Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-κB Signaling Pathway after Brain Injury

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
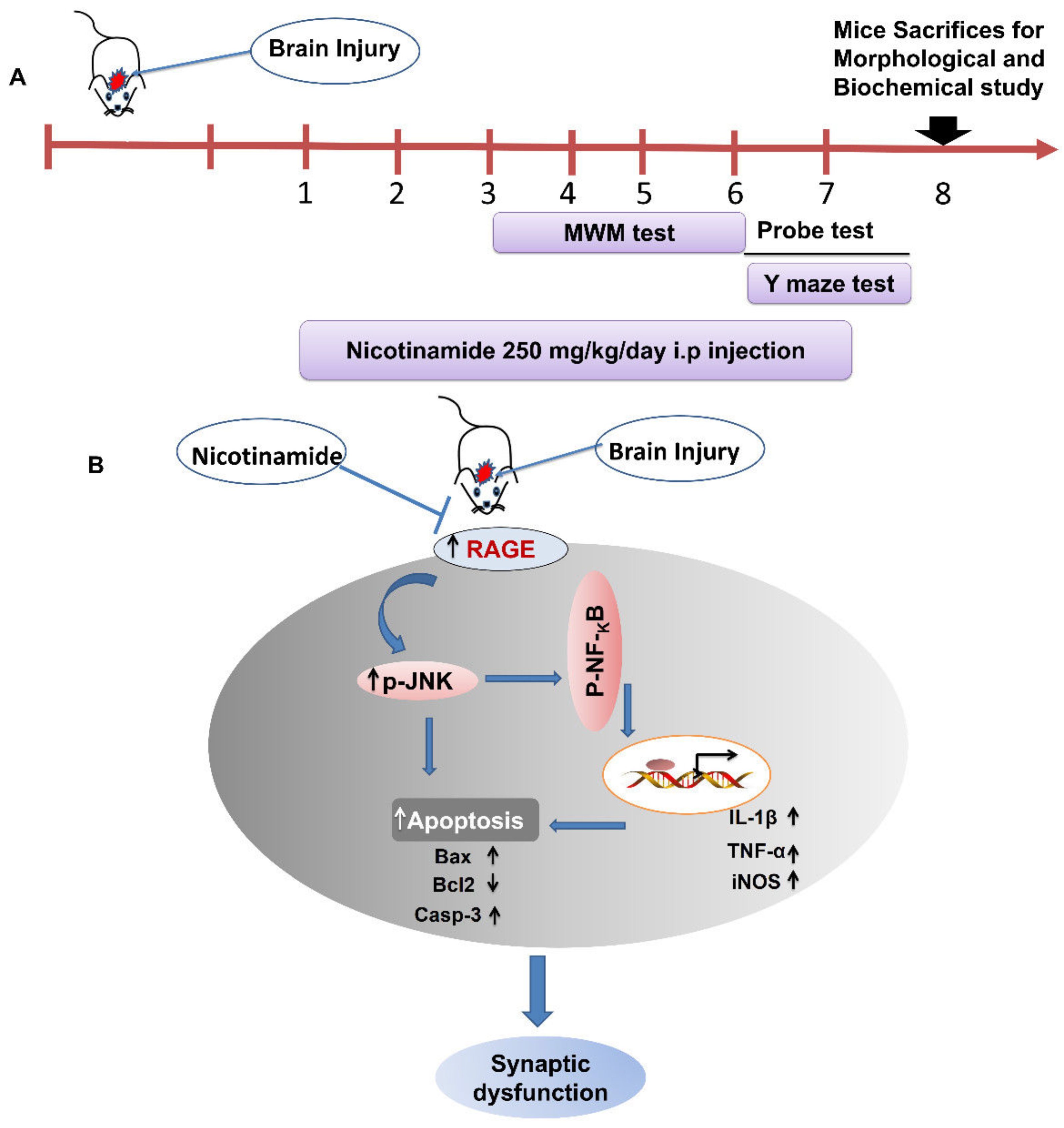
2.2. Stab Wound Cortical Injury (SWI)
2.3. Treatment
2.4. Morris Water Maze (MWM) Test
2.5. Y-maze Test
2.6. Beam Walking Test
2.7. Protein Extraction from Brain
2.8. Western Blot Analysis
2.9. Tissue Collection and Sample Preparation
2.10. Assessment of Lesion Volume
2.11. Immunofluorescence Staining
2.12. Nissl Staining
2.13. Fluoro-Jade B (FJB) Staining
2.14. Statistical Aanalysis
3. Results
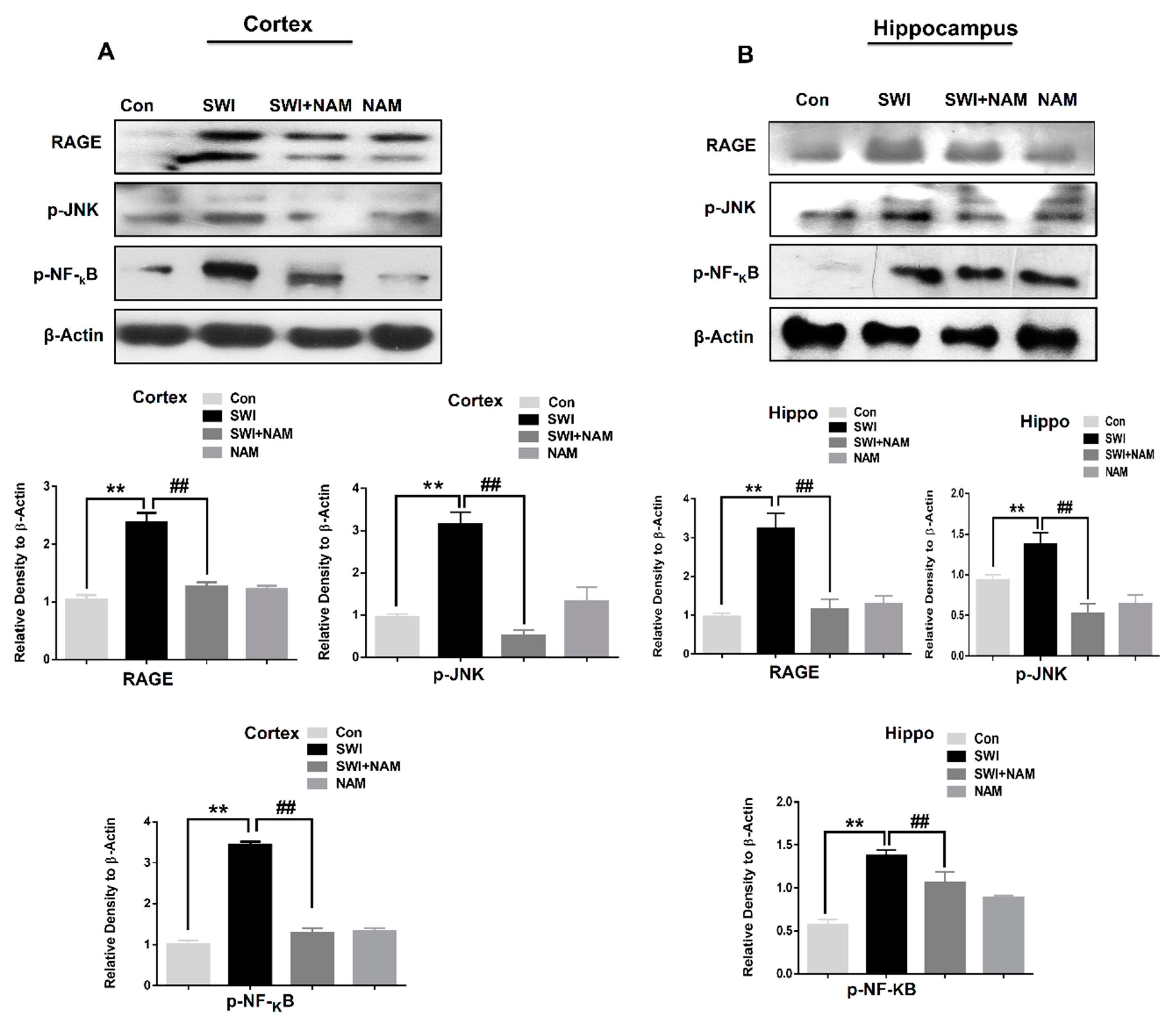
3.1. NAM Ameliorates RAGE, JNK, and NF-ΚB Signaling in Injured Mouse Brains
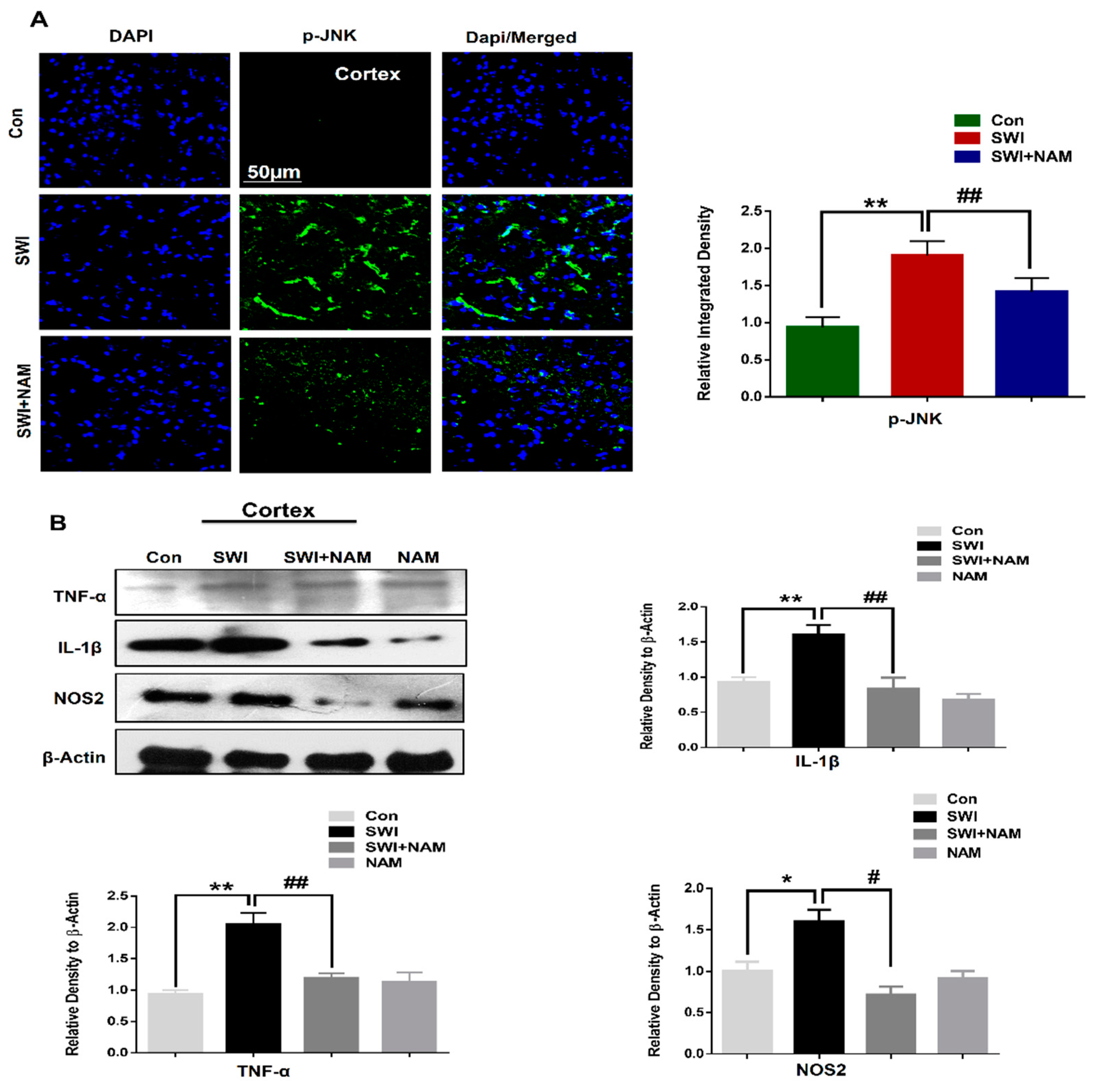
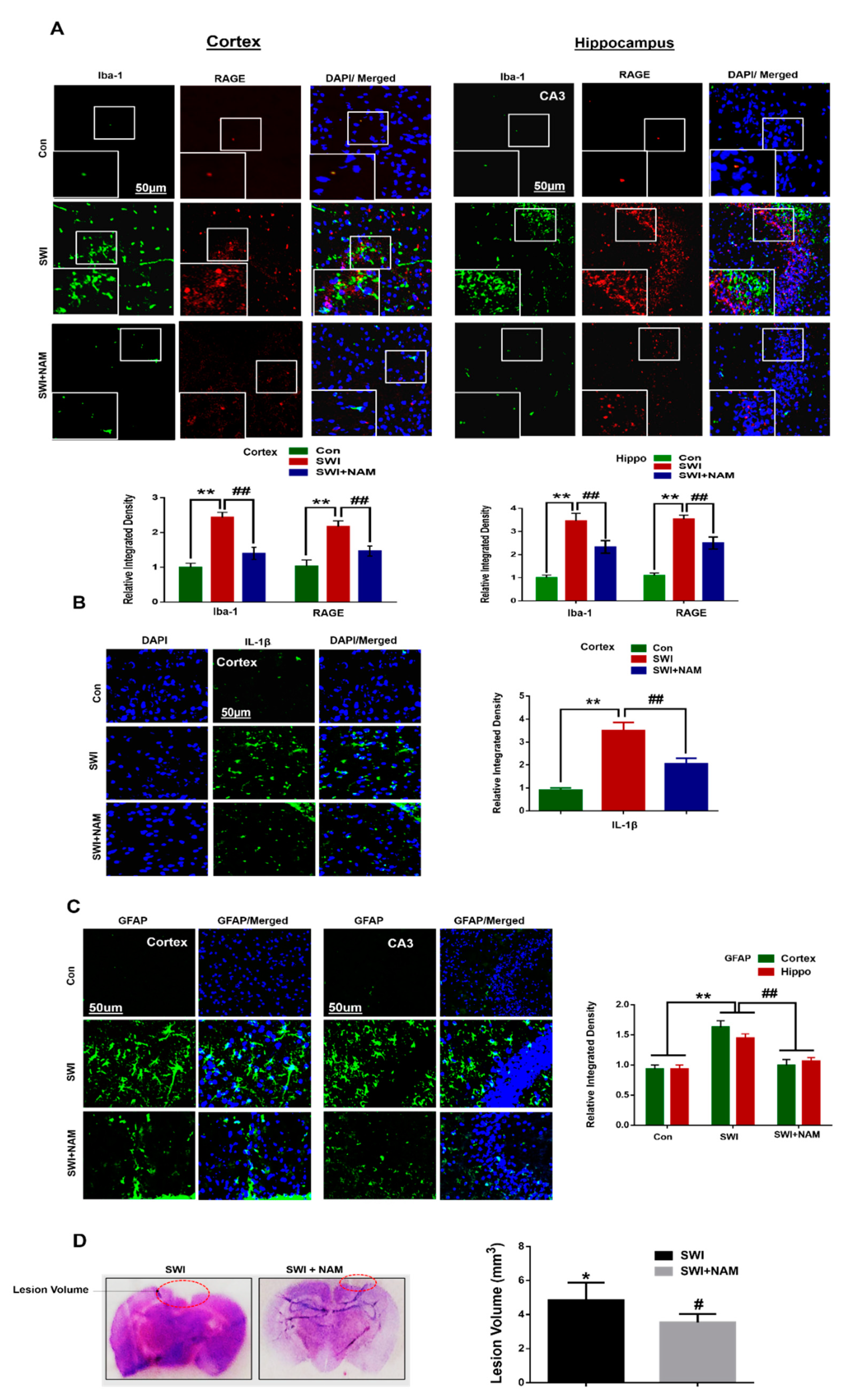
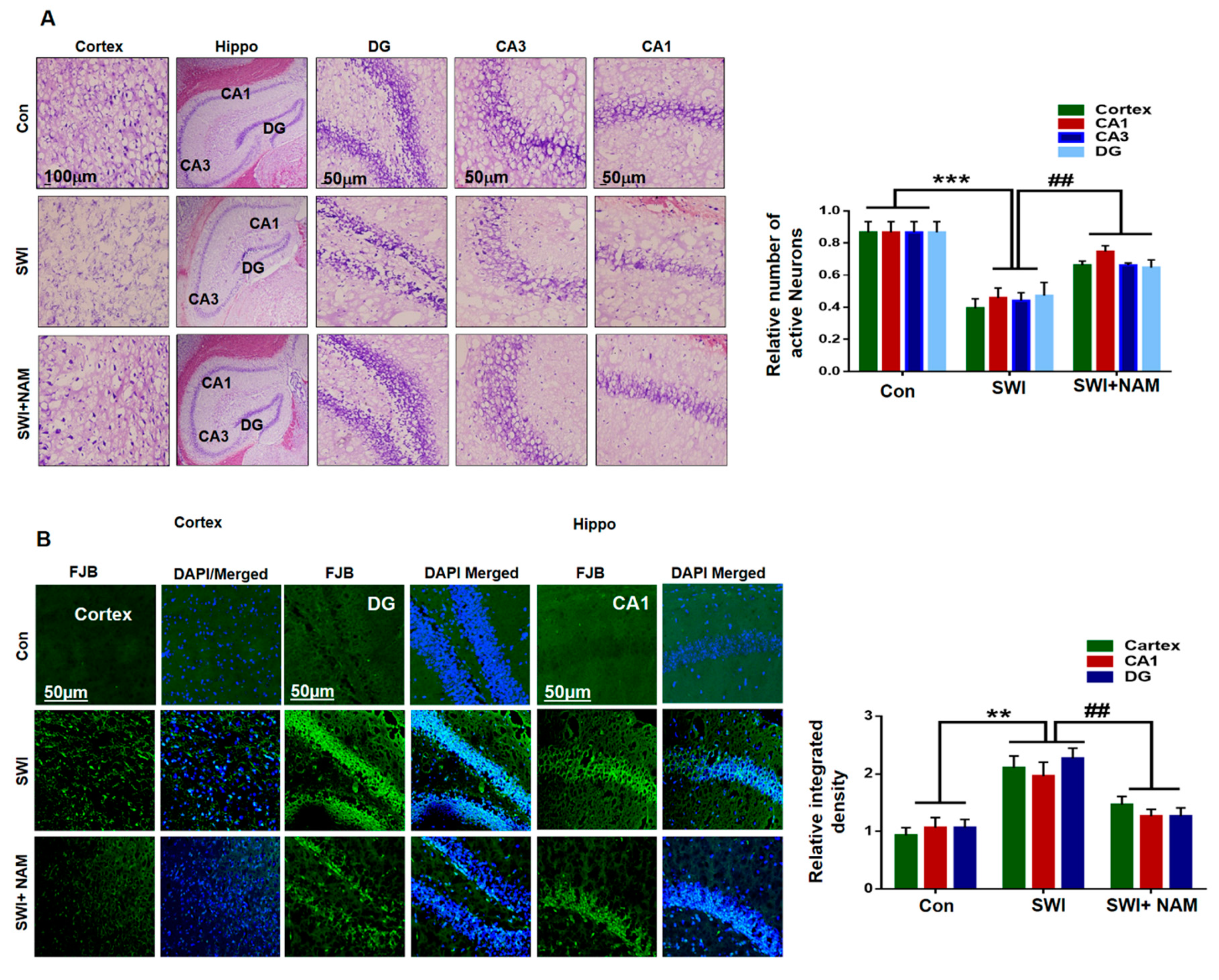
3.2. NAM Inhibited Neuroinflammation and Reduced Lesion Volume in The Cortex of the Injured Mouse Brains
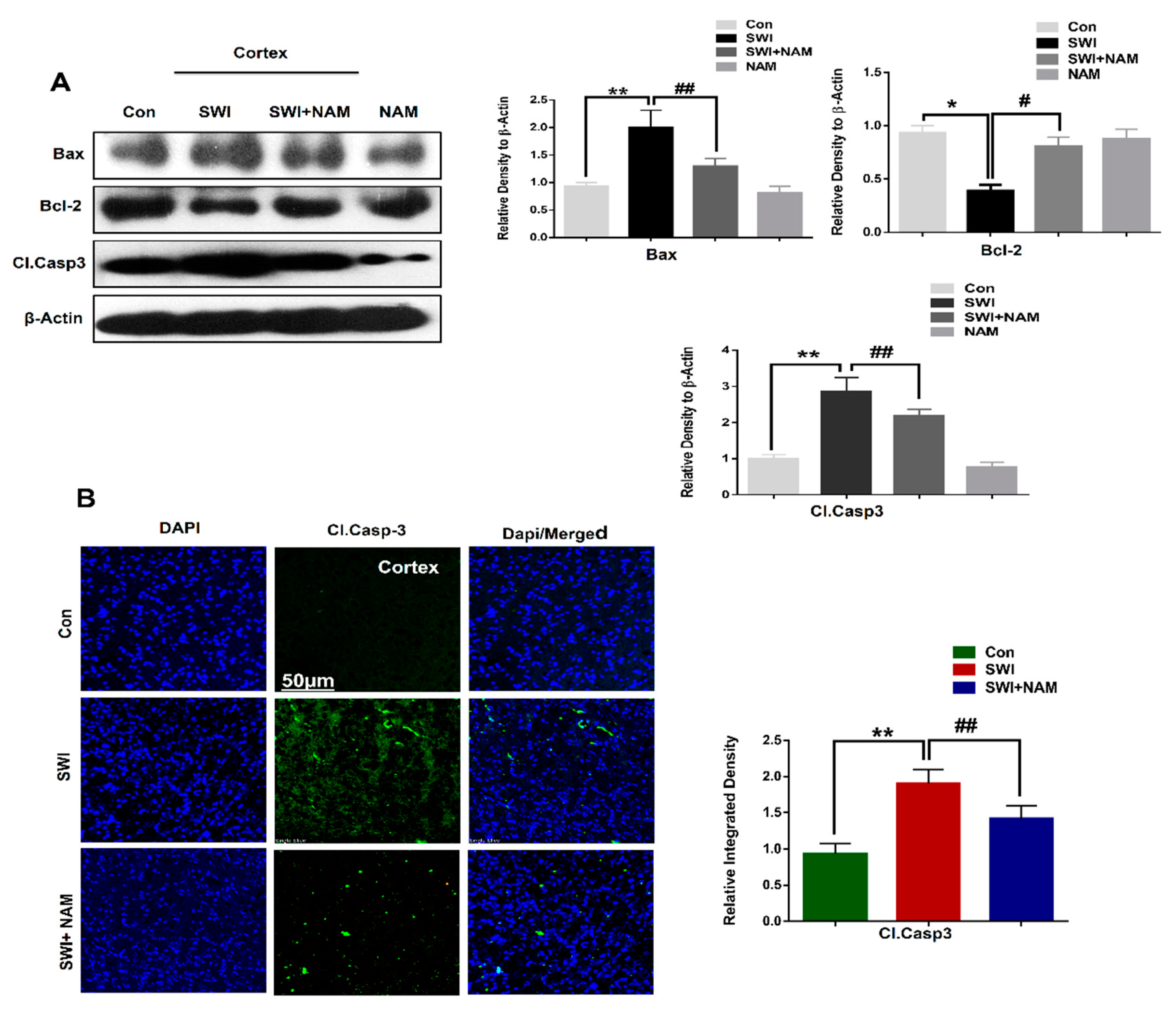
3.3. NAM Reduced Neuronal Apoptosis in TBI Mouse Brains
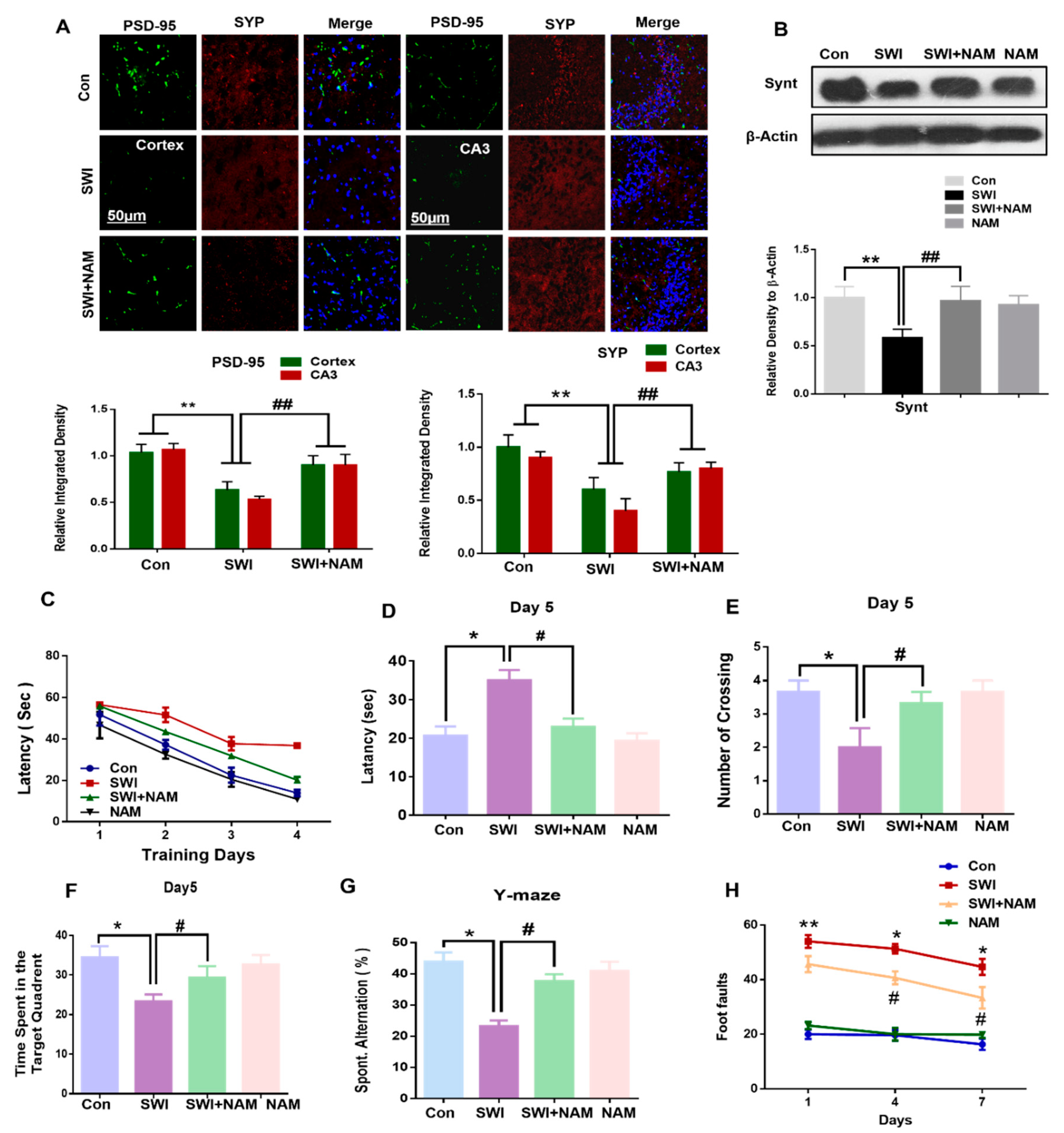
3.4. NAM Reversed the Synaptic Protein Loss and Improved Memory Dysfunction and Motor Neuronal Dysfunction
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tagge, C.A.; Fisher, A.M.; Minaeva, O.V.; Gaudreau-Balderrama, A.; Moncaster, J.A.; Zhang, X.L.; Wojnarowicz, M.W.; Casey, N.; Lu, H.; Kokiko-Cochran, O.N.; et al. Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain J. Neurol. 2018, 141, 422–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harch, P.G.; Andrews, S.R.; Fogarty, E.F.; Amen, D.; Pezzullo, J.C.; Lucarini, J.; Aubrey, C.; Taylor, D.V.; Staab, P.K.; Van Meter, K.W. A phase I study of low-pressure hyperbaric oxygen therapy for blast-induced post-concussion syndrome and post-traumatic stress disorder. J. Neurotrauma 2012, 29, 168–185. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.E. Mechanisms of traumatic brain injury: Biomechanical, structural and cellular considerations. Crit. Care Nurs. Q 2000, 23, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Chauhan, N.B. Chronic neurodegenerative consequences of traumatic brain injury. Restor. Neurol. Neurosci. 2014, 32, 337–365. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Axonal pathology in traumatic brain injury. Exp. Neurol. 2013, 246, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Alexander, M.P. Mild traumatic brain injury: Pathophysiology, natural history, and clinical management. Neurology 1995, 45, 1253–1260. [Google Scholar] [CrossRef]
