Association of Nicotine with Osteochondrogenesis and Osteoarthritis Development: The State of the Art of Preclinical Research




{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
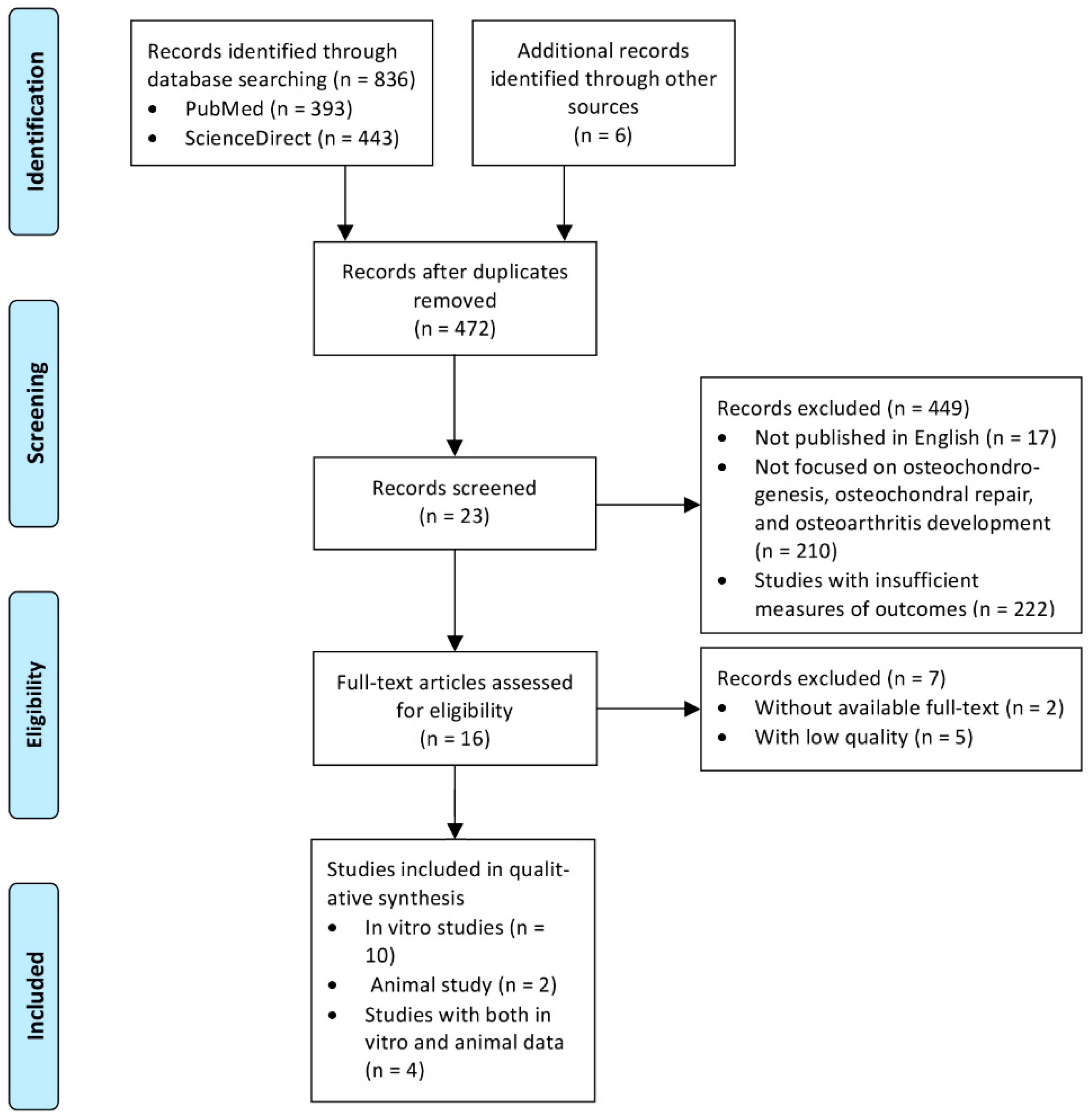
2.1. Search Strategy
2.2. Data Extraction and Critical Appraisal
3. Results
3.1. Search Results and the Characteristics of the Included Studies
3.2. In Vitro Studies
3.2.1. In Vitro Studies on the Effect of Nicotine on Chondrogenic Differentiation of Medicinal Signaling Cells
Bone Marrow-derived MSCs
Adipose-Derived MSCs
Wharton’s Jelly-Derived MSCs
3.2.2. In Vitro Studies on the Effects of Nicotine on Articular Chondrocytes
3.2.3. In Vitro Studies on the Effect of Nicotine on Osteogenic Differentiation of MSCs
Bone Marrow-Derived MSCs
Periodontal Ligament-Derived Signaling Cells
Adipose-Derived MSCs
3.2.4. In Vitro Studies of Effects of Smokeless Tobacco Extract (STE) on Osteoblast Differentiation
3.3. Animal Studies
3.3.1. Effect of Nicotine on Cartilage Formation in the Offspring
3.3.2. Effect of Nicotine on OA Susceptibility in Offspring
3.3.3. Effect of Nicotine in Animal Models of OA
3.3.4. Effect of Nicotine on Osteochondral Repair
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hunziker, E.B.; Lippuner, K.; Keel, M.J.; Shintani, N. An educational review of cartilage repair: precepts & practice-myths & misconceptions--progress & prospects. Osteoarthr. Cartil. 2015, 23, 334–350. [Google Scholar] [CrossRef] [PubMed]
- Cucchiarini, M.; Madry, H. Biomaterial-guided delivery of gene vectors for targeted articular cartilage repair. Nat. Rev. Rheumatol. 2019, 15, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Jeffries, M.A. Osteoarthritis year in review 2018: Genetics and epigenetics. Osteoarthr. Cartil. 2019, 27, 371–377. [Google Scholar] [CrossRef] [PubMed]
- DeFrate, L.E.; Kim-Wang, S.Y.; Englander, Z.A.; McNulty, A.L. Osteoarthritis year in review 2018: mechanics. Osteoarthr. Cartil. 2019, 27, 392–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhang, H.; Liang, N.; Fan, W.; Li, J.; Huang, Z.; Yin, Z.; Wu, Z.; Hu, J. Prevalence and associated factors of knee osteoarthritis in a rural Chinese adult population: an epidemiological survey. BMC Public Health 2016, 16, 94. [Google Scholar] [CrossRef] [PubMed]
- Elloumi, M.; Kallel, M.H. Which relationship does osteoarthritis share with smoking? Osteoarthr. Cartil. 2007, 15, 1097–1098. [Google Scholar] [CrossRef] [Green Version]
- Ying, X.; Cheng, S.; Shen, Y.; Cheng, X.; An Rompis, F.; Wang, W.; Lin, Z.; Chen, Q.; Zhang, W.; Kou, D.; et al. Nicotine promotes proliferation and collagen synthesis of chondrocytes isolated from normal human and osteoarthritis patients. Mol. Cell. Biochem. 2012, 359, 263–269. [Google Scholar] [CrossRef]
