An Integrative Systems Biology and Experimental Approach Identifies Convergence of Epithelial Plasticity, Metabolism, and Autophagy to Promote Chemoresistance

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. RNA Extraction, Reverse Transcription, and RT-qPCR
2.3. Western Blotting
2.4. Ammonia Production Assay
2.5. Cytoscape Analysis
2.6. Phylogenetic Reconstructions from Gene Expression Data
2.7. High-Throughput Screening
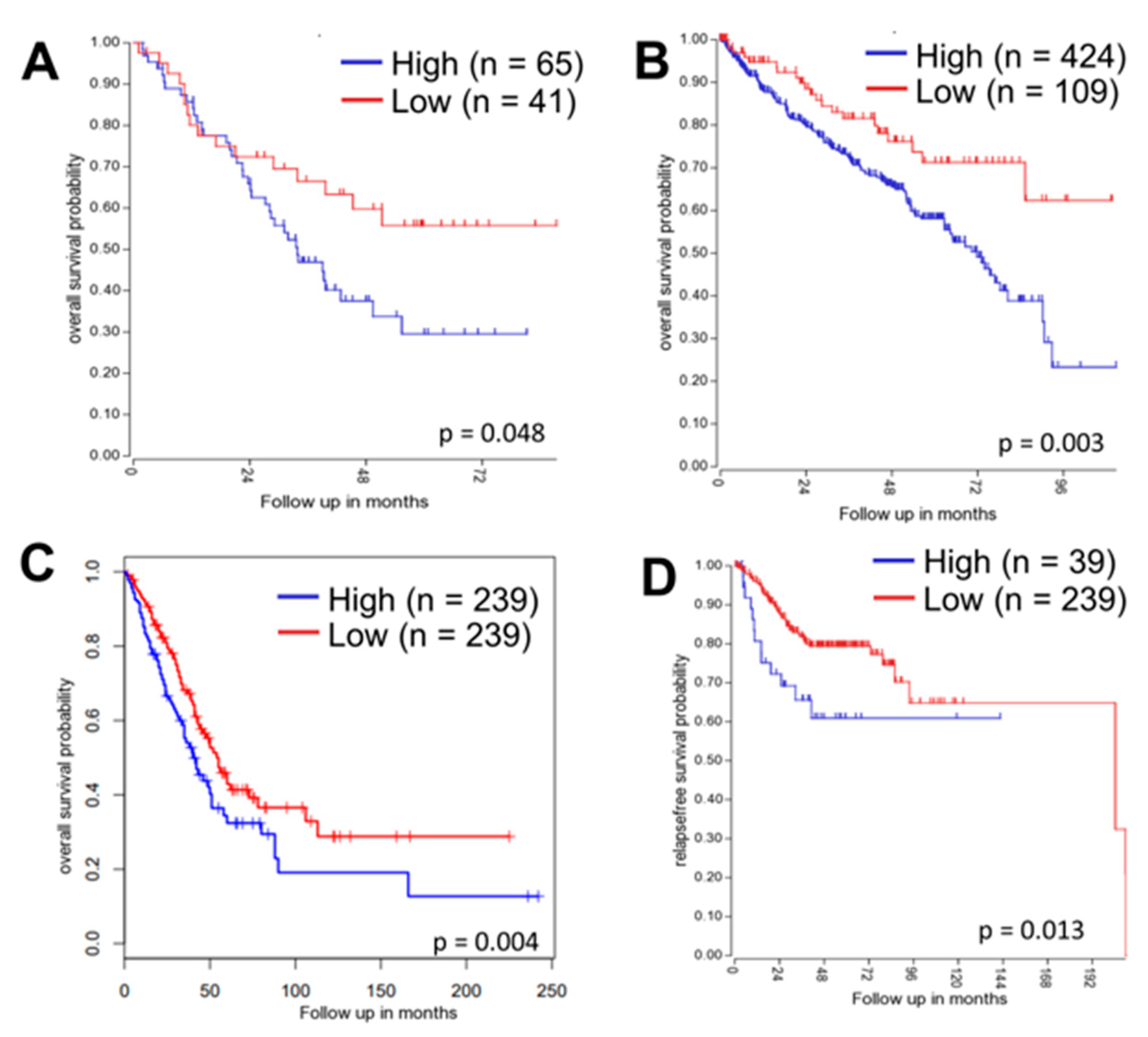
2.8. Correlation of ATG16L1 with Clinical Outcomes
2.9. Statistical Analyses
3. Results
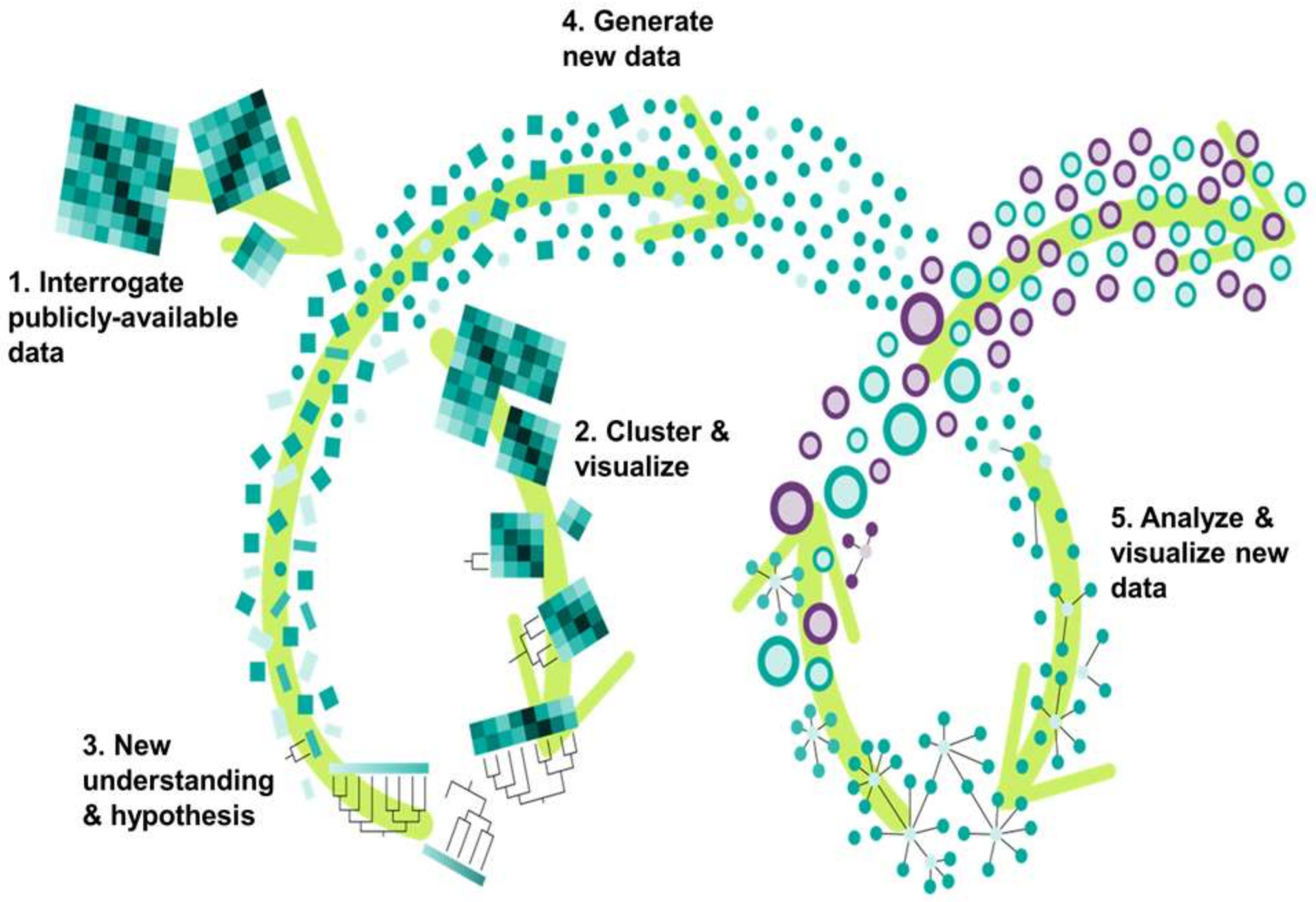
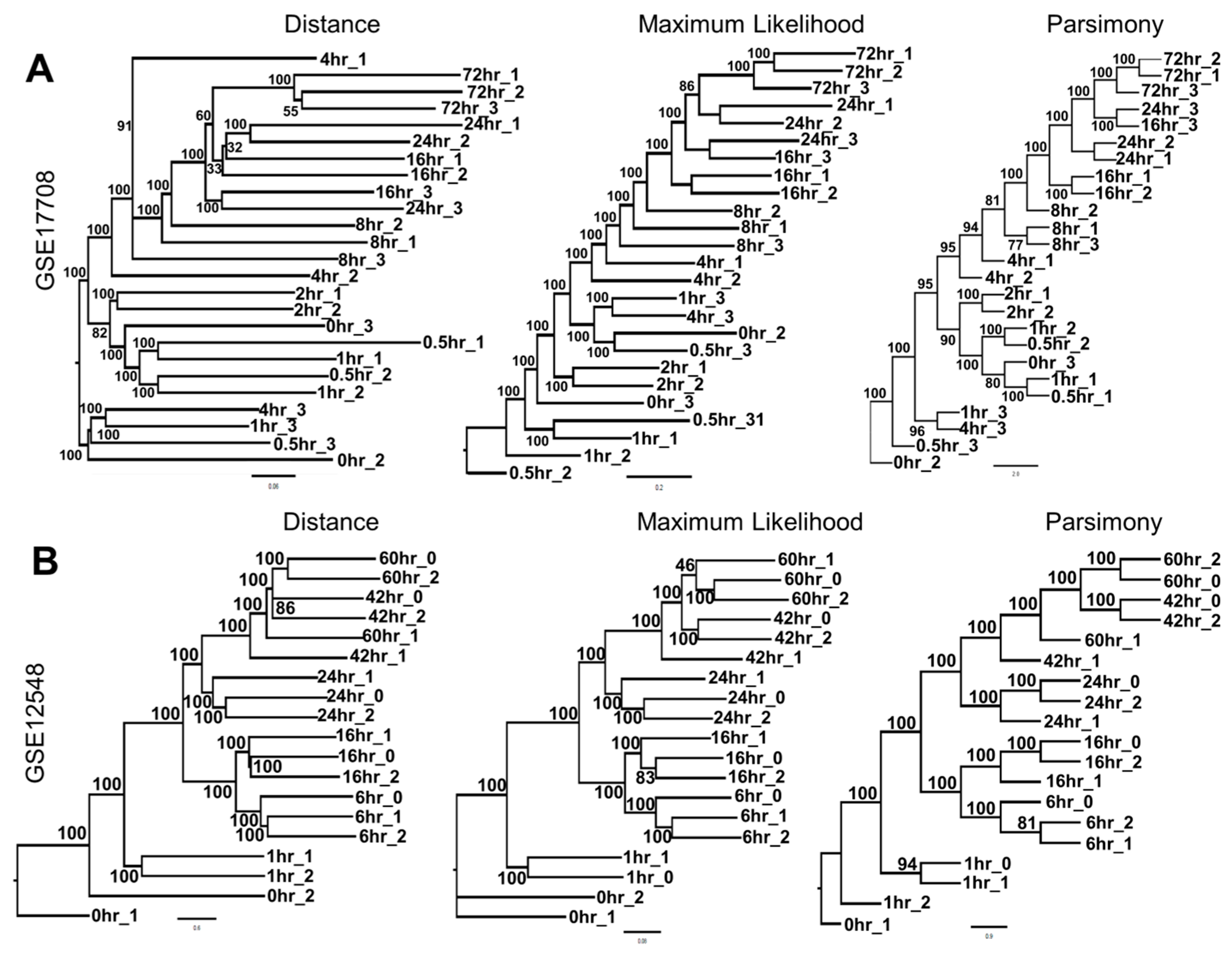
3.1. Phylogenetics Analyses Provide a Simple and Reliable Tool to Visualize Gene Expression Dynamics
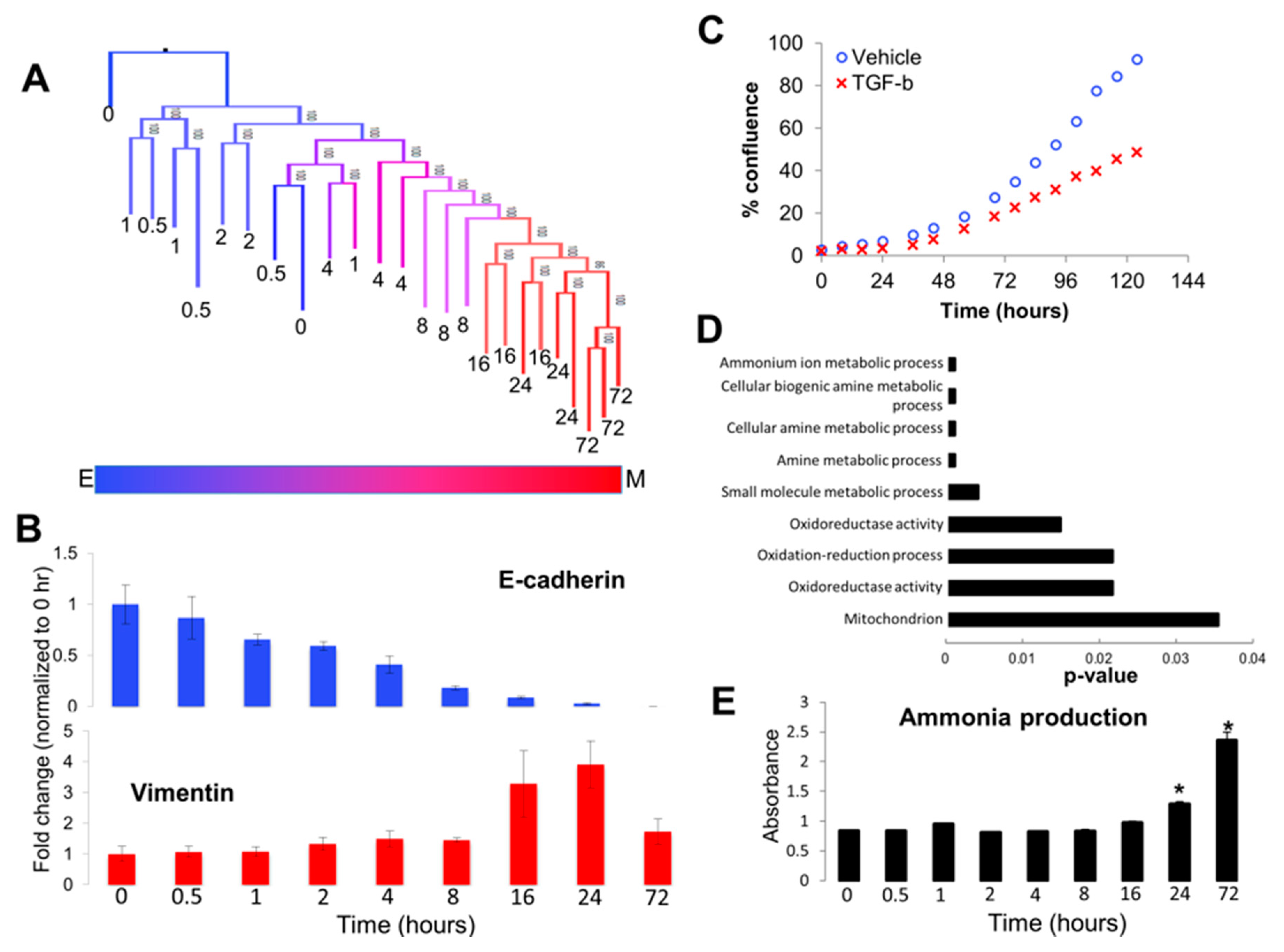
3.2. Analyzing Dynamics of TGF-β Treatment through Visualization of Tree Structure Reveals Two Distinct Temporally-Resolved Clades
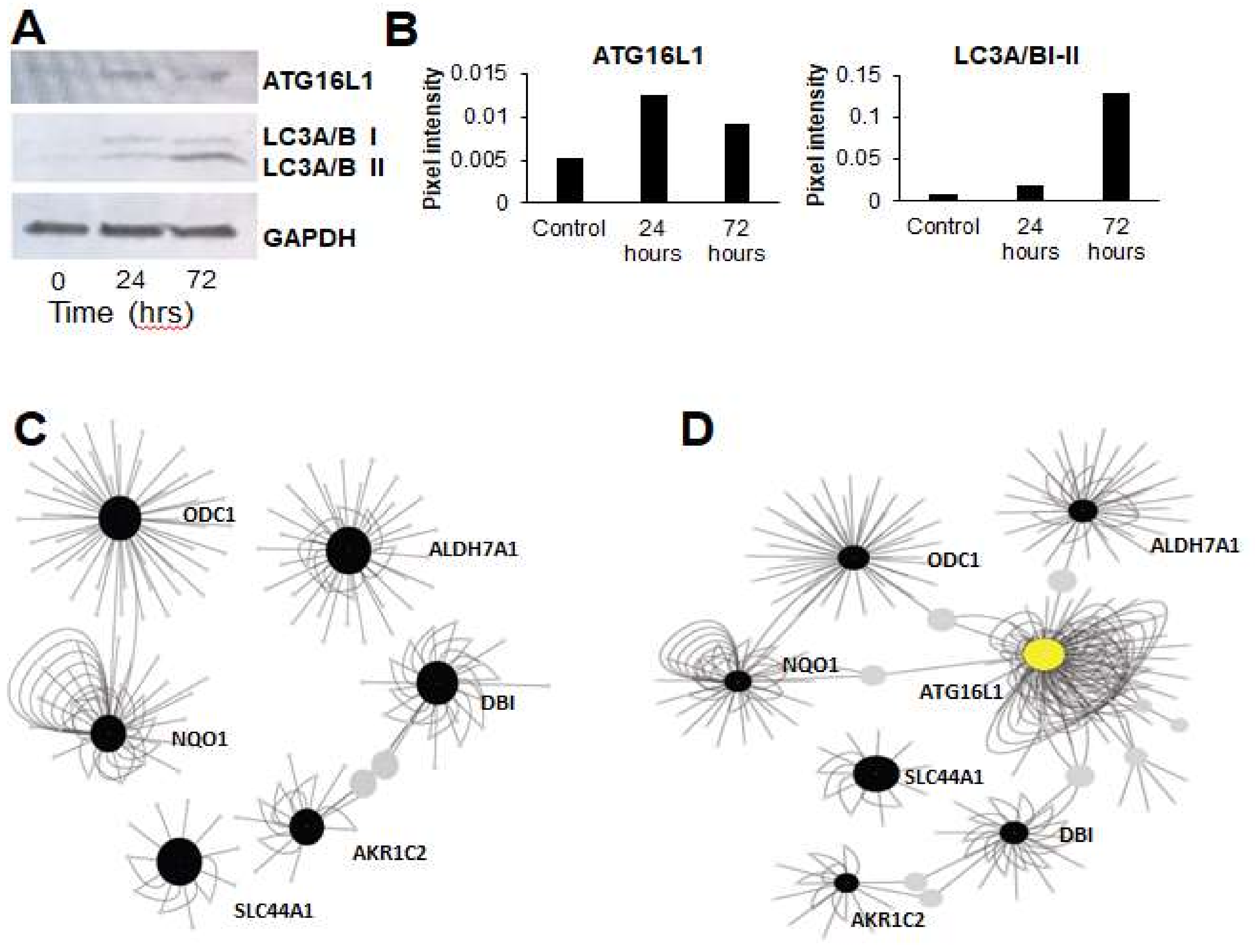
3.3. Gene Expression Networks Couple Ammonia Production to Autophagy
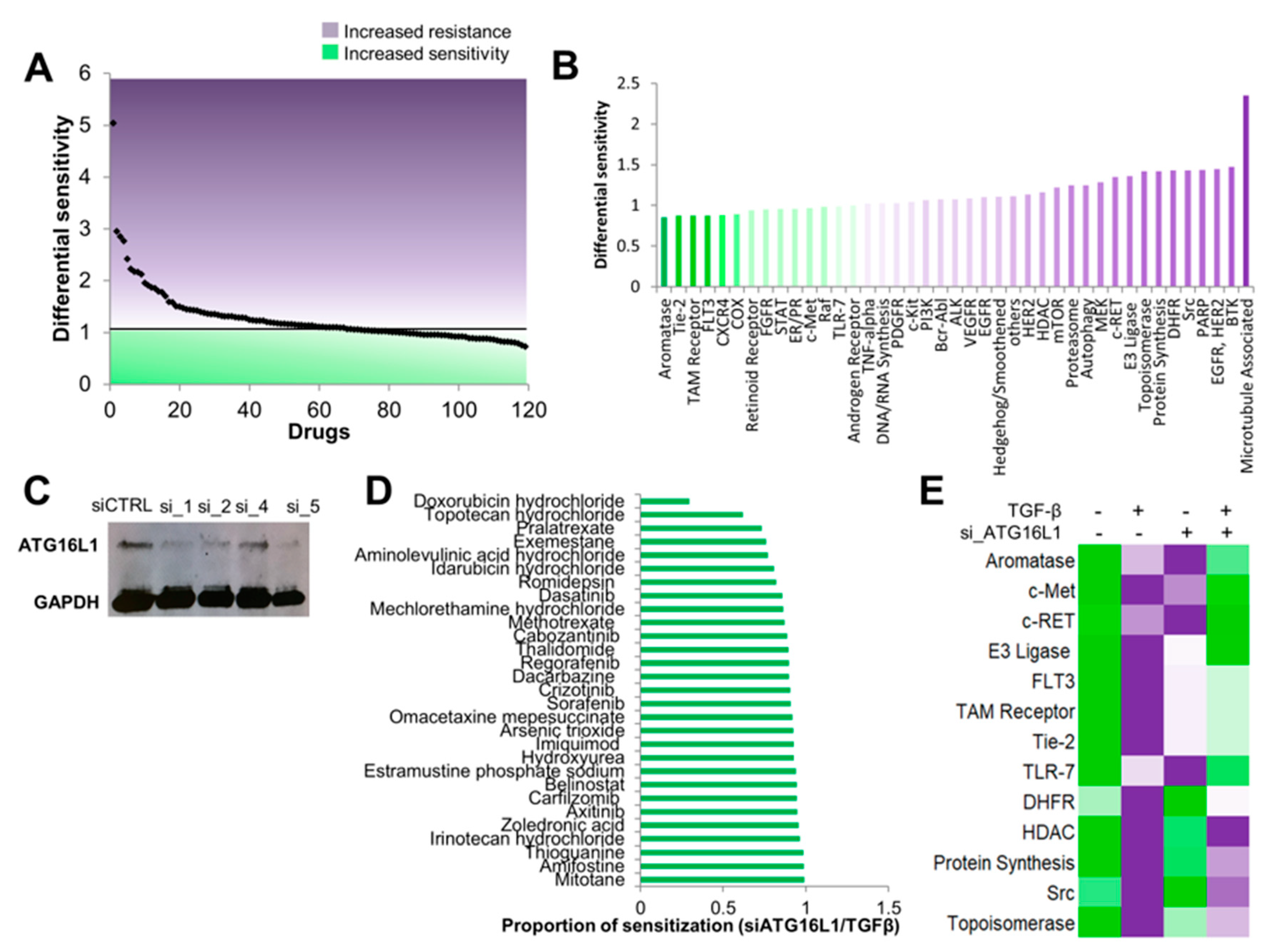
3.4. Autophagy Inhibition Re-sensitizes Cells to TGF-β-Induced Chemoresistance
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Chaiwanon, J.; Wang, W.; Zhu, J.Y.; Oh, E.; Wang, Z.Y. Information integration and communication in plant growth regulation. Cell 2016, 164, 1257–1268. [Google Scholar] [CrossRef]
- Pawson, C.T.; Scott, J.D. Signal integration through blending, bolstering and bifurcating of intracellular information. Nat. Struct. Mol. Biol. 2010, 17, 653–658. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Janda, E.; Litos, G.; Grunert, S.; Downward, J.; Beug, H. Oncogenic Ras/Her-2 mediate hyperproliferation of polarized epithelial cells in 3D cultures and rapid tumor growth via the PI3K pathway. Oncogene 2002, 21, 5148–5159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sever, R.; Brugge, J.S. Signal transduction in cancer. Cold Spring Harb Perspect. Med. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Van Golen, K.L.; Bao, L.W.; Pan, Q.; Miller, F.R.; Wu, Z.F.; Merajver, S.D. Mitogen activated protein kinase pathway is involved in RhoC GTPase induced motility, invasion and angiogenesis in inflammatory breast cancer. Clin. Exp. Metastasis 2002, 19, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, W.; Alkarim, S.; AlHejin, A.; Mukhtar, H.; Saini, K.S. Targeting signal transduction pathways of cancer stem cells for therapeutic opportunities of metastasis. Oncotarget 2016, 7, 76337–76353. [Google Scholar] [CrossRef]
- Janda, E.; Lehmann, K.; Killisch, I.; Jechlinger, M.; Herzig, M.; Downward, J.; Beug, H.; Grunert, S. Ras and TGF-β cooperatively regulate epithelial cell plasticity and metastasis: Dissection of Ras signaling pathways. J. Cell. Biol. 2002, 156, 299–313. [Google Scholar] [CrossRef]
- Brown, W.S.; Tan, L.; Smith, A.; Gray, N.S.; Wendt, M.K. Covalent targeting of fibroblast growth factor receptor inhibits metastatic breast cancer. Mol. Cancer Ther. 2016, 15, 2096–2106. [Google Scholar] [CrossRef]
- Kitisin, K.; Saha, T.; Blake, T.; Golestaneh, N.; Deng, M.; Kim, C.; Tang, Y.; Shetty, K.; Mishra, B.; Mishra, L. TGF-β signaling in development. Sci. STKE 2007, 2007. [Google Scholar] [CrossRef]
- Walton, K.L.; Johnson, K.E.; Harrison, C.A. Targeting TGF-β mediated SMAD signaling for the prevention of fibrosis. Front. Pharmacol. 2017, 8, 461. [Google Scholar] [CrossRef] [PubMed]
- Carthy, J.M. TGF-β signaling and the control of myofibroblast differentiation: Implications for chronic inflammatory disorders. J. Cell. Physiol. 2018, 233, 98–106. [Google Scholar] [CrossRef]
- Nawshad, A.; Lagamba, D.; Polad, A.; Hay, E.D. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: Implications for embryogenesis and tumor metastasis. Cells Tissues Organs. 2005, 179, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Papageorgis, P. TGF-β signaling in tumor initiation, epithelial-to-mesenchymal transition, and metastasis. J. Oncol. 2015, 2015, 587193. [Google Scholar] [CrossRef]