- Atkins, C.M.; Falo, M.C.; Alonso, O.F.; Bramlett, H.M.; Dietrich, W.D. Deficits in ERK and CREB activation in the hippocampus after traumatic brain injury. Neurosci. Lett. 2009, 459, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Riggio, S. Traumatic brain injury and its neurobehavioral sequelae. Psychiatr. Clin. North Am. 2010, 33, 807–819. [Google Scholar] [CrossRef]
- Saing, T.; Dick, M.; Nelson, P.T.; Kim, R.C.; Cribbs, D.H.; Head, E. Frontal cortex neuropathology in dementia pugilistica. J. Neurotrauma 2012, 29, 1054–1070. [Google Scholar] [CrossRef]
- Acosta, S.A.; Tajiri, N.; de la Pena, I.; Bastawrous, M.; Sanberg, P.R.; Kaneko, Y.; Borlongan, C.V. Alpha-synuclein as a pathological link between chronic traumatic brain injury and Parkinson’s disease. J. Cell Physiol. 2015, 230, 1024–1032. [Google Scholar] [CrossRef]
- Xiong, Y.; Mahmood, A.; Chopp, M. Animal models of traumatic brain injury. Nat. Rev. Neurosci. 2013, 14, 128–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tran, H.T.; LaFerla, F.M.; Holtzman, D.M.; Brody, D.L. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 2011, 31, 9513–9525. [Google Scholar] [CrossRef]
- Abrahamson, E.E.; Ikonomovic, M.D.; Ciallella, J.R.; Hope, C.E.; Paljug, W.R.; Isanski, B.A.; Flood, D.G.; Clark, R.S.; DeKosky, S.T. Caspase inhibition therapy abolishes brain trauma-induced increases in Abeta peptide: Implications for clinical outcome. Exp. Neurol. 2006, 197, 437–450. [Google Scholar] [CrossRef] [PubMed]
- Loane, D.J.; Pocivavsek, A.; Moussa, C.E.; Thompson, R.; Matsuoka, Y.; Faden, A.I.; Rebeck, G.W.; Burns, M.P. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 2009, 15, 377–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, O.I.; Heyde, C.E.; Ertel, W.; Stahel, P.F. Closed head injury—An inflammatory disease? Brain Res. Rev. 2005, 48, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Aungst, S.L.; Kabadi, S.V.; Thompson, S.M.; Stoica, B.A.; Faden, A.I. Repeated mild traumatic brain injury causes chronic neuroinflammation, changes in hippocampal synaptic plasticity, and associated cognitive deficits. J. Cereb. Blood Flow Metab. 2014, 34, 1223–1232. [Google Scholar] [CrossRef] [PubMed]
