Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene

 , ,
, ,  ,
, 
Abstract
:
1. Introduction
2. Material and Methods
2.1. Participants
2.2. DNA and RNA Samples Extraction
2.3. CNV Analysis
2.4. CNV Characterisation and Segregation Analysis
2.5. Gene Expression Analysis
2.6. Western Blot
2.7. Whole Exome Analysis
3. Results
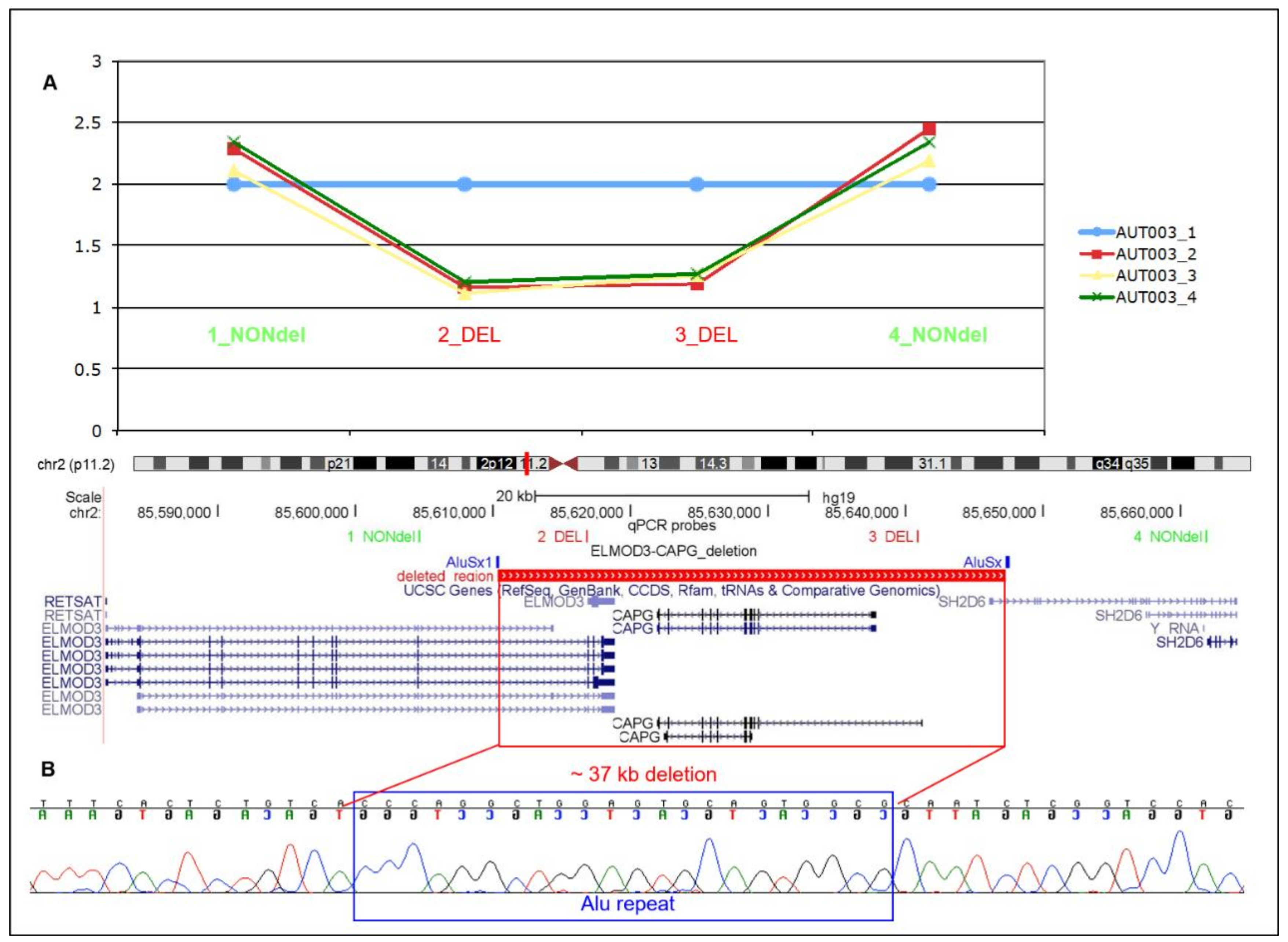
3.1. Identification and Characterisation of CAPG Deletion in Family AUT003
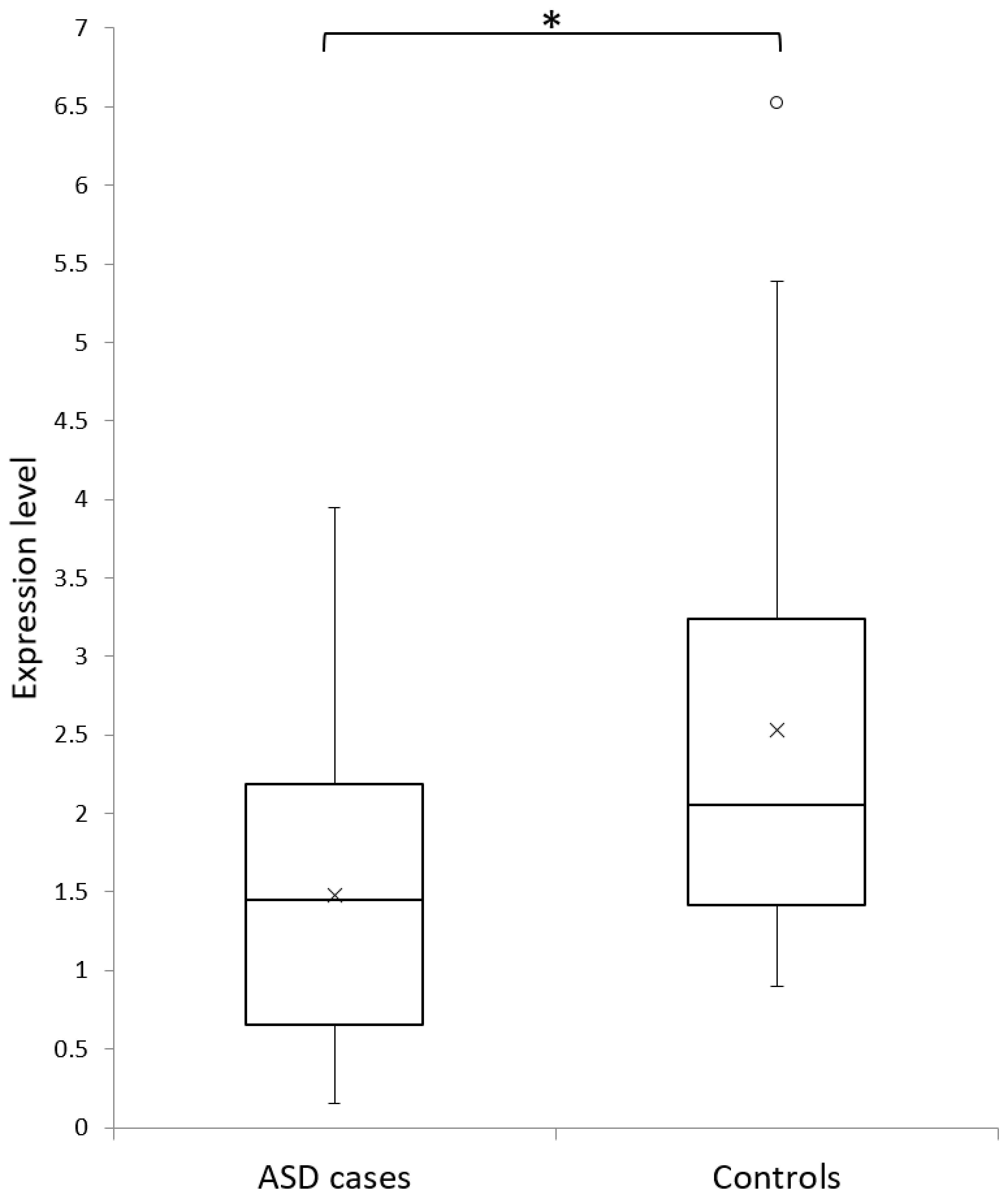
3.2. ELMOD3-CAPG-SH2D6 Deletion Frequency in ASD Cases and Controls
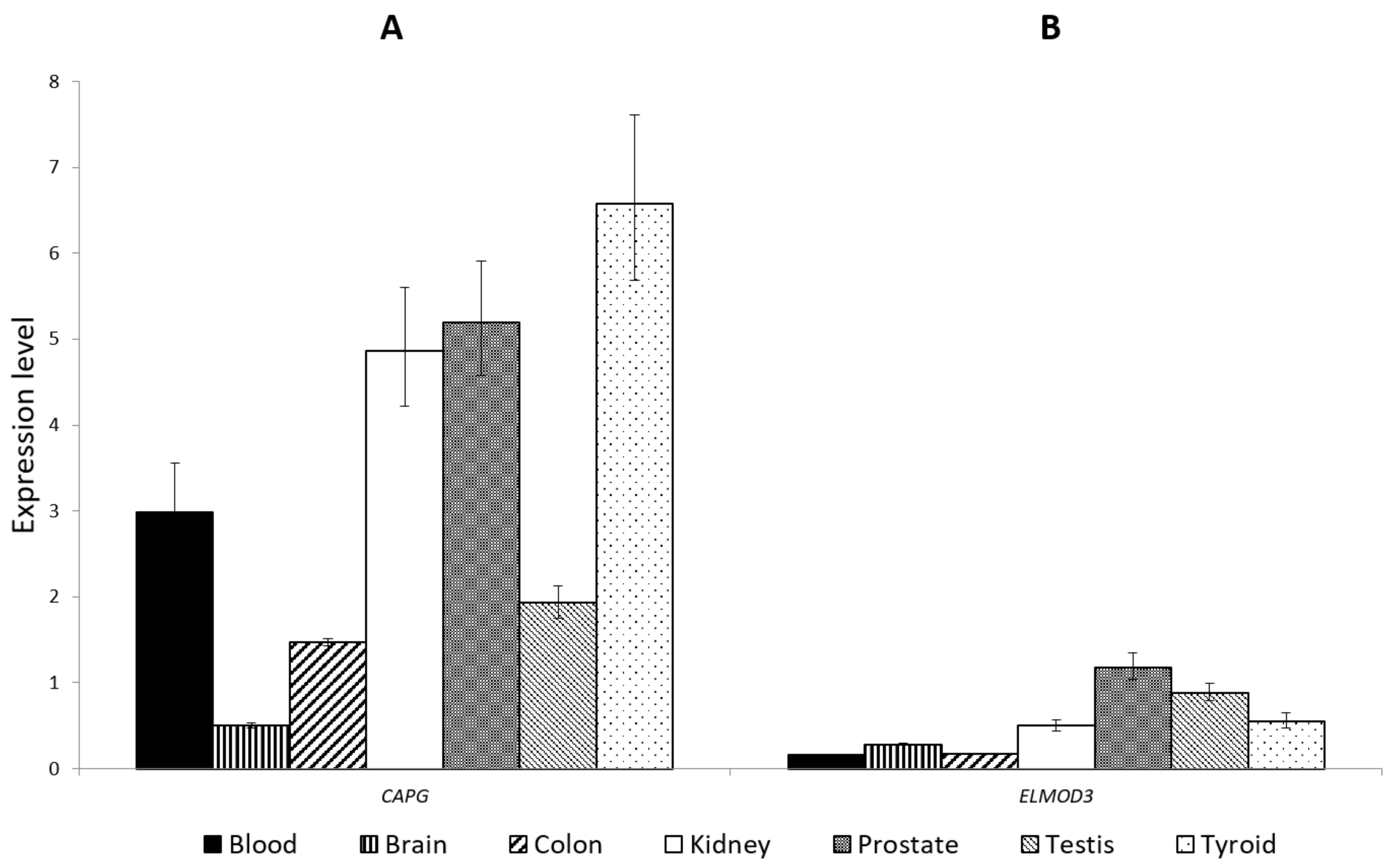