- Fuxe, J.; Vincent, T.; Garcia de Herreros, A. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: Role of EMT promoting SMAD complexes. Cell Cycle 2010, 9, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.S.; Huang, J.S. TGF-β control of cell proliferation. J. Cell. Biochem. 2005, 96, 447–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moustakas, A.; Pardali, K.; Gaal, A.; Heldin, C.H. Mechanisms of TGF-β signaling in regulation of cell growth and differentiation. Immunol. Lett. 2002, 82, 85–91. [Google Scholar] [CrossRef]
- Jiang, L.; Xiao, L.; Sugiura, H.; Huang, X.; Ali, A.; Kuro-o, M.; Deberardinis, R.J.; Boothman, D.A. Metabolic reprogramming during TGF-β-induced epithelial-to-mesenchymal transition. Oncogene 2015, 34, 3908–3916. [Google Scholar] [CrossRef]
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-β and potentiates TGF-β-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852. [Google Scholar] [CrossRef]
- Hazelbag, S.; Gorter, A.; Kenter, G.G.; van den Broek, L.; Fleuren, G. Transforming growth factor-beta1 induces tumor stroma and reduces tumor infiltrate in cervical cancer. Hum. Pathol. 2002, 33, 1193–1199. [Google Scholar] [CrossRef]
- Gigante, M.; Gesualdo, L.; Ranieri, E. TGF-β: A master switch in tumor immunity. Curr. Pharm. Des. 2012, 18, 4126–4134. [Google Scholar] [CrossRef] [PubMed]
- Yao, Z.; Fenoglio, S.; Gao, D.C.; Camiolo, M.; Stiles, B.; Lindsted, T.; Schlederer, M.; Johns, C.; Altorki, N.; Mittal, V.; et al. TGF-β il-6 axis mediates selective and adaptive mechanisms of resistance to molecular targeted therapy in lung cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 15535–15540. [Google Scholar] [CrossRef]
- Brunen, D.; Willems, S.M.; Kellner, U.; Midgley, R.; Simon, I.; Bernards, R. TGF-β: An emerging player in drug resistance. Cell Cycle 2013, 12, 2960–2968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magi, S.; Iwamoto, K.; Okada-Hatakeyama, M. Current status of mathematical modeling of cancer—From the viewpoint of cancer hallmarks. Curr. Opin. Chem. Biol. 2017, 2, 39–48. [Google Scholar]
- Anderson, A.R.; Quaranta, V. Integrative mathematical oncology. Nat. Rev. Cancer 2008, 8, 227–234. [Google Scholar] [CrossRef]