- Pierce, J.E.; Smith, D.H.; Trojanowski, J.Q.; McIntosh, T.K. Enduring cognitive, neurobehavioral and histopathological changes persist for up to one year following severe experimental brain injury in rats. Neuroscience 1998, 87, 359–369. [Google Scholar] [CrossRef]
- Lee, Y.K.; Hou, S.W.; Lee, C.C.; Hsu, C.Y.; Huang, Y.S.; Su, Y.C. Increased risk of dementia in patients with mild traumatic brain injury: A nationwide cohort study. PLoS ONE 2013, 8, e62422. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.P.; Shertzer, H.G.; Puga, A. Regulation of gene expression by reactive oxygen. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 67–101. [Google Scholar] [CrossRef] [PubMed]
- Glushakov, A.O.; Glushakova, O.Y.; Korol, T.Y.; Acosta, S.A.; Borlongan, C.V.; Valadka, A.B.; Hayes, R.L.; Glushakov, A.V. Chronic Upregulation of Cleaved-Caspase-3 Associated with Chronic Myelin Pathology and Microvascular Reorganization in the Thalamus after Traumatic Brain Injury in Rats. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef]
- Wilke, S.; Prehn, K.; Taud, B.; List, J.; Floel, A. Multimodal Assessment of Recurrent MTBI across the Lifespan. J. Clin. Med. 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Rehman, S.U.; Ali, T.; Alam, S.I.; Ullah, R.; Zeb, A.; Lee, K.W.; Rutten, B.P.F.; Kim, M.O. Ferulic Acid Rescues LPS-Induced Neurotoxicity via Modulation of the TLR4 Receptor in the Mouse Hippocampus. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mei, Z.; Zheng, P.; Tan, X.; Wang, Y.; Situ, B. Huperzine A alleviates neuroinflammation, oxidative stress and improves cognitive function after repetitive traumatic brain injury. Metab. Brain Dis. 2017, 32, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Yan, S.D.; Bierhaus, A.; Nawroth, P.P.; Stern, D.M. RAGE and Alzheimer’s disease: A progression factor for amyloid-beta-induced cellular perturbation? J. Alzheimers Dis. 2009, 16, 833–843. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.R.; Cho, W.H.; Kim, J.; Lee, H.J.; Chung, C.; Jeon, W.K.; Han, J.S. Increased expression of the receptor for advanced glycation end products in neurons and astrocytes in a triple transgenic mouse model of Alzheimer’s disease. Exp. Mol. Med. 2014, 46, e75. [Google Scholar] [CrossRef] [PubMed]