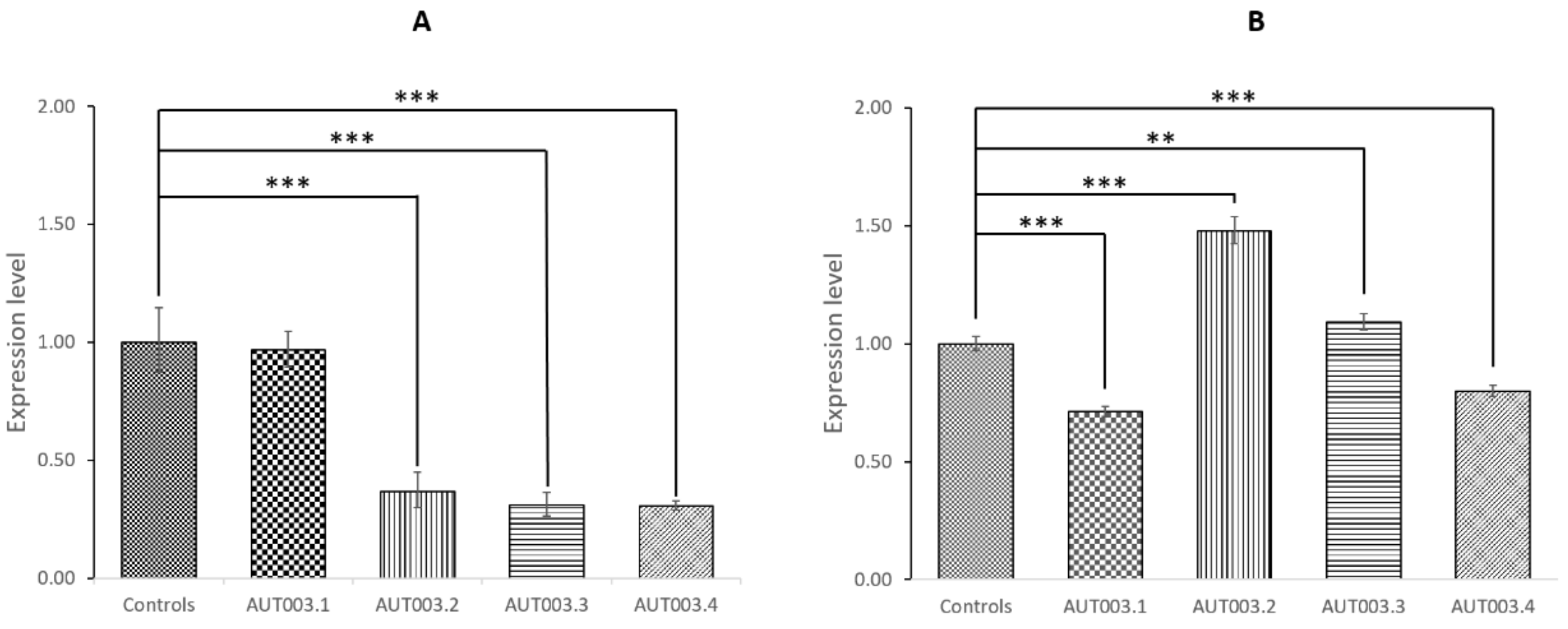
3.3. CAPG and ELMOD3 Gene Expression Analysis
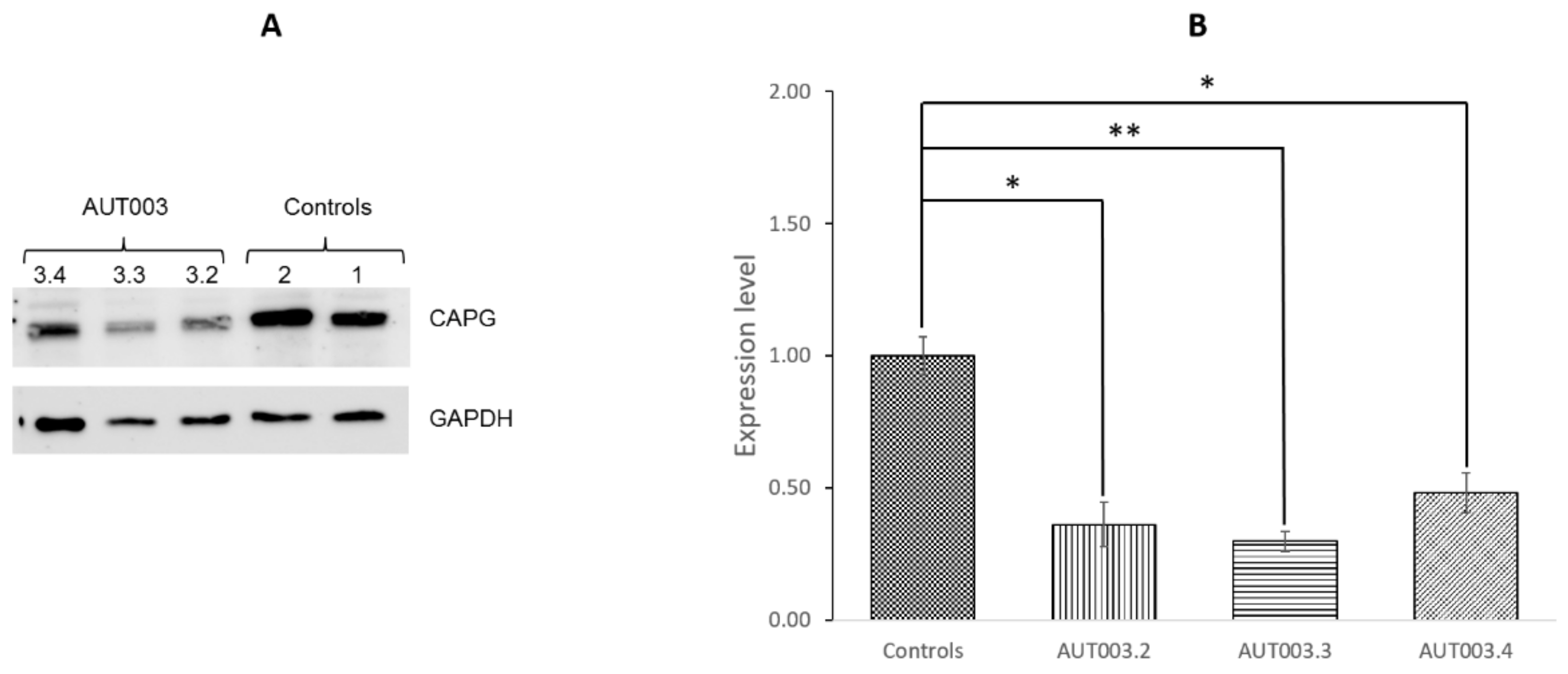
3.4. CAPG Protein Expression Analysis
3.5. Whole Exome Sequencing
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schaefer, G.B.; Mendelsohn, N.J. Professional practice and guidelines committee clinical genetics evaluation in identifying the etiology of autism spectrum disorders: 2013 guideline revisions. Genet. Med. 2013, 15, 399–407. [Google Scholar] [CrossRef] [PubMed]
- Caglayan, A.O. Genetic causes of syndromic and non-syndromic autism. Dev. Med. Child Neurol. 2010, 52, 130–138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Fishawy, P.; State, M.W. The genetics of autism: Key issues, recent findings and clinical implications. Psychiatry Clin North Am. 2011, 33, 83–105. [Google Scholar] [CrossRef] [PubMed]
- Geschwind, D.H. Genetics of autism spectrum disorders. Trends Cogn. Sci. 2011, 15, 409–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Health Organization: WHO. Available online: https://www.who.int/news-room/fact-sheets/detail/autism-spectrum-disorders (accessed on 7 December 2018).
