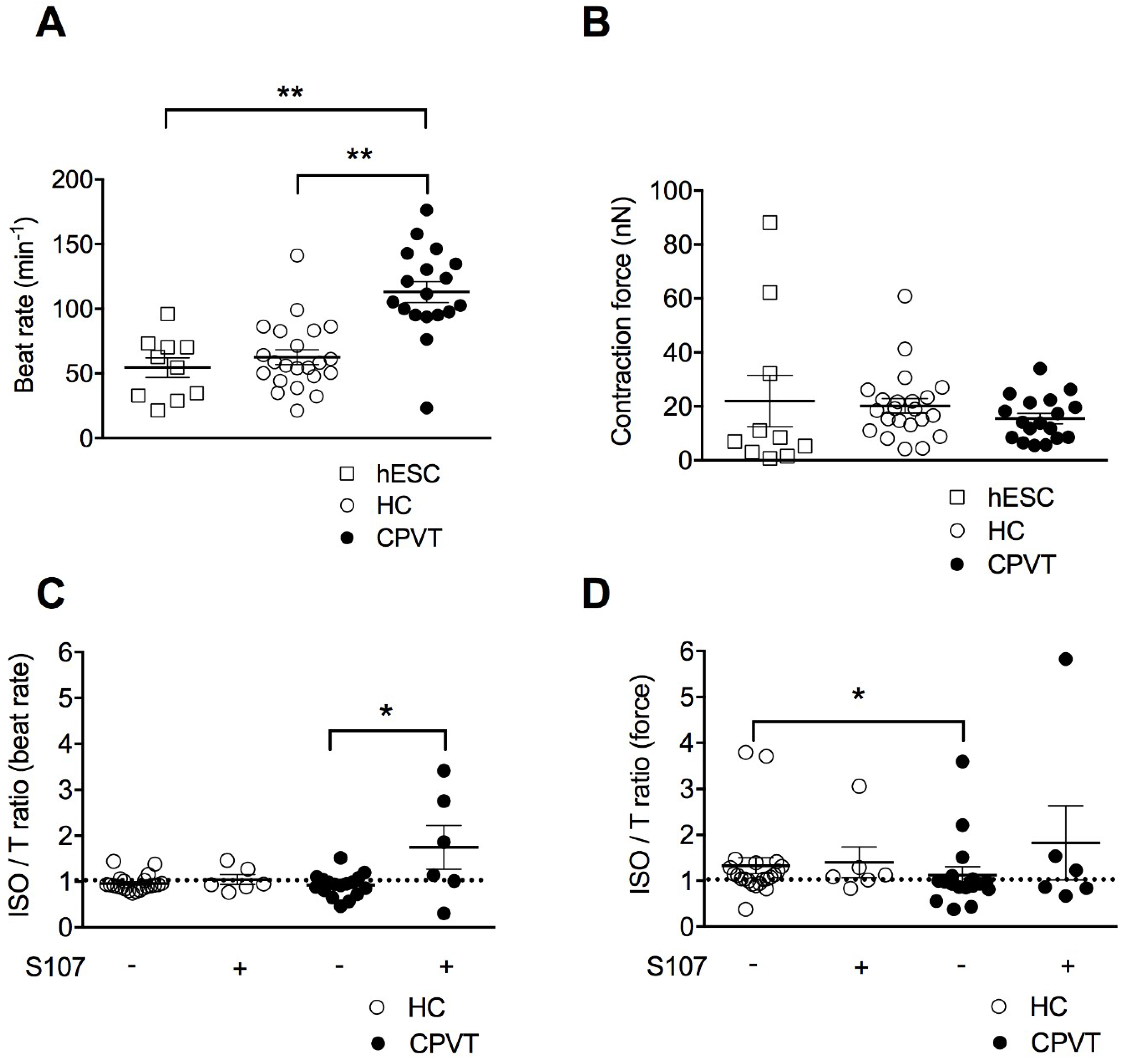
3.4. CPVT-EBs Present Higher Spontaneous Beats and Weaker Contraction Force Response Under Stress
3.5. Stabilizing the Closed State of RyR2 Using S107 Improves the Contractile Properties in CPVT-EBs
3.6. The CPVT Mutation D3638A Leads to Abnormal Intracellular Ca2+ Release under Stress
3.7. S107 Prevents the SR Ca2+ Leak in CPVT hiPSC-CMs and Does Not Affect HC hiPSC-CMs
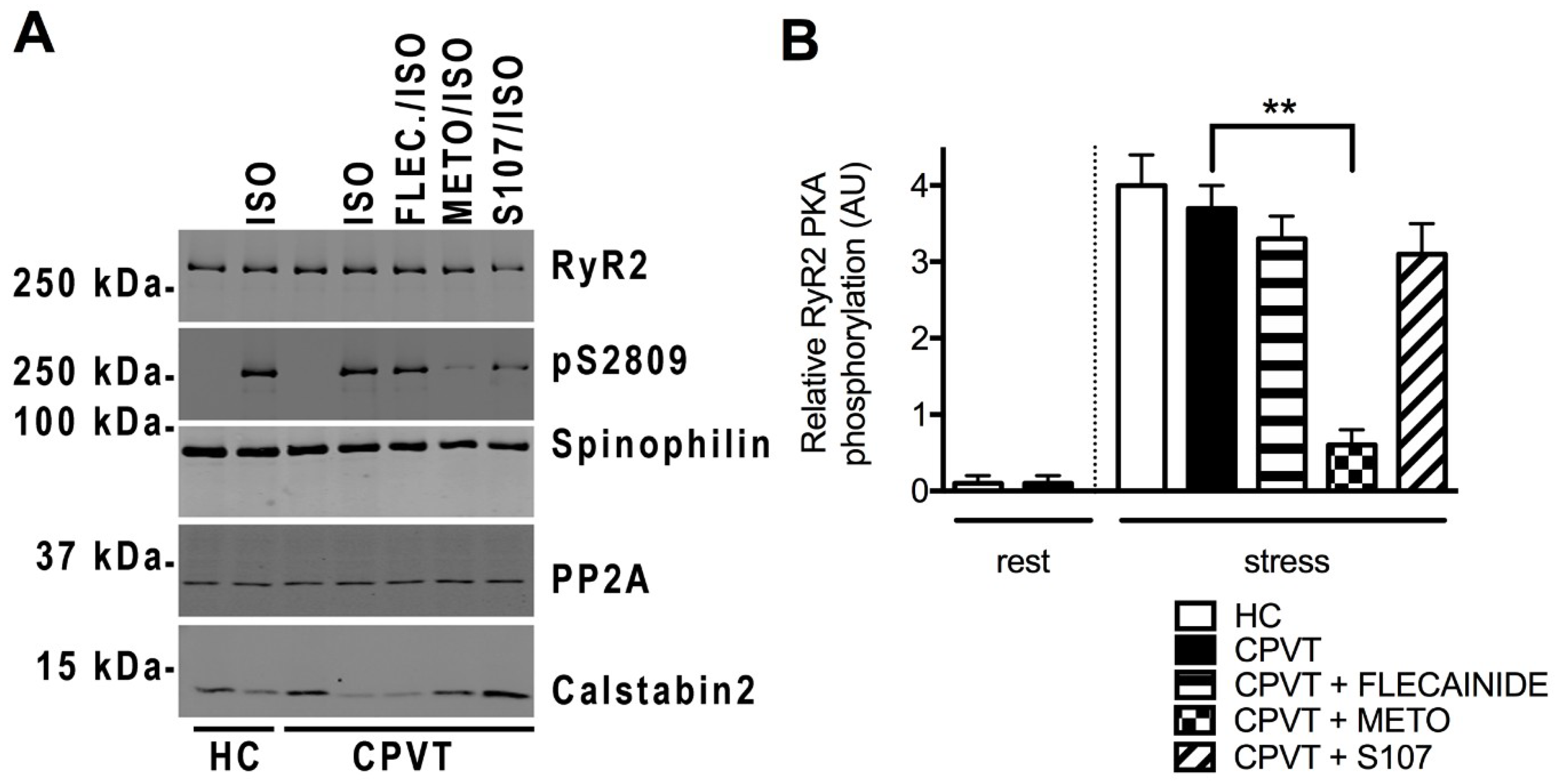
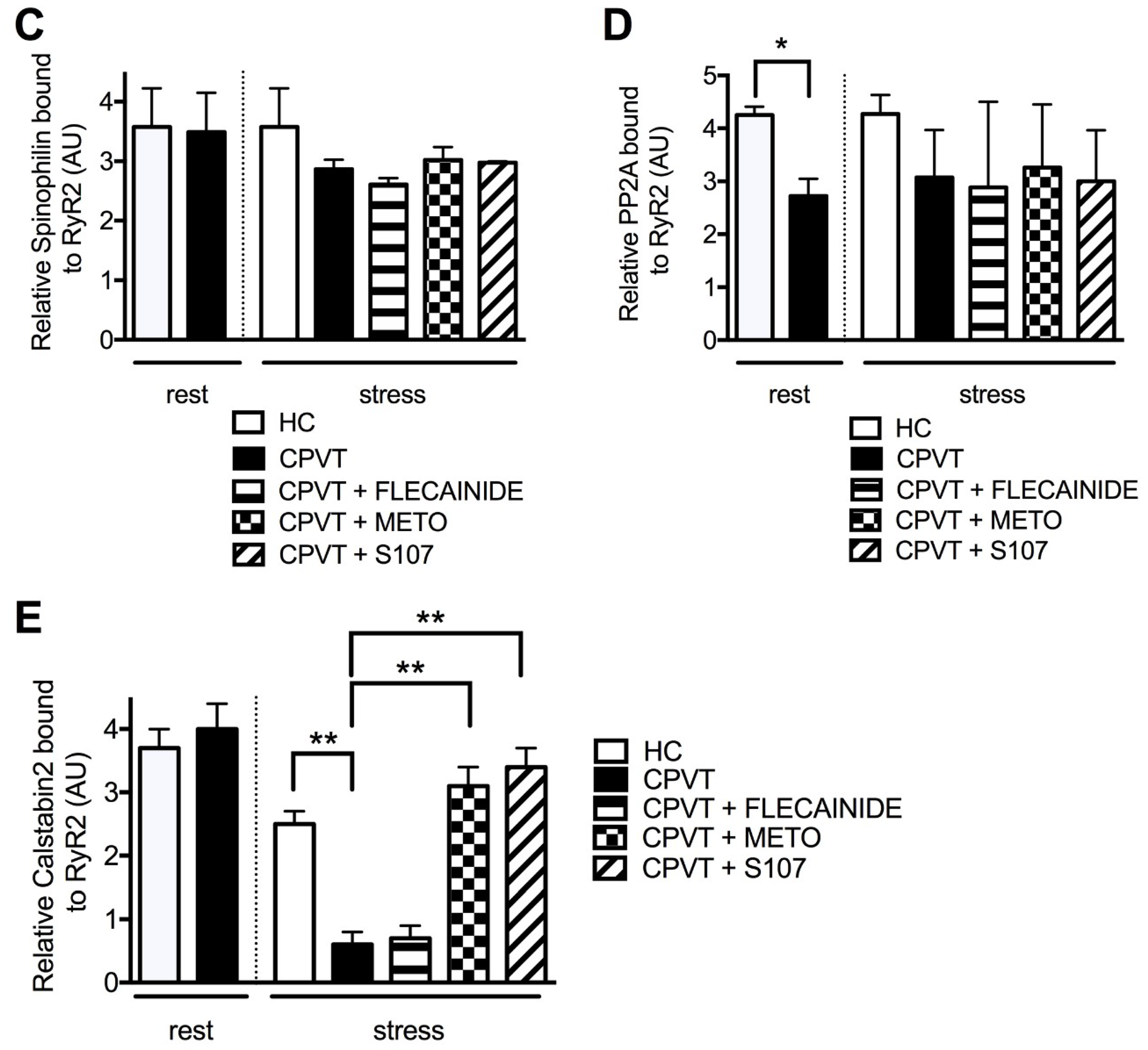
3.8. Unlike Flecainide, Metoprolol Does Not Prevent the Abnormal Ca2+ Release in CPVT hiPSC-CMs
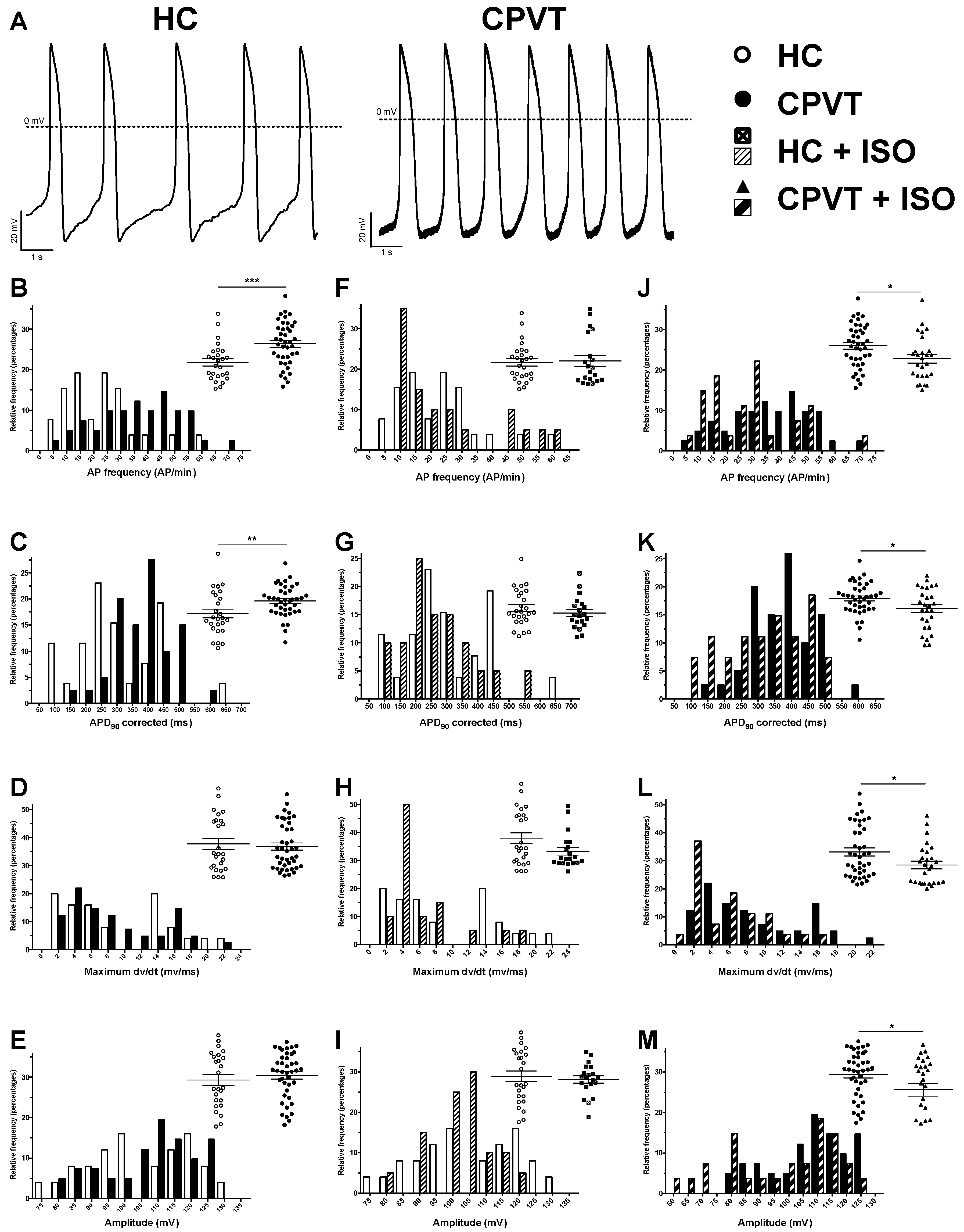
3.9. Electrical Activity Is Affected in CPVT RyR2-D3638A Patient Cells
3.10. Abnormal Electrical Activity Elicited with ISO Is Rescued with Flecainide and S107
3.11. Post-Translational Modifications Are Associated with the CPVT RyR2-D3638A Channels
4. Discussion
4.1. Insights from the Structure-Function Aspects
4.2. Modeling of CPVT Syndrome Using hiPSC-CMs in the Dish
4.3. The Inadequate Β-Blocker Response and Potent Effects of S107 and Flecainide
4.4. Study Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Napolitano, C.; Bloise, R.; Memmi, M.; Priori, S.G. Clinical utility gene card for: Catecholaminergic polymorphic ventricular tachycardia (CPVT). Eur. J. Hum. Genet. 2014, 22. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Sallam, K.; Wu, H.; Li, Y.; Itzhaki, I.; Garg, P.; Zhang, Y.; Termglichan, V.; Lan, F.; Gu, M.; et al. Patient-Specific and Genome-Edited Induced Pluripotent Stem Cell–Derived Cardiomyocytes Elucidate Single-Cell Phenotype of Brugada Syndrome. J. Am. Coll. Cardiol. 2016, 68, 2086–2096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, W.; Shen, H.; Wu, J.; Guo, W.; Pan, X.; Wang, R.; Chen, S.R.; Yan, N. Structural basis for the gating mechanism of the type 2 ryanodine receptor RyR2. Science 2016, 354, aah5324. [Google Scholar] [CrossRef] [PubMed]