- Ott, C.; Jacobs, K.; Haucke, E.; Navarrete Santos, A.; Grune, T.; Simm, A. Role of advanced glycation end products in cellular signaling. Redox. Biol. 2014, 2, 411–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, T.; Badshah, H.; Kim, T.H.; Kim, M.O. Melatonin attenuates d-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J. Pineal Res. 2015, 58, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Jing, J.; Yu, S.; Song, M.; Tan, H.; Cui, B.; Huang, L. Advanced glycation endproducts induce apoptosis of endothelial progenitor cells by activating receptor RAGE and NADPH oxidase/JNK signaling axis. Am. J. Transl. Res. 2016, 8, 2169–2178. [Google Scholar] [PubMed]
- Hoane, M.R.; Kaplan, S.A.; Ellis, A.L. The effects of nicotinamide on apoptosis and blood-brain barrier breakdown following traumatic brain injury. Brain Res. 2006, 1125, 185–193. [Google Scholar] [CrossRef]
- Kristian, T.; Balan, I.; Schuh, R.; Onken, M. Mitochondrial dysfunction and nicotinamide dinucleotide catabolism as mechanisms of cell death and promising targets for neuroprotection. J. Neurosci. Res. 2011, 89, 1946–1955. [Google Scholar] [CrossRef]
- Klaidman, L.; Morales, M.; Kem, S.; Yang, J.; Chang, M.L.; Adams, J.D., Jr. Nicotinamide offers multiple protective mechanisms in stroke as a precursor for NAD+, as a PARP inhibitor and by partial restoration of mitochondrial function. Pharmacology 2003, 69, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Gharavi, R.; Pitta, M.; Gleichmann, M.; Mattson, M.P. Nicotinamide prevents NAD+ depletion and protects neurons against excitotoxicity and cerebral ischemia: NAD+ consumption by SIRT1 may endanger energetically compromised neurons. Neuromol. Med. 2009, 11, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Kong, L.; Yao, Y.; Jiao, Y.; Song, J.; Tao, Z.; You, Z.; Yang, J. Osthole confers neuroprotection against cortical stab wound injury and attenuates secondary brain injury. J. Neuroinflamm. 2015, 12, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cernak, I.; Wing, I.D.; Davidsson, J.; Plantman, S. A novel mouse model of penetrating brain injury. Front. Neurol. 2014, 5, 209. [Google Scholar] [CrossRef]
- Ali, T.; Kim, T.; Rehman, S.U.; Khan, M.S.; Amin, F.U.; Khan, M.; Ikram, M.; Kim, M.O. Natural Dietary Supplementation of Anthocyanins via PI3K/Akt/Nrf2/HO-1 Pathways Mitigate Oxidative Stress, Neurodegeneration, and Memory Impairment in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 6076–6093. [Google Scholar] [CrossRef]
- Rehman, S.U.; Ahmad, A.; Yoon, G.H.; Khan, M.; Abid, M.N.; Kim, M.O. Inhibition of c-Jun N-Terminal Kinase Protects Against Brain Damage and Improves Learning and Memory After Traumatic Brain Injury in Adult Mice. Cereb. Cortex 2018, 28, 2854–2872. [Google Scholar] [CrossRef]