- Loomes, R.; Hull, L.; Mandy, W.P.L. What is the male-to-female ratio in autism spectrum disorder? A systematic review and meta-analysis. J. Am. Acad. Child Adolesc. Psychiatry 2017, 56, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Yin, J.; Schaaf, C.P. Autism genetics—An overview. Prenat. Diagn. 2016, 5, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Toro, R.; Konyukh, M.; Delorme, R.; Leblond, C.; Chaste, P.; Fauchereau, F.; Coleman, M.; Leboyer, M.; Gillberg, C.; Bourgeron, T. Key role for gene dosage and synaptic homeostasis in autism spectrum disorders. Trends Genet. 2010, 26, 363–372. [Google Scholar] [CrossRef]
- Gilman, S.R.; Iossifov, I.; Levy, D.; Ronemus, M.; Wigler, M.; Vitkup, D. Rare de novo variants associated with autism implicate a large functional network of genes involved in formation and function of synapses. Neuron 2011, 70, 898–907. [Google Scholar] [CrossRef]
- Zavattari, P.; Lampis, R.; Mulargia, A.; Loddo, M.; Angius, E.; Todd, J.A.; Cucca, F. Confirmation of the DRB1-DQB1 loci as the major component of IDDM1 in the isolated founder population of Sardinia. Hum. Mol. Genet. 2000, 9, 2967–2972. [Google Scholar] [CrossRef] [Green Version]
- Contu, D.; Morelli, L.; Zavattari, P.; Lampis, R.; Angius, E.; Frongia, P.; Murru, D.; Maioli, M.; Francalacci, P.; Todd, J.A.; et al. Sex-related bias and exclusion mapping of the nonrecombinant portion of chromosome Y in human type 1 diabetes in the isolated founder population of Sardinia. Diabetes 2002, 51, 3573–3576. [Google Scholar] [CrossRef]
- Pitzalis, M.; Zavattari, P.; Murru, R.; Deidda, E.; Zoledziewska, M.; Murru, D.; Moi, L.; Motzo, C.; Orrù, V.; Costa, G.; et al. Genetic loci linked to Type 1 Diabetes and Multiple Sclerosis families in Sardinia. BMC Med. Genet. 2008, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Zavattari, P.; Loche, A.; Civolani, P.; Pilia, S.; Moi, L.; Casini, M.R.; Minerba, L.; Loche, S. An INSIG2 polymorphism affects glucose homeostasis in Sardinian obese children and adolescents. Ann. Hum. Genet. 2010, 74, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Zavattari, P.; Loche, A.; Pilia, S.; Ibba, A.; Moi, L.; Guzzetti, C.; Casini, M.R.; Loche, S. rs9939609 in the FTO gene is associated with obesity but not with several biochemical parameters in Sardinian obese children. Ann. Hum. Genet. 2011, 75, 648–654. [Google Scholar] [CrossRef] [PubMed]
- Loudianos, G.; Dessi, V.; Lovicu, M.; Angius, A.; Cao, A.; Pirastu, M. The -75A-->C substitution in the 5′ UTR of the Wilson disease gene is a sequence polymorphism in the Mediterranean population. Am. J. Hum. Genet. 1998, 62, 484–485. [Google Scholar] [CrossRef]
- Crisponi, L.; Crisponi, G.; Meloni, A.; Toliat, M.R.; Nürnberg, G.; Usala, G.; Uda, M.; Masala, M.; Höhne, W.; Becker, C.; et al. Crisponi syndrome is caused by mutations in the CRLF1 gene and is allelic to cold-induced sweating syndrome Type 1. Am. J. Hum. Genet. 2007, 80, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Tammimies, K.; Marshall, C.R.; Walker, S.; Kaur, G.; Thiruvahindrapuram, B.; Lionel, A.C.; Yuen, R.K.C.; Uddin, M.; Roberts, W.; Weksberg, R.; et al. Molecular diagnostic yield of chromosomal microarray analysis and whole-exome sequencing in children with autism spectrum disorder. JAMA 2015, 314, 895. [Google Scholar] [CrossRef] [PubMed]
- Lord, C.; Rutter, M.; DiLavore, P.C.; Risi, S.; Gotham, K.B.S. (ADOS-2) Manual (Part I): Modules 1–4. In Autism Diagnostic Observation Schedule, 2nd ed.; Western Psychological Services: Torrance, CA, USA, 2012. [Google Scholar]
- Guo, Y.; He, J.; Zhao, S.; Wu, H.; Zhong, X.; Sheng, Q.; Samuels, D.C.; Shyr, Y.; Long, J. Illumina human exome genotyping array clustering and quality control. Nat. Protoc. 2014, 9, 2643–2662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, K.; Li, M.; Hadley, D.; Liu, R.; Glessner, J.; Grant, S.F.A.; Hakonarson, H.; Bucan, M. PennCNV: An integrated hidden Markov model designed for high-resolution copy number variation detection in whole-genome SNP genotyping data. Genome Res. 2007, 17, 1665–1674. [Google Scholar] [CrossRef] [Green Version]
- Colella, S.; Yau, C.; Taylor, J.M.; Mirza, G.; Butler, H.; Clouston, P.; Bassett, A.S.; Seller, A.; Holmes, C.C.; Ragoussis, J. QuantiSNP: An objective bayes hidden-markov model to detect and accurately map copy number variation using SNP genotyping data. Nucleic Acids Res. 2007, 35, 2013–2025. [Google Scholar] [CrossRef]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Chiara, M.; Gioiosa, S.; Chillemi, G.; D’Antonio, M.; Flati, T.; Picardi, E.; Zambelli, F.; Horner, D.S.; Pesole, G.; Castrignanò, T. CoVaCS: A consensus variant calling system. BMC Genomics 2018, 19, 120. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef] [PubMed]
- Genome aggregation database (gnomAD). Available online: http://gnomad.broadinstitute.org/ (accessed on 8 February 2018).