- Niemeyer, P.; Salzmann, G.M.; Hirschmuller, A.; Sudkamp, N.P. Factors that influence clinical outcome following autologous chondrocyte implantation for cartilage defects of the knee. Z. Fur Orthop. Und Unf. 2012, 150, 83–88. [Google Scholar] [CrossRef]
- Tie, K.; Wu, M.; Deng, Y.; Wen, Y.; Dan, X.; Chen, L.; Wang, H. Histone hypo-acetylation of Sox9 mediates nicotine-induced weak cartilage repair by suppressing BMSC chondrogenic differentiation. Stem Cell Res. Ther. 2018, 9, 98. [Google Scholar] [CrossRef] [Green Version]
- Porter, S.E.; Hanley, E.N., Jr. The musculoskeletal effects of smoking. J. Am. Acad. Orthop. Surg. 2001, 9, 9–17. [Google Scholar] [CrossRef]
- Jakoi, A.M.; Pannu, G.; D’Oro, A.; Buser, Z.; Pham, M.H.; Patel, N.N.; Hsieh, P.C.; Liu, J.C.; Acosta, F.L.; Hah, R.; et al. The Clinical Correlations between Diabetes, Cigarette Smoking and Obesity on Intervertebral Degenerative Disc Disease of the Lumbar Spine. Asian Spine J. 2017, 11, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wu, D.; Song, F.; Zhu, C.; Hui, Y.; Zhu, Q.; Wu, J.; Fan, W.; Hu, J. Activation of alpha7 nicotinic acetylcholine receptors prevents monosodium iodoacetate-induced osteoarthritis in rats. Cell. Physiol. Biochem. 2015, 35, 627–638. [Google Scholar] [CrossRef] [PubMed]
- Ding, C.; Cicuttini, F.; Blizzard, L.; Jones, G. Smoking interacts with family history with regard to change in knee cartilage volume and cartilage defect development. Arthritis Rheum. 2007, 56, 1521–1528. [Google Scholar] [CrossRef] [PubMed]
- Felson, D.T.; Zhang, Y. Smoking and osteoarthritis: A review of the evidence and its implications. Osteoarthr. Cartil. 2015, 23, 331–333. [Google Scholar] [CrossRef] [PubMed]
- Teng, P.; Liu, Y.; Dai, Y.; Zhang, H.; Liu, W.T.; Hu, J. Nicotine Attenuates Osteoarthritis Pain and Matrix Metalloproteinase-9 Expression via the alpha7 Nicotinic Acetylcholine Receptor. J. Immunol. 2019, 203, 485–492. [Google Scholar] [CrossRef] [PubMed]
- Sandmark, H.; Hogstedt, C.; Lewold, S.; Vingard, E. Osteoarthrosis of the knee in men and women in association with overweight, smoking, and hormone therapy. Ann. Rheum. Dis. 1999, 58, 151–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tie, K.; Zhang, X.; Tan, Y.; Deng, Y.; Li, J.; Ni, Q.; Wang, H.; Chen, L. Intrauterine low-functional programming of IGF1 by prenatal nicotine exposure mediates the susceptibility to osteoarthritis in female adult rat offspring. FASEB J. 2016, 30, 785–797. [Google Scholar] [CrossRef]
- Caplan, A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017, 6, 1445–1451. [Google Scholar] [CrossRef] [Green Version]
- Wahl, E.A.; Schenck, T.L.; Machens, H.-G.; Egaña, J.T. Acute stimulation of mesenchymal stem cells with cigarette smoke extract affects their migration, differentiation, and paracrine potential. Sci. Rep. 2016, 6, 22957. [Google Scholar] [CrossRef]