- Kim, M.J.; Rehman, S.U.; Amin, F.U.; Kim, M.O. Enhanced neuroprotection of anthocyanin-loaded PEG-gold nanoparticles against Abeta1-42-induced neuroinflammation and neurodegeneration via the NF-KB /JNK/GSK3beta signaling pathway. Nanomed. Nanotech. Biol. Med. 2017, 13, 2533–2544. [Google Scholar] [CrossRef]
- Wang, B.J.; Chiu, H.W.; Lee, Y.L.; Li, C.Y.; Wang, Y.J.; Lee, Y.H. Pterostilbene Attenuates Hexavalent Chromium-Induced Allergic Contact Dermatitis by Preventing Cell Apoptosis and Inhibiting IL-1beta-Related NLRP3 Inflammasome Activation. J. Clin. Med. 2018, 7, 489. [Google Scholar] [CrossRef]
- Tsai, Y.C.; Kuo, P.L.; Kuo, M.C.; Hung, W.W.; Wu, L.Y.; Chang, W.A.; Wu, P.H.; Lee, S.C.; Chen, H.C.; Hsu, Y.L. The Interaction of miR-378i-Skp2 Regulates Cell Senescence in Diabetic Nephropathy. J. Clin. Med. 2018, 7, 468. [Google Scholar] [CrossRef]
- Ali, T.; Rehman, S.U.; Shah, F.A.; Kim, M.O. Acute dose of melatonin via Nrf2 dependently prevents acute ethanol-induced neurotoxicity in the developing rodent brain. J. Neuroinflamm. 2018, 15, 119. [Google Scholar] [CrossRef]
- Kazuno, A.; Maki, D.; Yamato, I.; Nakajima, N.; Seta, H.; Soeda, S.; Ozawa, S.; Uchiyama, Y.; Tamaki, T. Regeneration of Transected Recurrent Laryngeal Nerve Using Hybrid-Transplantation of Skeletal Muscle-Derived Stem Cells and Bioabsorbable Scaffold. J. Clin. Med. 2018, 7, 276. [Google Scholar] [CrossRef] [PubMed]
- Badshah, H.; Ali, T.; Kim, M.O. Osmotin attenuates LPS-induced neuroinflammation and memory impairments via the TLR4/NFkappaB signaling pathway. Sci. Rep. 2016, 6, 24493. [Google Scholar] [CrossRef] [PubMed]
- Ikram, M.; Muhammad, T.; Rehman, S.U.; Khan, A.; Jo, M.G.; Ali, T.; Kim, M.O. Hesperetin Confers Neuroprotection by Regulating Nrf2/TLR4/NF-kappaB Signaling in an Abeta Mouse Model. Mol. Neurobiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Muhammad, T.; Ali, T.; Ikram, M.; Khan, A.; Alam, S.I.; Kim, M.O. Melatonin Rescue Oxidative Stress-Mediated Neuroinflammation/Neurodegeneration and Memory Impairment in Scopolamine-Induced Amnesia Mice Model. J. Neuroimmune Pharmacol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ali, T.; Kim, M.J.; Rehman, S.U.; Ahmad, A.; Kim, M.O. Anthocyanin-Loaded PEG-Gold Nanoparticles Enhanced the Neuroprotection of Anthocyanins in an Abeta1-42 Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 6490–6506. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Amin, F.U.; Khan, M.; Abid, M.N.; Rehman, S.U.; Kim, T.H.; Kim, M.W.; Kim, M.O. Anthocyanins abrogate glutamate-induced AMPK activation, oxidative stress, neuroinflammation, and neurodegeneration in postnatal rat brain. J. Neuroinflamm. 2016, 13, 286. [Google Scholar] [CrossRef]