- 1000 Genomes Project. Available online: http://www.1000genomes.org/ (accessed on 8 February 2018).
- Yuen, R.K.; Merico, D.; Cao, H.; Pellecchia, G.; Alipanahi, B.; Thiruvahindrapuram, B.; Tong, X.; Sun, Y.; Cao, D.; Zhang, T.; et al. Genome-wide characteristics of de novo mutations in autism. NPJ Genomic Med. 2016, 1, 16027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jansen, A.; Dieleman, G.C.; Smit, A.B.; Verhage, M.; Verhulst, F.C.; Polderman, T.J.C.; Posthuma, D. Gene-set analysis shows association between FMRP targets and autism spectrum disorder. Eur. J. Hum. Genet. 2017, 25, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Bayés, A.; van de Lagemaat, L.N.; Collins, M.O.; Croning, M.D.R.; Whittle, I.R.; Choudhary, J.S.; Grant, S.G.N. Characterization of the proteome, diseases and evolution of the human postsynaptic density. Nat. Neurosci. 2011, 14, 19–21. [Google Scholar] [CrossRef] [PubMed]
- Pinto, D.; Delaby, E.; Merico, D.; Barbosa, M.; Merikangas, A.; Klei, L.; Thiruvahindrapuram, B.; Xu, X.; Ziman, R.; Wang, Z.; et al. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am. J. Hum. Genet. 2014, 94, 677–694. [Google Scholar] [CrossRef] [PubMed]
- Lahbib, S.; Leblond, C.S.; Hamza, M.; Regnault, B.; Lemée, L.; Mathieu, A.; Jaouadi, H.; Mkaouar, R.; Youssef-Turki, I.; Belhadj, A.B.; et al. Homozygous 2p11.2 deletion supports the implication of ELMOD3 in hearing loss and reveals the potential association of CAPG with ASD/ID etiology. J. Appl. Genet. 2018, 60, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Bierut, L.J.; Agrawal, A.; Bucholz, K.K.; Doheny, K.F.; Laurie, C.; Pugh, E.; Fisher, S.; Fox, L.; Howells, W.; Bertelsen, S.; et al. A genome-wide association study of alcohol dependence. Proc. Natl. Acad. Sci. USA 2010, 107, 5082–5087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krawczak, M.; Nikolaus, S.; von Eberstein, H.; Croucher, P.J.P.; El Mokhtari, N.E.; Schreiber, S. PopGen: Population-based recruitment of patients and controls for the analysis of complex genotype-phenotype relationships. Public Health Genomics 2006, 9, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, T.H.; Gai, X.; Perin, J.C.; Glessner, J.T.; Xie, H.; Murphy, K.; O’Hara, R.; Casalunovo, T.; Conlin, L.K.; D’Arcy, M.; et al. High-resolution mapping and analysis of copy number variations in the human genome: A data resource for clinical and research applications. Genome Res. 2009, 19, 1682–1690. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, A.F.R.; Dandona, S.; Chen, L.; Assogba, O.; Belanger, M.; Ewart, G.; LaRose, R.; Doelle, H.; Williams, K.; Wells, G.A.; et al. Kinesin family member 6 variant Trp719Arg does not associate with angiographically defined coronary artery disease in the Ottawa heart genomics study. J. Am. Coll. Cardiol. 2009, 53, 1471–1472. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.C.; Lewinger, J.P.; Song, C.; Campbell, P.T.; Conti, D.V.; Edlund, C.K.; Duggan, D.J.; Rangrej, J.; Lemire, M.; Hudson, T.; et al. Genotype-environment interactions in microsatellite stable/microsatellite instability-low colorectal cancer: Results from a genome-wide association study. Cancer Epidemiol. Biomark. Prev. 2011, 20, 758–766. [Google Scholar] [CrossRef] [PubMed]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Y.; Tang, X.; Vitriol, E.; Chen, G.; Zheng, J.Q. Actin capping protein is required for dendritic spine development and synapse formation. J. Neurosci. 2011, 31, 10228–10233. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, M.; Hoogenraad, C.C. Synapse pathology in psychiatric and neurologic disease. Curr. Neurol. Neurosci. Rep. 2010, 10, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Korobova, F.; Svitkina, T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell 2010, 21, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Bar-shira, O.; Chechik, G. Predicting protein–protein interactions in the post synaptic density. Mol. Cell. Neurosci. 2013, 56, 128–139. [Google Scholar] [CrossRef]
- Ehrhart, F.; Coort, S.L.; Eijssen, L.; Cirillo, E.; Smeets, E.; Sangani, N.B.; Evelo, C.; Curfs, L.M.G. Integrated analysis of human transcriptome data for Rett syndrome finds a network of involved genes. BioRxiv 2018. [Google Scholar]
- Matarazzo, V.; Cohen, D.; Palmer, A.M.; Simpson, P.J.; Khokhar, B.; Pan, S.-J.; Ronnett, G.V. The transcriptional repressor Mecp2 regulates terminal neuronal differentiation. Mol. Cell. Neurosci. 2004, 27, 44–58. [Google Scholar] [CrossRef]