- Gerlinger, M.; McGranahan, N.; Dewhurst, S.M.; Burrell, R.A.; Tomlinson, I.; Swanton, C. Cancer: Evolution within a lifetime. Annu. Rev. Genet. 2014, 48, 215–236. [Google Scholar] [CrossRef] [PubMed]
- Maley, C.C.; Aktipis, A.; Graham, T.A.; Sottoriva, A.; Boddy, A.M.; Janiszewska, M.; Silva, A.S.; Gerlinger, M.; Yuan, Y.; Pienta, K.J.; et al. Classifying the evolutionary and ecological features of neoplasms. Nat. Rev. Cancer 2017, 17, 605–619. [Google Scholar] [CrossRef] [Green Version]
- Somarelli, J.A.; Ware, K.E.; Kostadinov, R.; Robinson, J.M.; Amri, H.; Abu-Asab, M.; Fourie, N.; Diogo, R.; Swofford, D.; Townsend, J.P. Phylooncology: Understanding cancer through phylogenetic analysis. Biochim. Biophys. Acta 2017, 1867, 101–108. [Google Scholar] [CrossRef]
- Somarelli, J.A.; Shetler, S.; Jolly, M.K.; Wang, X.; Bartholf Dewitt, S.; Hish, A.J.; Gilja, S.; Eward, W.C.; Ware, K.E.; Levine, H.; et al. Mesenchymal-epithelial transition in sarcomas is controlled by the combinatorial expression of MicroRNA 200s and GRHL2. Mol. Cell. Biol. 2016, 36, 2503–2513. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Hillis, D.M.; Bull, J.J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Wiens, J.J. Polymorphic characters in phylogenetic systematics. Syst. Biol. 1995, 44, 482–500. [Google Scholar] [CrossRef]
- Hillis, D.M.; Huelsenbeck, J.P.; Cunningham, C.W. Application and accuracy of molecular phylogenies. Science 1994, 264, 671–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, E.; Claude, J.; Strimmer, K. Ape: Analyses of phylogenetics and evolution in R language. Bioinformatics 2004, 20, 289–290. [Google Scholar] [CrossRef]
- Schliep, K.P. Phangorn: Phylogenetic analysis in R. Bioinformatics 2011, 27, 592–593. [Google Scholar] [CrossRef] [PubMed]
- Berriz, G.F.; King, O.D.; Bryant, B.; Sander, C.; Roth, F.P. Characterizing gene sets with funcassociate. Bioinformatics 2003, 19, 2502–2504. [Google Scholar] [CrossRef] [PubMed]