- Cai, Z.; Liu, N.; Wang, C.; Qin, B.; Zhou, Y.; Xiao, M.; Chang, L.; Yan, L.J.; Zhao, B. Role of RAGE in Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 483–495. [Google Scholar] [CrossRef]
- Rehman, S.U.; Shah, S.A.; Ali, T.; Chung, J.I.; Kim, M.O. Anthocyanins Reversed d-Galactose-Induced Oxidative Stress and Neuroinflammation Mediated Cognitive Impairment in Adult Rats. Mol. Neurobiol. 2017, 54, 255–271. [Google Scholar] [CrossRef]
- Ojala, J.O.; Sutinen, E.M. The Role of Interleukin-18, Oxidative Stress and Metabolic Syndrome in Alzheimer’s Disease. J. Clin. Med. 2017, 6, 55. [Google Scholar] [CrossRef]
- Kim, E.J.; Yang, S.J. Nicotinamide Reduces Amyloid Precursor Protein and Presenilin 1 in Brain Tissues of Amyloid Beta-Tail Vein Injected Mice. Clin. Nutr. Res. 2017, 6, 130–135. [Google Scholar] [CrossRef]
- Yao, Z.; Yang, W.; Gao, Z.; Jia, P. Nicotinamide mononucleotide inhibits JNK activation to reverse Alzheimer disease. Neurosci. Lett. 2017, 647, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Turunc Bayrakdar, E.; Uyanikgil, Y.; Kanit, L.; Koylu, E.; Yalcin, A. Nicotinamide treatment reduces the levels of oxidative stress, apoptosis, and PARP-1 activity in Abeta(1-42)-induced rat model of Alzheimer’s disease. Free Radic. Res. 2014, 48, 146–158. [Google Scholar] [CrossRef]
- Ansari, M.A.; Roberts, K.N.; Scheff, S.W. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic. Biol. Med. 2008, 45, 443–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lenzlinger, P.M.; Morganti-Kossmann, M.C.; Laurer, H.L.; McIntosh, T.K. The duality of the inflammatory response to traumatic brain injury. Mol. Neurobiol. 2001, 24, 169–181. [Google Scholar] [CrossRef]
- Salminen, A.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Inflammation in Alzheimer’s disease: amyloid-beta oligomers trigger innate immunity defence via pattern recognition receptors. Prog. Neurobiol. 2009, 87, 181–194. [Google Scholar] [CrossRef]
- Schmidt, A.M.; Sahagan, B.; Nelson, R.B.; Selmer, J.; Rothlein, R.; Bell, J.M. The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer’s disease. Curr. Opin. Investig. Drugs 2009, 10, 672–680. [Google Scholar] [PubMed]
