Current Progress in Pharmacogenetics of Second-Line Antidiabetic Medications: Towards Precision Medicine for Type 2 Diabetes

Abstract
:1. Introduction
2. DPP-4 Inhibitors
2.1. Adenosine Triphosphate-Binding Cassette Subfamily B Member 1 (ABCB1)
2.2. DPP4
2.3. GLP-1 Receptor Gene (GLP1R)
2.4. Transcription Factor 7-Like 2 (TCF7L2)
2.5. Patatin-Like Phospholipase 3 (PNPLA3)
2.6. Cyclin-Dependent Kinase 5 Regulatory Subunit Associated Protein 1-Like 1 (CDKAL1)
2.7. Potassium Channels (KCN) Gene Family
2.8. Protein Kinase D1 (PRKD1)
3. GLP-1 Receptor Agonists
3.1. GLP1R
3.2. Cannabinoid Type 1 Receptor (CNR1)
3.3. Sortilin Related VPS10 Domain Containing Receptor 1 (SORCS1)
3.4. WFS1 and TCF7L2
4. SGLT2 Inhibitors
4.1. Uridine Diphosphate-Glucuronosyltransferase (UGT) Gene Family
4.2. Solute Carrier Family 5 Member 2 (SLC5A2)
4.3. PNPLA3
5. Future Perspectives and Challenges
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- The Role of Pharmacogenomics in Precision Medicine. Available online: https://www.mlo-online.com/role-pharmacogenomics-precision-medicine (accessed on 29 January 2019).
- National Research Council (US) Committee on A Framework for Developing a New Taxonomy of Disease. Toward Precision Medicine: Building a Knowledge Network for Biomedical Research and a New Taxonomy of Disease; National Academies Press: Washington, DC, USA, 2011; p. 125.
- FACT SHEET: President Obama’s Precision Medicine Initiative. Available online: https://obamawhitehouse.archives.gov/the-press-office/2015/01/30/fact-sheet-president-obama-s-precision-medicine-initiative (accessed on 7 February 2019).
- Nimmesgern, E.; Norstedt, I.; Draghia-Akli, R. Enabling personalized medicine in Europe by the European Commission’s funding activities. Pers. Med. 2017, 14, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Carrasco-Ramiro, F.; Pieró-Pastor, R.; Aguado, B. Human genomics projects and precision medicine. Gene Ther. 2017, 24, 551–561. [Google Scholar] [CrossRef] [PubMed]
- Katz, D.A.; Murray, B.; Bhathena, A.; Sahelijo, L. Defining drug disposition determinants: A pharmacogenetic-pharmacokinetic strategy. Nat. Rev. Drug Discov. 2008, 7, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Table of Pharmacogenomic Biomarkers in Drug Labeling. Available online: https://www.fda.gov/Drugs/ScienceResearch/ucm572698.htm (accessed on 26 February 2019).
- Baynes, J.W. Role of oxidative stress in development of complications in diabetes. Diabetes 1991, 40, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gutierrez, R.; Gionfriddo, M.R.; Ospina, N.S.; Maraka, S.; Tamhane, S.; Montori, V.M.; Brito, J.P. Shared decision making in endocrinology: Present and future directions. Lancet Diabetes Endocrinol. 2016, 4, 706–716. [Google Scholar] [CrossRef]
- Davies, M.J.; D’Alessio, D.A.; Fradkin, J.; Kernan, W.N.; Mathieu, C.; Mingrone, G.; Rossing, P.; Tsapas, A.; Wexler, D.J.; Buse, J.B. Management of hyperglycemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia 2018, 61, 2461–2498. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.C. The pharmacogenetics of metformin. Diabetologia 2017, 60, 1648–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srinivasan, S.; Yee, S.W.; Giacomini, K.M. Pharmacogenetics of Antidiabetic Drugs. Adv. Pharmacol. 2018, 83, 361–389. [Google Scholar] [PubMed]
- Chan, P.; Shao, L.; Tomlinson, B.; Zhang, Y.; Liu, Z.M. Metformin transporter pharmacogenomics: Insights into drug disposition-where are we now? Expert Opin. Drug Metab. Toxicol. 2018, 14, 1149–1159. [Google Scholar] [CrossRef]
- International Transporter Consortium; Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar]