- Gonzalez-Gronow, M.; Cuchacovich, M.; Francos, R.; Cuchacovich, S.; del Fernandez, M.P.; Blanco, A.; Bowers, E.V.; Kaczowka, S.; Pizzo, S.V. Antibodies against the voltage-dependent anion channel (VDAC) and its protective ligand hexokinase-I in children with autism. J. Neuroimmunol. 2010, 227, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Gvozdjáková, A.; Low, H.; Sun, I.; Navas, P.; Crane, F. Plasma membrane coenzyme Q: Evidence for a role in autism. Biol. Targets Ther. 2014, 8, 199. [Google Scholar] [CrossRef] [PubMed]
- Thinnes, F.P. Plasmalemmal VDAC-1 corroborated as amyloid Aß-receptor. Front. Aging Neurosci. 2015, 7, 188. [Google Scholar] [CrossRef] [PubMed]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G.; et al. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suda, S.; Iwata, K.; Shimmura, C.; Kameno, Y.; Anitha, A.; Thanseem, I.; Nakamura, K.; Matsuzaki, H.; Tsuchiya, K.J.; Sugihara, G.; et al. Decreased expression of axon-guidance receptors in the anterior cingulate cortex in autism. Mol. Autism 2011, 2, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mah, S.; Nelson, M.R.; DeLisi, L.E.; Reneland, R.H.; Markward, N.; James, M.R.; Nyholt, D.R.; Hayward, N.; Handoko, H.; Mowry, B.; et al. Identification of the semaphorin receptor PLXNA2 as a candidate for susceptibility to schizophrenia. Mol. Psychiatry 2006, 11, 471–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.-F.; Kohen, R.; Parent, R.; Duan, Y.; Fisher, G.L.; Korn, M.J.; Ji, L.; Wan, G.; Jin, J.; Püschel, A.W.; et al. PlexinA2 forward signaling through Rap1 GTPases regulates dentate gyrus development and schizophrenia-like behaviors. Cell Rep. 2018, 22, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Lilja, J.; Zacharchenko, T.; Georgiadou, M.; Jacquemet, G.; Franceschi, N.D.; Peuhu, E.; Hamidi, H.; Pouwels, J.; Martens, V.; Nia, F.H.; et al. SHANK proteins limit integrin activation by directly interacting with Rap1 and R-Ras. Nat. Cell Biol. 2017, 19, 292–305. [Google Scholar] [CrossRef]
- Booker, S.A.; Althof, D.; Gross, A.; Loreth, D.; Müller, J.; Unger, A.; Fakler, B.; Varro, A.; Watanabe, M.; Gassmann, M.; et al. KCTD12 auxiliary proteins modulate kinetics of GABA B receptor-mediated inhibition in cholecystokinin-containing interneurons. Cereb. Cortex 2016, 27, bhw090. [Google Scholar] [CrossRef]
- Fritzius, T.; Turecek, R.; Seddik, R.; Kobayashi, H.; Tiao, J.; Rem, P.D.; Metz, M.; Kralikova, M.; Bouvier, M.; Gassmann, M.; et al. KCTD hetero-oligomers confer unique kinetic properties on hippocampal GABAB receptor-induced K+ Currents. J. Neurosci. 2017, 37, 1162–1175. [Google Scholar] [CrossRef]
- Schwenk, J.; Pérez-Garci, E.; Schneider, A.; Kollewe, A.; Gauthier-Kemper, A.; Fritzius, T.; Raveh, A.; Dinamarca, M.C.; Hanuschkin, A.; Bildl, W.; et al. Modular composition and dynamics of native GABAB receptors identified by high-resolution proteomics. Nat. Neurosci. 2016, 19, 233–242. [Google Scholar] [CrossRef]
- Schwenk, J.; Metz, M.; Zolles, G.; Turecek, R.; Fritzius, T.; Bildl, W.; Tarusawa, E.; Kulik, A.; Unger, A.; Ivankova, K.; et al. Native GABAB receptors are heteromultimers with a family of auxiliary subunits. Nature 2010, 465, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Angelicheva, D.; Tournev, I.; Guergueltcheva, V.; Mihaylova, V.; Azmanov, D.N.; Morar, B.; Radionova, M.; Smith, S.J.; Zlatareva, D.; Stevens, J.M.; et al. Partial epilepsy syndrome in a Gypsy family linked to 5q31.3-q32. Epilepsia 2009, 50, 1679–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hentschke, M.; Wiemann, M.; Hentschke, S.; Kurth, I.; Hermans-Borgmeyer, I.; Seidenbecher, T.; Jentsch, T.J.; Gal, A.; Hübner, C.A. Mice with a targeted disruption of the Cl-/HCO3- exchanger AE3 display a reduced seizure threshold. Mol. Cell. Biol. 2006, 26, 182–191. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.; Ruusuvuori, E.; Sipila, S.T.; Haapanen, A.; Damkier, H.H.; Kurth, I.; Hentschke, M.; Schweizer, M.; Rudhard, Y.; Laatikainen, L.M.; et al. Mice with targeted Slc4a10 gene disruption have small brain ventricles and show reduced neuronal excitability. Proc. Natl. Acad. Sci. USA 2008, 105, 311–316. [Google Scholar] [CrossRef] [PubMed]
- Sinning, A.; Liebmann, L.; Hübner, C.A. Disruption of Slc4a10 augments neuronal excitability and modulates synaptic short-term plasticity. Front. Cell. Neurosci. 2015, 9, 223. [Google Scholar] [CrossRef]
- Sinning, A.; Liebmann, L.; Kougioumtzes, A.; Westermann, M.; Bruehl, C.; Hübner, C.A. Synaptic glutamate release is modulated by the Na+ -driven Cl-/HCO₃− exchanger Slc4a8. J. Neurosci. 2011, 31, 7300–7311. [Google Scholar] [CrossRef]
- Sander, T.; Toliat, M.R.; Heils, A.; Leschik, G.; Becker, C.; Rüschendorf, F.; Rohde, K.; Mundlos, S.; Nürnberg, P. Association of the 867Asp variant of the human anion exchanger 3 gene with common subtypes of idiopathic generalized epilepsy. Epilepsy Res. 2002, 51, 249–255. [Google Scholar] [CrossRef]