- Lee, E.J.; Park, J.H. Receptor for Advanced Glycation Endproducts (RAGE), Its Ligands, and Soluble RAGE: Potential Biomarkers for Diagnosis and Therapeutic Targets for Human Renal Diseases. Genomics Inform. 2013, 11, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Piras, S.; Furfaro, A.L.; Domenicotti, C.; Traverso, N.; Marinari, U.M.; Pronzato, M.A.; Nitti, M. RAGE Expression and ROS Generation in Neurons: Differentiation versus Damage. Oxid. Med. Cell Longev. 2016, 2016, 9348651. [Google Scholar] [CrossRef]
- Marshall, J.; Szmydynger-Chodobska, J.; Rioult-Pedotti, M.S.; Lau, K.; Chin, A.T.; Kotla, S.K.R.; Tiwari, R.K.; Parang, K.; Threlkeld, S.W.; Chodobski, A. TrkB-enhancer facilitates functional recovery after traumatic brain injury. Sci. Rep. 2017, 7, 10995. [Google Scholar] [CrossRef]
- Hellewell, S.; Semple, B.D.; Morganti-Kossmann, M.C. Therapies negating neuroinflammation after brain trauma. Brain Res. 2016, 1640, 36–56. [Google Scholar] [CrossRef]
- Lee, S.H.; Choi, B.Y.; Lee, S.H.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Suh, S.W. Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. Int. J. Mol. Sci. 2017, 18, 2510. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Sun, P.; Zhang, J.C.; Zhang, Q.; Yao, S.L. Proinflammatory effects of S100A8/A9 via TLR4 and RAGE signaling pathways in BV-2 microglial cells. Int. J. Mol. Med. 2017, 40, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Khodeer, D.M.; Zaitone, S.A.; Farag, N.E.; Moustafa, Y.M. Cardioprotective effect of pioglitazone in diabetic and non-diabetic rats subjected to acute myocardial infarction involves suppression of AGE-RAGE axis and inhibition of apoptosis. Can. J. Physiol. Pharmacol. 2016, 94, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 321. [Google Scholar] [CrossRef] [PubMed]
- Son, M.; Oh, S.; Park, H.; Ahn, H.; Choi, J.; Kim, H.; Lee, H.S.; Lee, S.; Park, H.J.; Kim, S.U.; et al. Protection against RAGE-mediated neuronal cell death by sRAGE-secreting human mesenchymal stem cells in 5xFAD transgenic mouse model. Brain Behav. Immun. 2017, 66, 347–358. [Google Scholar] [CrossRef] [PubMed]
- Wei, C.C.; Kong, Y.Y.; Li, G.Q.; Guan, Y.F.; Wang, P.; Miao, C.Y. Nicotinamide mononucleotide attenuates brain injury after intracerebral hemorrhage by activating Nrf2/HO-1 signaling pathway. Sci. Rep. 2017, 7, 717. [Google Scholar] [CrossRef] [PubMed]