- Moreau, A.; Mercier, A.; Theriault, O.; Boutjdir, M.; Burger, B.; Keller, D.I.; Chahine, M. Biophysical, Molecular, and Pharmacological Characterization of Voltage-Dependent Sodium Channels from Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Can. J. Cardiol. 2017, 33, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Pesl, M.; Acimovic, I.; Pribyl, J.; Hezova, R.; Vilotic, A.; Fauconnier, J.; Vrbsky, J.; Kruzliak, P.; Skladal, P.; Kara, T.; et al. Forced aggregation and defined factors allow highly uniform-sized embryoid bodies and functional cardiomyocytes from human embryonic and induced pluripotent stem cells. Heart Vessels 2014, 29, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Thomas, N.L.; George, C.H.; Lai, F.A. Role of ryanodine receptor mutations in cardiac pathology: More questions than answers? Biochem. Soc. Trans. 2006, 34, 913–918. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Puttonen, K.A.; Lindeberg, H.; Ruponen, M.; Hovatta, O.; Koistinaho, J.; Lammi, M.J. Chondrogenic differentiation of human pluripotent stem cells in chondrocyte co-culture. Int. J. Biochem. Cell Biol. 2013, 45, 1802–1812. [Google Scholar] [CrossRef] [PubMed]
- Leenhardt, A.; Lucet, V.; Denjoy, I.; Grau, F.; Ngoc, D.D.; Coumel, P. Catecholaminergic polymorphic ventricular tachycardia in children. A 7-year follow-up of 21 patients. Circulation 1995, 91, 1512–1519. [Google Scholar] [CrossRef] [PubMed]