- Kogan-Sakin, I.; Tabach, Y.; Buganim, Y.; Molchadsky, A.; Solomon, H.; Madar, S.; Kamer, I.; Stambolsky, P.; Shelly, A.; Goldfinger, N.; et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell. Death Differ. 2011, 18, 271–281. [Google Scholar] [CrossRef]
- Sartor, M.A.; Mahavisno, V.; Keshamouni, V.G.; Cavalcoli, J.; Wright, Z.; Karnovsky, A.; Kuick, R.; Jagadish, H.V.; Mirel, B.; Weymouth, T.; et al. ConceptGen: A gene set enrichment and gene set relation mapping tool. Bioinformatics 2010, 26, 456–463. [Google Scholar] [CrossRef]
- Takahashi, E.; Nagano, O.; Ishimoto, T.; Yae, T.; Suzuki, Y.; Shinoda, T.; Nakamura, S.; Niwa, S.; Ikeda, S.; Koga, H.; et al. Tumor necrosis factor-alpha regulates transforming growth factor-beta-dependent epithelial-mesenchymal transition by promoting hyaluronan-CD44-moesin interaction. J. Biol. Chem. 2010, 285, 4060–4073. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T.; et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef] [Green Version]
- Vega, S.; Morales, A.V.; Ocana, O.H.; Valdes, F.; Fabregat, I.; Nieto, M.A. Snail blocks the cell cycle and confers resistance to cell death. Genes Dev. 2004, 18, 1131–1143. [Google Scholar] [CrossRef] [Green Version]
- Cheong, H.; Lindsten, T.; Thompson, C.B. Autophagy and ammonia. Autophagy 2012, 8, 122–123. [Google Scholar] [CrossRef] [Green Version]
- Du, B.; Shim, J.S. Targeting epithelial-mesenchymal transition (EMT) to overcome drug resistance in cancer. Molecules 2016, 21. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. Emt, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef]
- Fischer, K.R.; Durrans, A.; Lee, S.; Sheng, J.; Li, F.; Wong, S.T.; Choi, H.; El Rayes, T.; Ryu, S.; Troeger, J.; et al. Epithelial-to-mesenchymal transition is not required for lung metastasis but contributes to chemoresistance. Nature 2015, 527, 472–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; Le Bleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sui, X.; Chen, R.; Wang, Z.; Huang, Z.; Kong, N.; Zhang, M.; Han, W.; Lou, F.; Yang, J.; Zhang, Q.; et al. Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell. Death Dis 2013, 4, e838. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.J.; Chee, C.E.; Huang, S.; Sinicrope, F.A. The role of autophagy in cancer: Therapeutic implications. Mol. Cancer Ther. 2011, 10, 1533–1541. [Google Scholar] [CrossRef] [PubMed]