- Ng, T.K.; Carballosa, C.M.; Pelaez, D.; Wong, H.K.; Choy, K.W.; Pang, C.P.; Cheung, H.S. Nicotine alters MicroRNA expression and hinders human adult stem cell regenerative potential. Stem Cells Dev. 2013, 22, 781–790. [Google Scholar] [CrossRef]
- Ng, T.K.; Huang, L.; Cao, D.; Yip, Y.W.-Y.; Tsang, W.M.; Yam, G.H.-F.; Pang, C.P.; Cheung, H.S. Cigarette smoking hinders human periodontal ligament-derived stem cell proliferation, migration and differentiation potentials. Sci. Rep. 2015, 5, 7828. [Google Scholar] [CrossRef] [PubMed]
- Ying, X.; Zhang, W.; Cheng, S.; Nie, P.; Cheng, X.; Shen, Y.; Wang, W.; Xue, E.; Chen, Q.; Kou, D.; et al. Nicotine-induced chondrogenic differentiation of human bone marrow stromal cells in vitro. Knee Surg. Sports Traumatol. Arthrosc. 2012, 20, 2329–2336. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Li, T.Q.; Yan, Y.E.; Magdalou, J.; Wang, H.; Chen, L.B. Effect of nicotine on chondrogenic differentiation of rat bone marrow mesenchymal stem cells in alginate bead culture. Bio-Med. Mater. Eng. 2012, 22, 81–87. [Google Scholar] [CrossRef]
- Yang, X.; Qi, Y.; Avercenc-Leger, L.; Vincourt, J.B.; Hupont, S.; Huselstein, C.; Wang, H.; Chen, L.; Magdalou, J. Effect of nicotine on the proliferation and chondrogenic differentiation of the human Wharton’s jelly mesenchymal stem cells. Bio-Med. Mater. Eng. 2017, 28, S217–S228. [Google Scholar] [CrossRef]
- Shaito, A.; Saliba, J.; Husari, A.; El-Harakeh, M.; Chhouri, H.; Hashem, Y.; Shihadeh, A.; El-Sabban, M. Electronic Cigarette Smoke Impairs Normal Mesenchymal Stem Cell Differentiation. Sci. Rep. 2017, 7, 14281. [Google Scholar] [CrossRef]
- Henderson, J.S.; Johnson, R.B. The effects of smokeless tobacco extract on bone nodule formation and mineralization by chick osteoblasts in vitro. Arch. Oral Biol. 1995, 40, 615–621. [Google Scholar] [CrossRef]
- Lenz, L.G.; Ramp, W.K.; Galvin, R.J.; Pierce, W.M., Jr. Inhibition of cell metabolism by a smokeless tobacco extract: Tissue and species specificity. Proc. Soc. Exp. Biol. Med. 1992, 199, 211–217. [Google Scholar] [CrossRef]
- Xie, Z.; Zhao, Z.; Yang, X.; Pei, L.; Luo, H.; Ni, Q.; Li, B.; Qi, Y.; Tie, K.; Magdalou, J.; et al. Prenatal nicotine exposure intergenerationally programs imperfect articular cartilage via histone deacetylation through maternal lineage. Toxicol. Appl. Pharmacol. 2018, 352, 107–118. [Google Scholar] [CrossRef]
- Tie, K.; Tan, Y.; Deng, Y.; Li, J.; Ni, Q.; Magdalou, J.; Chen, L.; Wang, H. Prenatal nicotine exposure induces poor articular cartilage quality in female adult offspring fed a high-fat diet and the intrauterine programming mechanisms. Reprod. Toxicol. 2016, 60, 11–20. [Google Scholar] [CrossRef]