- Pesl, M.; Pribyl, J.; Acimovic, I.; Vilotic, A.; Jelinkova, S.; Salykin, A.; Lacampagne, A.; Dvorak, P.; Meli, A.C.; Skladal, P.; et al. Atomic force microscopy combined with human pluripotent stem cell derived cardiomyocytes for biomechanical sensing. Biosens. Bioelectr. 2016, 85, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Sun, N.; Yazawa, M.; Liu, J.; Han, L.; Sanchez-Freire, V.; Abilez, O.J.; Navarrete, E.G.; Hu, S.; Wang, L.; Lee, A.; et al. Patient-specific induced pluripotent stem cells as a model for familial dilated cardiomyopathy. Sci. Transl. Med. 2012, 4, 130ra147. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sun, N.; Bruce, M.A.; Wu, J.C.; Butte, M.J. Atomic force mechanobiology of pluripotent stem cell-derived cardiomyocytes. PLoS ONE 2012, 7, e37559. [Google Scholar] [CrossRef] [PubMed]
- Di Pasquale, E.; Lodola, F.; Miragoli, M.; Denegri, M.; Avelino-Cruz, J.E.; Buonocore, M.; Nakahama, H.; Portararo, P.; Bloise, R.; Napolitano, C.; et al. CaMKII inhibition rectifies arrhythmic phenotype in a patient-specific model of catecholaminergic polymorphic ventricular tachycardia. Cell Death Dis. 2013, 4, e843. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, A.M.; Reiken, S.; Dura, M.; Murphy, P.W.; Deng, S.X.; Landry, D.W.; Nieman, D.; Lehnart, S.E.; Samaru, M.; LaCampagne, A.; et al. Remodeling of ryanodine receptor complex causes “leaky” channels: A molecular mechanism for decreased exercise capacity. Proc. Natl. Acad. Sci. USA 2008, 105, 2198–2202. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Xie, W.; Betzenhauser, M.; Reiken, S.; Chen, B.X.; Wronska, A.; Marks, A.R. Calcium leak through ryanodine receptors leads to atrial fibrillation in 3 mouse models of catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2012, 111, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Fauconnier, J.; Meli, A.C.; Thireau, J.; Roberge, S.; Shan, J.; Sassi, Y.; Reiken, S.R.; Rauzier, J.M.; Marchand, A.; Chauvier, D.; et al. Ryanodine receptor leak mediated by caspase-8 activation leads to left ventricular injury after myocardial ischemia-reperfusion. Proc. Natl. Acad. Sci. USA 2011, 108, 13258–13263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wehrens, X.H.; Lehnart, S.E.; Huang, F.; Vest, J.A.; Reiken, S.R.; Mohler, P.J.; Sun, J.; Guatimosim, S.; Song, L.S.; Rosemblit, N.; et al. FKBP12.6 deficiency and defective calcium release channel (ryanodine receptor) function linked to exercise-induced sudden cardiac death. Cell 2003, 113, 829–840. [Google Scholar] [CrossRef]
- Fernandez-Velasco, M.; Rueda, A.; Rizzi, N.; Benitah, J.P.; Colombi, B.; Napolitano, C.; Priori, S.G.; Richard, S.; Gomez, A.M. Increased Ca2+ sensitivity of the ryanodine receptor mutant RyR2R4496C underlies catecholaminergic polymorphic ventricular tachycardia. Circ. Res. 2009, 104, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Meli, A.C.; Refaat, M.M.; Dura, M.; Reiken, S.; Wronska, A.; Wojciak, J.; Carroll, J.; Scheinman, M.M.; Marks, A.R. A novel ryanodine receptor mutation linked to sudden death increases sensitivity to cytosolic calcium. Circ. Res. 2011, 109, 281–290. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Betzenhauser, M.J.; Kushnir, A.; Reiken, S.; Meli, A.C.; Wronska, A.; Dura, M.; Chen, B.X.; Marks, A.R. Role of chronic ryanodine receptor phosphorylation in heart failure and beta-adrenergic receptor blockade in mice. J. Clin. Investig. 2010, 120, 4375–4387. [Google Scholar] [CrossRef] [PubMed]