- Schiller, J.H.; Larson, T.; Ou, S.H.; Limentani, S.; Sandler, A.; Vokes, E.; Kim, S.; Liau, K.; Bycott, P.; Olszanski, A.J.; et al. Efficacy and safety of axitinib in patients with advanced non-small-cell lung cancer: Results from a phase ii study. J. Clin. Oncol. 2009, 27, 3836–3841. [Google Scholar] [CrossRef] [PubMed]
- Mross, K.; Frost, A.; Steinbild, S.; Hedbom, S.; Buchert, M.; Fasol, U.; Unger, C.; Kratzschmar, J.; Heinig, R.; Boix, O.; et al. A phase I dose-escalation study of regorafenib (BAY 73-4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin. Cancer Res. 2012, 18, 2658–2667. [Google Scholar] [CrossRef]
- Neal, J.W.; Dahlberg, S.E.; Wakelee, H.A.; Aisner, S.C.; Bowden, M.; Huang, Y.; Carbone, D.P.; Gerstner, G.J.; Lerner, R.E.; Rubin, J.L.; et al. Erlotinib, cabozantinib, or erlotinib plus cabozantinib as second-line or third-line treatment of patients with EGFR wild-type advanced non-small-cell lung cancer (ECOG-ACRIN 1512): A randomised, controlled, open-label, multicentre, phase 2 trial. Lancet Oncol. 2016, 17, 1661–1671. [Google Scholar] [CrossRef]
- Nowell, P.C. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, K.A.; Barber, L.J.; Davies, M.N.; Ashenden, M.; Sottoriva, A.; Gerlinger, M. Cancer evolution and the limits of predictability in precision cancer medicine. Trends Cancer 2016, 2, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, A.P. Epigenetic stochasticity, nuclear structure and cancer: The implications for medicine. J. Intern. Med. 2014, 276, 5–11. [Google Scholar] [CrossRef]
- Ostrow, S.L.; Barshir, R.; DeGregori, J.; Yeger-Lotem, E.; Hershberg, R. Cancer evolution is associated with pervasive positive selection on globally expressed genes. PLoS Genet. 2014, 10, e1004239. [Google Scholar] [CrossRef] [PubMed]