- Gourgari, E.; Wilhelm, E.E.; Hassanzadeh, H.; Aroda, V.R.; Shoulson, I. A comprehensive review of the FDA-approved labels of diabetes drugs: Indications, safety, and emerging cardiovascular safety data. J. Diabetes Complicat. 2017, 31, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Elrick, H.; Stimmler, L.; Hlad, C.J.; Arai, Y. Plasma insulin response to oral and intravenous glucose administration. J. Clin. Endocrinol. Metab. 1964, 24, 1076–1082. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, R.; Matsuyama, T.; Namba, M.; Watanabe, N.; Itoh, H.; Kono, N.; Tarui, S. Glucagonostatic and insulinotropic action of glucagonlike peptide I-(7–36)-amide. Diabetes 1989, 38, 902–905. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Kleine, N.; Orskov, C.; Holst, J.J.; Willims, B.; Creutzfeldt, W. Normalization of fasting hyperglycaemia by exogenous glucagon-like peptide 1 (7–36 amide) in type 2 (non-insulin-dependent) diabetic patients. Diabetologia 1993, 36, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Drucker, D.J.; Philippe, J.; Mojsov, S.; Chick, W.L.; Habener, J.F. Glucagon-like peptide I stimulates insulin gene expression and increases cyclic AMP levels in a rat islet cell line. Proc. Natl. Acad. Sci. USA 1987, 84, 3434–3438. [Google Scholar] [CrossRef]
- Drucker, D.J. Glucagon-like peptide-1 and the islet beta-cell: Augmentation of cell proliferation and inhibition of apoptosis. Endocrinology 2003, 144, 5145–5148. [Google Scholar] [CrossRef] [PubMed]
- Weber, A.E. Dipeptidyl peptidase IV inhibitors for the treatment of diabetes. J. Med. Chem. 2004, 47, 4135–4141. [Google Scholar] [CrossRef]
- Brubaker, P.L.; Drucker, D.J. Minireview: Glucagon-like peptides regulate cell proliferation and apoptosis in the pancreas, gut, and central nervous system. Endocrinology 2004, 145, 2653–2659. [Google Scholar] [CrossRef]
- Meier, J.J.; Gallwitz, B.; Salmen, S.; Goetze, O.; Holst, J.J.; Schmidt, W.E.; Nauck, M.A. Normalization of glucose concentrations and deceleration of gastric emptying after solid meals during intravenous glucagon-like peptide 1 in patients with type 2 diabetes. J. Clin. Endocrinol. Metab. 2003, 88, 2719–2725. [Google Scholar] [CrossRef]
- Drucker, D.J.; Nauck, M.A. The incretin system: Glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors in type 2 diabetes. Lancet 2006, 368, 1696–1705. [Google Scholar] [CrossRef]
- Deacon, C.F.; Nauck, M.A.; Toft-Nielsen, M.; Pridal, L.; Willms, B.; Holst, J.J. Both subcutaneously and intravenously administered glucagon-like peptide I are rapidly degraded from the NH2-terminus in type II diabetic patients and in healthy subjects. Diabetes 1995, 44, 1126–1131. [Google Scholar] [CrossRef] [PubMed]
- Choy, M.; Lam, S. Sitagliptin: A novel drug for the treatment of type 2 diabetes. Cardiol. Rev. 2007, 15, 264–271. [Google Scholar] [CrossRef]
- Li, N.; Wang, L.J.; Jiang, B.; Li, X.Q.; Guo, C.L.; Guo, S.J.; Shi, D.Y. Recent progress of the development of dipeptidyl peptidase-4 inhibitors for the treatment of type 2 diabetes mellitus. Eur. J. Med. Chem. 2018, 151, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Deacon, C.F. Dipeptidyl peptidase-4 inhibitors in the treatment of type 2 diabetes: A comparative review. Diabetes Obes. Metab. 2011, 13, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Morales, J. The pharmacologic basis for clinical differences among GLP-1 receptor agonists and DPP-4 inhibitors. Postgrad. Med. 2011, 123, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Scirica, B.M.; Bhatt, D.L.; Braunwald, E.; Steg, P.G.; Davidson, J.; Hirshberg, B.; Ohman, P.; Frederich, R.; Wiviott, S.D.; Hoffman, E.B.; et al. SAVOR-TIMI 53 Steering Committee and Investigators. Saxagliptin and cardiovascular outcomes in patients with type 2 diabetes mellitus. N. Engl. J. Med. 2013, 369, 1317–1326. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.X.; Evans, M. Choosing between GLP-1 Receptor Agonists and DPP-4 Inhibitors: A Pharmacological Perspective. J. Nutr. Metab. 2012, 2012, 381713. [Google Scholar] [CrossRef]