- Novak, A.; Barad, L.; Zeevi-Levin, N.; Shick, R.; Shtrichman, R.; Lorber, A.; Itskovitz-Eldor, J.; Binah, O. Cardiomyocytes generated from CPVTD307H patients are arrhythmogenic in response to beta-adrenergic stimulation. J. Cell. Mol. Med. 2012, 16, 468–482. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Gepstein, A.; Arbel, G.; Caspi, O.; Miller, L.; Belhassen, B.; Nof, E.; Glikson, M.; et al. Modeling of catecholaminergic polymorphic ventricular tachycardia with patient-specific human-induced pluripotent stem cells. J. Am. Coll. Cardiol. 2012, 60, 990–1000. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, K.; Makiyama, T.; Yoshida, Y.; Wuriyanghai, Y.; Kamakura, T.; Nishiuchi, S.; Hayano, M.; Harita, T.; Yamamoto, Y.; Kohjitani, H.; et al. Patient-Specific Human Induced Pluripotent Stem Cell Model Assessed with Electrical Pacing Validates S107 as a Potential Therapeutic Agent for Catecholaminergic Polymorphic Ventricular Tachycardia. PLoS ONE 2016, 11, e0164795. [Google Scholar] [CrossRef] [PubMed]
- Preininger, M.K.; Jha, R.; Maxwell, J.T.; Wu, Q.; Singh, M.; Wang, B.; Dalal, A.; Mceachin, Z.T.; Rossoll, W.; Hales, C.M.; et al. A human pluripotent stem cell model of catecholaminergic polymorphic ventricular tachycardia recapitulates patient-specific drug responses. Dis. Models Mech. 2016, 9, 927–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zalk, R.; Clarke, O.B.; des Georges, A.; Grassucci, R.A.; Reiken, S.; Mancia, F.; Hendrickson, W.A.; Frank, J.; Marks, A.R. Structure of a mammalian ryanodine receptor. Nature 2015, 517, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Marx, S.O.; Reiken, S.; Hisamatsu, Y.; Gaburjakova, M.; Gaburjakova, J.; Yang, Y.M.; Rosemblit, N.; Marks, A.R. Phosphorylation-dependent regulation of ryanodine receptors: A novel role for leucine/isoleucine zippers. J. Cell Biol. 2001, 153, 699–708. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.B.; Moretti, A.; Mederos y Schnitzler, M.; Iop, L.; Storch, U.; Bellin, M.; Dorn, T.; Ruppenthal, S.; Pfeiffer, S.; Goedel, A.; et al. Dantrolene rescues arrhythmogenic RYR2 defect in a patient-specific stem cell model of catecholaminergic polymorphic ventricular tachycardia. EMBO Mol. Med. 2012, 4, 180–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neco, P.; Torrente, A.G.; Mesirca, P.; Zorio, E.; Liu, N.; Priori, S.G.; Napolitano, C.; Richard, S.; Benitah, J.P.; Mangoni, M.E.; et al. Paradoxical effect of increased diastolic Ca(2+) release and decreased sinoatrial node activity in a mouse model of catecholaminergic polymorphic ventricular tachycardia. Circulation 2012, 126, 392–401. [Google Scholar] [CrossRef] [PubMed]
- Lymperopoulos, A.; McCrink, K.A.; Brill, A. Impact of CYP2D6 Genetic Variation on the Response of the Cardiovascular Patient to Carvedilol and Metoprolol. Curr. Drug Metab. 2015, 17, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Cheung, J.W.; Meli, A.C.; Xie, W.; Mittal, S.; Reiken, S.; Wronska, A.; Xu, L.; Steinberg, J.S.; Markowitz, S.M.; Iwai, S.; et al. Short-coupled polymorphic ventricular tachycardia at rest linked to a novel ryanodine receptor (RyR2) mutation: Leaky RyR2 channels under non-stress conditions. Int. J. Cardiol. 2015, 180, 228–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fauconnier, J.; Thireau, J.; Reiken, S.; Cassan, C.; Richard, S.; Matecki, S.; Marks, A.R.; Lacampagne, A. Leaky RyR2 trigger ventricular arrhythmias in Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2010, 107, 1559–1564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehnart, S.E.; Mongillo, M.; Bellinger, A.; Lindegger, N.; Chen, B.X.; Hsueh, W.; Reiken, S.; Wronska, A.; Drew, L.J.; Ward, C.W.; et al. Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in mice. J. Clin. Investig. 2008, 118, 2230–2245. [Google Scholar] [CrossRef] [PubMed]