- Casasent, A.K.; Edgerton, M.; Navin, N.E. Genome evolution in ductal carcinoma in situ: Invasion of the clones. J. Pathol. 2017, 241, 208–218. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Huysentruyt, L.C. On the origin of cancer metastasis. Crit. Rev. Oncog. 2013, 18, 43–73. [Google Scholar] [CrossRef]
- Gatenby, R.; Brown, J. The evolution and ecology of resistance in cancer therapy. Cold Spring Harb. Perspect. Med. 2018, 8. [Google Scholar] [CrossRef]
- Kurtz, I.; Dass, P.D.; Cramer, S. The importance of renal ammonia metabolism to whole body acid-base balance: A reanalysis of the pathophysiology of renal tubular acidosis. Miner. Electrolyte Metab. 1990, 16, 331–340. [Google Scholar]
- Eng, C.H.; Yu, K.; Lucas, J.; White, E.; Abraham, R.T. Ammonia derived from glutaminolysis is a diffusible regulator of autophagy. Sci. Signal. 2010, 3. [Google Scholar] [CrossRef]
- Guido, C.; Whitaker-Menezes, D.; Capparelli, C.; Balliet, R.; Lin, Z.; Pestell, R.G.; Howell, A.; Aquila, S.; Ando, S.; Martinez-Outschoorn, U.; et al. Metabolic reprogramming of cancer-associated fibroblasts by TGF-β drives tumor growth: Connecting TGF-β signaling with "warburg-like" cancer metabolism and l-lactate production. Cell Cycle 2012, 11, 3019–3035. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Zhang, X.L.; Bitzer, M.; von Gersdorff, G.; Bottinger, E.P.; Lisanti, M.P. Caveolin-1 regulates transforming growth factor TGF-β /SMAD signaling through an interaction with the TGF-β type I receptor. J. Biol. Chem. 2001, 276, 6727–6738. [Google Scholar] [CrossRef] [PubMed]
- Shiroto, T.; Romero, N.; Sugiyama, T.; Sartoretto, J.L.; Kalwa, H.; Yan, Z.; Shimokawa, H.; Michel, T. Caveolin-1 is a critical determinant of autophagy, metabolic switching, and oxidative stress in vascular endothelium. PLoS ONE 2014, 9, e87871. [Google Scholar] [CrossRef]
- Pasquini, G.; Giaccone, G. C-met inhibitors for advanced non-small cell lung cancer. Expert Opin. Investig. Drugs 2018, 27, 363–375. [Google Scholar] [CrossRef]