- Moher, D.; Liberati, A.; Tetzlaff, J.; Altman, D.G. Preferred reporting items for systematic reviews and meta-analyses: The PRISMA statement. PLoS Med. 2009, 6, e1000097. [Google Scholar] [CrossRef]
- Crump, K.S.; Hoel, D.G.; Langley, C.H.; Peto, R. Fundamental carcinogenic processes and their implications for low dose risk assessment. Cancer Res. 1976, 36, 2973–2979. [Google Scholar] [PubMed]
- Wesson, D.R.; Smith, D.E. Buprenorphine in the treatment of opiate dependence. J. Psychoact. Drugs 2010, 42, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Kawakita, A.; Sato, K.; Makino, H.; Ikegami, H.; Takayama, S.; Toyama, Y.; Umezawa, A. Nicotine acts on growth plate chondrocytes to delay skeletal growth through the alpha7 neuronal nicotinic acetylcholine receptor. PLoS ONE 2008, 3, e3945. [Google Scholar] [CrossRef] [PubMed]
- Schneider, N.G.; Olmstead, R.E.; Franzon, M.A.; Lunell, E. The nicotine inhaler: clinical pharmacokinetics and comparison with other nicotine treatments. Clin. Pharmacokinet. 2001, 40, 661–684. [Google Scholar] [CrossRef] [PubMed]
- Maatta, A.J.; Paananen, M.; Marttila, R.; Auvinen, J.; Miettunen, J.; Karppinen, J. Maternal Smoking During Pregnancy Is Associated With Offspring’s Musculoskeletal Pain in Adolescence: Structural Equation Modeling. Nicotine Tob. Res. 2017, 19, 797–803. [Google Scholar] [CrossRef]
- Gao, L.; Goebel, L.K.H.; Orth, P.; Cucchiarini, M.; Madry, H. Subchondral drilling for articular cartilage repair: A systematic review of translational research. Dis. Models Mech. 2018, 11. [Google Scholar] [CrossRef]
- Madry, H.; Gao, L.; Eichler, H.; Orth, P.; Cucchiarini, M. Bone Marrow Aspirate Concentrate-Enhanced Marrow Stimulation of Chondral Defects. Stem Cells Int. 2017, 2017, 1609685. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cai, X.; Gao, L.; Cucchiarini, M.; Madry, H. Association of Nicotine with Osteochondrogenesis and Osteoarthritis Development: The State of the Art of Preclinical Research. J. Clin. Med. 2019, 8, 1699. https://doi.org/10.3390/jcm8101699
Cai X, Gao L, Cucchiarini M, Madry H. Association of Nicotine with Osteochondrogenesis and Osteoarthritis Development: The State of the Art of Preclinical Research. Journal of Clinical Medicine. 2019; 8(10):1699. https://doi.org/10.3390/jcm8101699
Chicago/Turabian StyleCai, Xiaoyu, Liang Gao, Magali Cucchiarini, and Henning Madry. 2019. "Association of Nicotine with Osteochondrogenesis and Osteoarthritis Development: The State of the Art of Preclinical Research" Journal of Clinical Medicine 8, no. 10: 1699. https://doi.org/10.3390/jcm8101699
APA StyleCai, X., Gao, L., Cucchiarini, M., & Madry, H. (2019). Association of Nicotine with Osteochondrogenesis and Osteoarthritis Development: The State of the Art of Preclinical Research. Journal of Clinical Medicine, 8(10), 1699. https://doi.org/10.3390/jcm8101699