- Lehnart, S.E.; Wehrens, X.H.; Laitinen, P.J.; Reiken, S.R.; Deng, S.X.; Cheng, Z.; Landry, D.W.; Kontula, K.; Swan, H.; Marks, A.R. Sudden death in familial polymorphic ventricular tachycardia associated with calcium release channel (ryanodine receptor) leak. Circulation 2004, 109, 3208–3214. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Betzenhauser, M.J.; Reiken, S.; Meli, A.C.; Xie, W.; Chen, B.X.; Arancio, O.; Marks, A.R. Role of leaky neuronal ryanodine receptors in stress- induced cognitive dysfunction. Cell 2012, 150, 1055–1067. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.; Kushnir, A.; Betzenhauser, M.J.; Reiken, S.; Li, J.; Lehnart, S.E.; Lindegger, N.; Mongillo, M.; Mohler, P.J.; Marks, A.R. Phosphorylation of the ryanodine receptor mediates the cardiac fight or flight response in mice. J. Clin. Investig. 2010, 120, 4388–4398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Syeda, F.; Holmes, A.P.; Yu, T.Y.; Tull, S.; Kuhlmann, S.M.; Pavlovic, D.; Betney, D.; Riley, G.; Kucera, J.P.; Jousset, F.; et al. PITX2 Modulates Atrial Membrane Potential and the Antiarrhythmic Effects of Sodium-Channel Blockers. J. Am. Coll. Cardiol. 2016, 25, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Van der Werf, C.; Kannankeril, P.J.; Sacher, F.; Krahn, A.D.; Viskin, S.; Leenhardt, A.; Shimizu, W.; Sumitomo, N.; Fish, F.A.; Bhuiyan, Z.A.; et al. Flecainide therapy reduces exercise-induced ventricular arrhythmias in patients with catecholaminergic polymorphic ventricular tachycardia. J. Am. Coll. Cardiol. 2011, 57, 2244–2254. [Google Scholar] [CrossRef] [PubMed]