- Rastogi, I.; Rajanna, S.; Webb, A.; Chhabra, G.; Foster, B.; Webb, B.; Puri, N. Mechanism of c-met and EGFR tyrosine kinase inhibitor resistance through epithelial mesenchymal transition in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2016, 477, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Barrow-McGee, R.; Kishi, N.; Joffre, C.; Menard, L.; Hervieu, A.; Bakhouche, B.A.; Noval, A.J.; Mai, A.; Guzman, C.; Robert-Masson, L.; et al. Beta 1-integrin-c-met cooperation reveals an inside-in survival signalling on autophagy-related endomembranes. Nat. Commun. 2016, 7, 11942. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, S.; Ware, K.E.; Ding, Y.; Kim, S.Y.; Sheth, M.U.; Rao, S.; Chan, W.; Armstrong, A.J.; Eward, W.C.; Jolly, M.K.; et al. An Integrative Systems Biology and Experimental Approach Identifies Convergence of Epithelial Plasticity, Metabolism, and Autophagy to Promote Chemoresistance. J. Clin. Med. 2019, 8, 205. https://doi.org/10.3390/jcm8020205
Xu S, Ware KE, Ding Y, Kim SY, Sheth MU, Rao S, Chan W, Armstrong AJ, Eward WC, Jolly MK, et al. An Integrative Systems Biology and Experimental Approach Identifies Convergence of Epithelial Plasticity, Metabolism, and Autophagy to Promote Chemoresistance. Journal of Clinical Medicine. 2019; 8(2):205. https://doi.org/10.3390/jcm8020205
Chicago/Turabian StyleXu, Shengnan, Kathryn E. Ware, Yuantong Ding, So Young Kim, Maya U. Sheth, Sneha Rao, Wesley Chan, Andrew J. Armstrong, William C. Eward, Mohit Kumar Jolly, and et al. 2019. "An Integrative Systems Biology and Experimental Approach Identifies Convergence of Epithelial Plasticity, Metabolism, and Autophagy to Promote Chemoresistance" Journal of Clinical Medicine 8, no. 2: 205. https://doi.org/10.3390/jcm8020205
APA StyleXu, S., Ware, K. E., Ding, Y., Kim, S. Y., Sheth, M. U., Rao, S., Chan, W., Armstrong, A. J., Eward, W. C., Jolly, M. K., & Somarelli, J. A. (2019). An Integrative Systems Biology and Experimental Approach Identifies Convergence of Epithelial Plasticity, Metabolism, and Autophagy to Promote Chemoresistance. Journal of Clinical Medicine, 8(2), 205. https://doi.org/10.3390/jcm8020205