- Demirci, F.Y.K.; Chang, M.-H.; Mah, T.S.; Romero, M.F.; Gorin, M.B. Proximal renal tubular acidosis and ocular pathology: A novel missense mutation in the gene (SLC4A4) for sodium bicarbonate cotransporter protein (NBCe1). Mol. Vis. 2006, 12, 324–330. [Google Scholar]
- Sebat, J.; Lakshmi, B.; Malhotra, D.; Troge, J.; Lese-Martin, C.; Walsh, T.; Yamrom, B.; Yoon, S.; Krasnitz, A.; Kendall, J.; et al. Strong association of de novo copy number mutations with autism. Science 2007, 316, 445–449. [Google Scholar] [CrossRef]
- Gurnett, C.A.; Veile, R.; Zempel, J.; Blackburn, L.; Lovett, M.; Bowcock, A. Disruption of sodium bicarbonate transporter SLC4A10 in a patient with complex partial epilepsy and mental retardation. Arch. Neurol. 2008, 65, 550. [Google Scholar] [CrossRef]
- Krepischi, A.C.V.; Knijnenburg, J.; Bertola, D.R.; Kim, C.A.; Pearson, P.L.; Bijlsma, E.; Szuhai, K.; Kok, F.; Vianna-Morgante, A.M.; Rosenberg, C. Two distinct regions in 2q24.2-q24.3 associated with idiopathic epilepsy. Epilepsia 2010, 51, 2457–2460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, M.; Van Paesschen, W.; Stalmans, I.; Horita, S.; Yamada, H.; Bergmans, B.A.; Legius, E.; Riant, F.; De Jonghe, P.; Li, Y.; et al. Defective membrane expression of the Na+ -HCO3- cotransporter NBCe1 is associated with familial migraine. Proc. Natl. Acad. Sci. USA 2010, 107, 15963–15968. [Google Scholar] [CrossRef] [PubMed]
- Bassères, D.S.; Tizzei, E.V.; Duarte, A.A.; Costa, F.F.; Saad, S.T.O. ARHGAP10, a novel human gene coding for a potentially cytoskeletal Rho-GTPase activating protein. Biochem. Biophys. Res. Commun. 2002, 294, 579–585. [Google Scholar] [CrossRef]
- Dubois, T.; Paléotti, O.; Mironov, A.A.; Fraisier, V.; Stradal, T.E.B.; De Matteis, M.A.; Franco, M.; Chavrier, P. Golgi-localized GAP for Cdc42 functions downstream of ARF1 to control Arp2/3 complex and F-actin dynamics. Nat. Cell Biol. 2005, 7, 353–364. [Google Scholar] [CrossRef] [PubMed]
- Akin, O.; Mullins, R.D. Capping protein increases the rate of actin-based motility by promoting filament nucleation by the Arp2/3 complex. Cell 2008, 133, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Iwasa, J.H.; Mullins, R.D. Spatial and temporal relationships between actin-filament nucleation, capping, and disassembly. Curr. Biol. 2007, 17, 395–406. [Google Scholar] [CrossRef] [PubMed]
- Mejillano, M.R.; Kojima, S.; Applewhite, D.A.; Gertler, F.B.; Svitkina, T.M.; Borisy, G.G. Lamellipodial versus filopodial mode of the actin nanomachinery. Cell 2004, 118, 363–373. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Measure | Mother | Father |
|---|---|---|
| BAPQ Scores | ||
| Aloof | 2.4 | 3.5 |
| Pragmatic Language | 2.2 | 2.7 |
| Rigidity | 2.2 | 2.7 |
| BAPQ Total | 2.4 | 3 |
| SRS: ARV scores | ||
| Awareness | 10 | 8 |
| Mannerism | 8 | 10 |
| Communication | 9 | 21 |
| Cognition | 4 | 10 |
| Motivation | 3 | 10 |
| SRS:ARV Total | 34 | 59 |
| ADOS—IV Module Scores | ||
| Communication | 0 | 0 |
| Reciprocal Social Interaction | 0 | 0 |
| Identified Variant | pLi Score | Synaptic Genes [28,29] | Genotype | |||
|---|---|---|---|---|---|---|
| Gene Base Change | AminoAcid Change | Gene | AUT003.3 | AUT003.4 | ||
| chr1:g.19559158delA NM_001271428:c.1739delT | p.F580Sfs*10 | EMC1 | 0/1 | 0/0 | ||
| chr1:g.24779922delT NM_001322854:c.565delT | p.L189Cfs*15 | NIPAL3 | 0/1 | 0/0 | ||
| chr1:g.108771659G>A NM_001143989:c.C1543T | p.R515X | NBPF4 | 0/1 | 0/1 | ||
| chr1:g.158299688A>T NM_001764:c.T561A | p.Y187X | CD1B | 0/1 | 0/1 | ||
| chr2:g.30961265C>T NM_144575:c.1594+1G>A | CAPN13 | 0/1 | 0/0 | |||
| chr2:g.58386928-58386929insTAAT NM_001114636:c.1114_1115insATTA | p.T372Nfs*12 | FANCL | 0/1 | 0/1 | ||
| chr2:g.120439282C>T NM_001105198:c.C853T | p.R285X | TMEM177 | 0/1 | 0/1 | ||
| chr3:g.38770224G>A NM_001293306:c.C2449T | p.R817X | SCN10A | 0/0 | 0/1 | ||
| chr3:g.188327410T>A NM_001167671:c.T891A | p.Y297X | LPP | 0/0 | 0/1 | ||
| chr4:g.8235241G>A NM_001318480:c.3054+1G>A | SH3TC1 | 0/1 | 0/1 | |||
| chr6:43306235-43306238delTCTT NM_014345:c.5498_5501del | p.K1833Rfs*6 | ZNF318 | 0/0 | 0/1 | ||
| chr7:g.150028118C>T NM_138434:c.C625T | p.Q209X | ZBED6CL | 0/1 | 0/0 | ||
| chr7:g.150864372G>T NM_001098834:c.C264A | p.C88X | GBX1 | 0/0 | 0/1 | ||
| chr8:g.42259530-42259531delAT NM_001135694:c.551_552del | p.H184Rfs*5 | VDAC3 | 0.97 | PSD; Excitability | 0/1 | 0/1 |
| chr8:g.145140986delC NM_003801:c.1824delC | p.C610Afs*43 | GPAA1 | 0/0 | 0/1 | ||
| chr10:g.102240866-102240872del NM_003393:c.353_359del | p.G120Wfs*37 | WNT8B | 0/1 | 0/0 | ||