- Islamoglu, H.; Cao, R.; Teskey, G.; Gyurjian, K.; Lucar, S.; Fraix, M.P.; Sathananthan, A.; Chan, J.K.; Venketaraman, V. Effects of ReadiSorb L-GSH in Altering Granulomatous Responses against Mycobacterium tuberculosis Infection. J. Clin. Med. 2018, 7, 40. [Google Scholar] [CrossRef]
- Jia, H.; Li, X.; Gao, H.; Feng, Z.; Li, X.; Zhao, L.; Jia, X.; Zhang, H.; Liu, J. High doses of nicotinamide prevent oxidative mitochondrial dysfunction in a cellular model and improve motor deficit in a Drosophila model of Parkinson’s disease. J. Neurosci. Res. 2008, 86, 2083–2090. [Google Scholar] [CrossRef]
- Liu, D.; Pitta, M.; Jiang, H.; Lee, J.H.; Zhang, G.; Chen, X.; Kawamoto, E.M.; Mattson, M.P. Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: Evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol. Aging 2013, 34, 1564–1580. [Google Scholar] [CrossRef]
- Peterson, T.C.; Hoane, M.R.; McConomy, K.S.; Farin, F.M.; Bammler, T.K.; MacDonald, J.W.; Kantor, E.D.; Anderson, G.D. A Combination Therapy of Nicotinamide and Progesterone Improves Functional Recovery following Traumatic Brain Injury. J. Neurotrauma 2015, 32, 765–779. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hu, X.; Yang, Y.; Takata, T.; Sakurai, T. Nicotinamide mononucleotide protects against beta-amyloid oligomer-induced cognitive impairment and neuronal death. Brain Res. 2016, 1643, 1–9. [Google Scholar] [CrossRef] [PubMed]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alam, S.I.; Ur Rehman, S.; Ok Kim, M. Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-κB Signaling Pathway after Brain Injury. J. Clin. Med. 2019, 8, 271. https://doi.org/10.3390/jcm8020271
Alam SI, Ur Rehman S, Ok Kim M. Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-κB Signaling Pathway after Brain Injury. Journal of Clinical Medicine. 2019; 8(2):271. https://doi.org/10.3390/jcm8020271
Chicago/Turabian StyleAlam, Sayed Ibrar, Shafiq Ur Rehman, and Myeong Ok Kim. 2019. "Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-κB Signaling Pathway after Brain Injury" Journal of Clinical Medicine 8, no. 2: 271. https://doi.org/10.3390/jcm8020271
APA StyleAlam, S. I., Ur Rehman, S., & Ok Kim, M. (2019). Nicotinamide Improves Functional Recovery via Regulation of the RAGE/JNK/NF-κB Signaling Pathway after Brain Injury. Journal of Clinical Medicine, 8(2), 271. https://doi.org/10.3390/jcm8020271