- Bannister, M.L.; Thomas, N.L.; Sikkel, M.B.; Mukherjee, S.; Maxwell, C.; MacLeod, K.T.; George, C.H.; Williams, A.J. The mechanism of flecainide action in CPVT does not involve a direct effect on RyR2. Circ. Res. 2015, 116, 1324–1335. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acimovic, I.; Refaat, M.M.; Moreau, A.; Salykin, A.; Reiken, S.; Sleiman, Y.; Souidi, M.; Přibyl, J.; Kajava, A.V.; Richard, S.; et al. Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes. J. Clin. Med. 2018, 7, 423. https://doi.org/10.3390/jcm7110423
Acimovic I, Refaat MM, Moreau A, Salykin A, Reiken S, Sleiman Y, Souidi M, Přibyl J, Kajava AV, Richard S, et al. Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes. Journal of Clinical Medicine. 2018; 7(11):423. https://doi.org/10.3390/jcm7110423
Chicago/Turabian StyleAcimovic, Ivana, Marwan M. Refaat, Adrien Moreau, Anton Salykin, Steve Reiken, Yvonne Sleiman, Monia Souidi, Jan Přibyl, Andrey V. Kajava, Sylvain Richard, and et al. 2018. "Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes" Journal of Clinical Medicine 7, no. 11: 423. https://doi.org/10.3390/jcm7110423
APA StyleAcimovic, I., Refaat, M. M., Moreau, A., Salykin, A., Reiken, S., Sleiman, Y., Souidi, M., Přibyl, J., Kajava, A. V., Richard, S., Lu, J. T., Chevalier, P., Skládal, P., Dvořak, P., Rotrekl, V., Marks, A. R., Scheinman, M. M., Lacampagne, A., & Meli, A. C. (2018). Post-Translational Modifications and Diastolic Calcium Leak Associated to the Novel RyR2-D3638A Mutation Lead to CPVT in Patient-Specific hiPSC-Derived Cardiomyocytes. Journal of Clinical Medicine, 7(11), 423. https://doi.org/10.3390/jcm7110423