| chr10:g.123845143-123845144delCC NM_001291876:c.3128_3129del | p.P1044Tfs*4 | TACC2 | 0/0 | 0/1 | ||
| chr11:g.59577372dupC NM_017840:c.76dupG | p.A26Gfs*14 | MRPL16 | 0/1 | 0/0 | ||
| chr11:g.116701354G>A NM_000040:c.55+1G>A | APOC3 | 0/0 | 0/1 | |||
| chr12:g.25705804C>T NM_001145728:c.89+1G>A | LMNTD1 | 0/0 | 0/1 | |||
| chr12:g.54757526delC NM_020370:c.110delG | p.G37Afs*22 | GPR84 | 0/1 | 0/0 | ||
| chr12:g.64436690C>T NM_001346201:c.C610T | R204X | SRGAP1 | 0.99 | 0/1 | 0/0 | |
| chr12:g.133198246dupA NM_012226:c.889dupA | p.N298Kfs*6 | P2RX2 | 0/0 | 0/1 | ||
| chr15:g.75641682T>C NM_024608:c.434+2T>C | NEIL1 | 0/0 | 0/1 | |||
| chr16:g.107113-107114insTC NM_024571:c.369_370insTC | p.F125Pfs*5 | SNRNP25 | 0/1 | 0/1 | ||
| chr18:g.71822640delT NM_014177:c.462+2T>- | TIMM21 | 0/0 | 0/1 | |||
| chr18:g.77917844dupG NM_032510:c.940dupC | p.Q314Pfs*72 | PARD6G | 0/0 | 0/1 | ||
| chr19:g.10204081dupA NM_001321411:c.1165dupT | p.S389Ffs*4 | ANGPTL6 | 0/0 | 0/1 | ||
| chr19:g.43859812G>C NM_020406:c.380-1G>C | CD177 | 0/1 | 0/1 | |||
| chr20:g.61591931delT NM_022082:c.473delT | p.I158Tfs*11 | SLC17A9 | 0/0 | 0/1 | ||
| chr21:g.44839738C>A NM_001320643:c.1119+1G>T | SIK1 | 0.99 | 0/1 | 0/1 | ||
| chr22:g.25124143C>T NM_001255975:c.1905+1G>A | PIWIL3 | 0/0 | 0/1 | |||
| chrX:g.23019507dupT NM_182699:c.1334dupT | p.I446Nfs*35 | DDX53 | 1/1 | 1/1 | ||
| Identified Variant | pLi Score | Synaptic Genes [28,29] | Genotype | |||
|---|---|---|---|---|---|---|
| Gene Base Change | AminoAcid Change | Gene | AUT003.3 | AUT003.4 | ||
| chr1:g.109801543A>G NM_001408:c.A3800G | p.H1267R | CELSR2 | 0.9999992 | 0/1 | 0/1 | |
| chr1:g.117146504G>A NM_001007237:c.C1366T | p.R456C | IGSF3 | 0.987084919 | 0/1 | 0/1 | |
| chr1:g.162725022G>A NM_006182:c.G494A | p.R165Q | DDR2 | 0.990992372 | 0/1 | 0/1 | |
| chr1:g.208390903G>T NM_025179:c.C365A | p.S122Y | PLXNA2 | 0.993981182 | PSD | 0/1 | 0/1 |
| chr2:g.96956078C>T NM_014014:c.G2728A | p.V910I | SNRNP200 | 1 | 0/1 | 0/1 | |
| chr2:g.239975199C>T NM_006037:c.G3172A | p.A1058T | HDAC4 | 0.999989727 | 0/1 | 0/1 | |
| chr4:g.126238072T>C NM_001291285:c.T506C | p.V169A | FAT4 | 0.999999998 | 0/1 | 0/1 | |
| chr5:g.141052986C>T NM_022481:c.G954A | p.M318I | ARAP3 | 0.967505029 | 0/1 | 0/1 | |
| chr5:g.143587022G>A NM_020768:c.G745A | p.A249T | KCTD16 | 0.959972608 | PSD; Excitability | 0/1 | 0/1 |
| chr7:g.100491451G>C NM_000665:c.C403G | p.P135A | ACHE | 0.996674942 | 0/1 | 0/1 | |
| chr9:g.88938095T>A NM_001185074:c.A2201T | p.E734V | ZCCHC6 | 0.999999563 | 0/1 | 0/1 | |
| chr10:g.70726833C>A NM_001256910:c.C760A | p.H254N | DDX21 | 0.998039216 | 0/1 | 0/1 | |
| chr10:g.79581043G>A NM_004747:c.C3199T | p.R1067C | DLG5 | 0.999998542 | 0/1 | 0/1 | |
| chr10:g.79590510C>G NM_004747:c.G1870C | p.E624Q | DLG5 | 0.999998542 | 0/1 | 0/1 | |
| chr10:g.24874455G>T NM_020824:c.C4763A | p.S1588Y | ARHGAP21 | 0.999946673 | PSD | 0/1 | 0/1 |
| chr11:g.94918634T>G NM_144665:c.A548C | p.N183T | SESN3 | 0.994296083 | 0/1 | 0/1 | |
| chr11:g.114027109G>A NM_001018011:c.G1319A | p.R440Q | ZBTB16 | 0.980087306 | 0/1 | 0/1 | |
| chr12:g.112699118C>T NM_001109662:c.G2432A | p.R811H | HECTD4 | 1 | 0/1 | 0/1 | |
| chr17:g.42335485C>T NM_000342:c.G1151A | p.R384H | SLC4A1 | 0.909484295 | PSD; Excitability | 0/1 | 0/1 |
| chr19:g.50100192G>A NM_020719:c.G2600A | p.R867H | PRR12 | 0.999969577 | 0/1 | 0/1 | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bacchelli, E.; Loi, E.; Cameli, C.; Moi, L.; Vega Benedetti, A.F.; Blois, S.; Fadda, A.; Bonora, E.; Mattu, S.; Fadda, R.; et al. Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene. J. Clin. Med. 2019, 8, 212. https://doi.org/10.3390/jcm8020212
Bacchelli E, Loi E, Cameli C, Moi L, Vega Benedetti AF, Blois S, Fadda A, Bonora E, Mattu S, Fadda R, et al. Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene. Journal of Clinical Medicine. 2019; 8(2):212. https://doi.org/10.3390/jcm8020212
Chicago/Turabian StyleBacchelli, Elena, Eleonora Loi, Cinzia Cameli, Loredana Moi, Ana Florencia Vega Benedetti, Sylvain Blois, Antonio Fadda, Elena Bonora, Sandra Mattu, Roberta Fadda, and et al. 2019. "Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene" Journal of Clinical Medicine 8, no. 2: 212. https://doi.org/10.3390/jcm8020212
APA StyleBacchelli, E., Loi, E., Cameli, C., Moi, L., Vega Benedetti, A. F., Blois, S., Fadda, A., Bonora, E., Mattu, S., Fadda, R., Chessa, R., Maestrini, E., Doneddu, G., & Zavattari, P. (2019). Analysis of a Sardinian Multiplex Family with Autism Spectrum Disorder Points to Post-Synaptic Density Gene Variants and Identifies CAPG as a Functionally Relevant Candidate Gene. Journal of Clinical Medicine, 8(2), 212. https://doi.org/10.3390/jcm8020212