- Wilson, J.R.; Shuey, M.M.; Brown, N.J.; Devin, J.K. Hypertension and Type 2 Diabetes Are Associated with Decreased Inhibition of Dipeptidyl Peptidase-4 by Sitagliptin. J. Endocr. Soc. 2017, 1, 1168–1178. [Google Scholar] [CrossRef] [PubMed]
- Han, E.; Park, H.S.; Kwon, O.; Choe, E.Y.; Wang, H.J.; Lee, Y.H.; Lee, S.H.; Kim, C.H.; Kim, L.K.; Kwak, S.H.; et al. A genetic variant in GLP1R is associated with response to DPP-4 inhibitors in patients with type 2 diabetes. Medicine 2016, 95, e5155. [Google Scholar] [CrossRef] [Green Version]
- Javorský, M.; Gotthardová, I.; Klimčáková, L.; Kvapil, M.; Židzik, J.; Schroner, Z.; Doubravová, P.; Gala, I.; Dravecká, I.; Tkáč, I. A missense variant in GLP1R gene is associated with the glycaemic response to treatment with gliptins. Diabetes Obes. Metab. 2016, 18, 941–944. [Google Scholar] [CrossRef]
- Zimdahl, H.; Ittrich, C.; Graefe-Mody, U.; Boehm, B.O.; Mark, M.; Woerle, H.J.; Dugi, K.A. Influence of TCF7L2 gene variants on the therapeutics response to the dipeptidylpeptidase-4 inhibitor linagliptin. Diabetologia 2014, 57, 1869–1875. [Google Scholar] [CrossRef]
- Kan, H.; Hyogo, H.; Ochi, H.; Hotta, K.; Fukuhara, T.; Kobayashi, T.; Naeshiro, N.; Honda, Y.; Kawaoka, T.; Tsuge, M.; et al. Influence of the rs738409 polymorphism in patatin-like phospholipase 3 on the treatment efficacy of non-alcoholic fatty liver disease with type 2 diabetes mellitus. Hepatol. Res. 2016, 46, E146–E153. [Google Scholar] [CrossRef]
- Osada, U.N.; Sunagawa, H.; Terauchi, Y.; Ueda, S. A Common Susceptibility Gene for Type 2 Diabetes Is Associated with Drug Response to a DPP-4 Inhibitor: Pharmacogenomic Cohort in Okinawa Japan. PLoS ONE 2016, 11, e0154821. [Google Scholar] [CrossRef]
- Jamaluddin, J.L.; Huri, H.Z.; Vethakkan, S.R. Clinical and genetic predictors of dipeptidyl peptidase-4 inhibitor treatment response in Type 2 diabetes mellitus. Pharmacogenomics 2016, 17, 867–881. [Google Scholar] [CrossRef]
- Gotthardová, I.; Javorský, M.; Klimčáková, L.; Kvapil, M.; Schroner, Z.; Kozárová, M.; Malachovská, Z.; Ürgeová, A.; Židzik, J.; Tkáč, I. KCNQ1 gene polymorphism is associated with glycaemic response to treatment with DPP-4 inhibitors. Diabetes Res. Clin. Pract. 2017, 130, 142–147. [Google Scholar] [CrossRef]
- Liao, W.L.; Lee, W.J.; Chen, C.C.; Lu, C.H.; Chen, C.H.; Chou, Y.C.; Lee, I.T.; Sheu, W.H.H.; Wu, J.Y.; Yang, C.F.; et al. Pharmacogenetics of dipeptidyl peptidase 4 inhibitors in a Taiwananse population with type 2 diabetes. Oncotarget 2017, 8, 18050–18058. [Google Scholar] [CrossRef]
- Aquilante, C.L.; Wempe, M.F.; Sidhom, M.S.; Kosmiski, L.A.; Predhomme, J.A. Effect of ABCB1 polymorphisms and atorvastatin on sitagliptin pharmacokinetics in healthy volunteers. Eur. J. Clin. Pharmacol. 2013, 69, 1401–1409. [Google Scholar] [CrossRef] [Green Version]
- Chu, X.Y.; Bleasby, K.; Yabut, J.; Cai, X.; Chan, G.H.; Hafey, M.J.; Xu, S.; Bergman, A.J.; Braun, M.P.; Dean, D.C.; Evers, R. Transport of the dipeptidyl peptidase-4 inhibitor sitagliptin by human organic anion transporter 3, organic anion transporting polypeptide 4C1, and multidrug resistance P-glycoprotein. J. Pharmacol. Exp. Ther. 2007, 321, 673–683. [Google Scholar] [CrossRef]
- Krishna, R.; Bergman, A.; Larson, P.; Cote, J.; Lasseter, K.; Dilzer, S.; Wang, A.; Zeng, W.; Chen, L.; Wagner, J.; Herman, G. Effect of a single cyclosporine dose on the single-dose pharmacokinetics of sitagliptin (MK-0431), a dipeptidyl peptidase-4 inhibitor, in healthy male subjects. J. Clin. Pharmacol. 2007, 47, 165–174. [Google Scholar] [CrossRef]
- Kwon, O.; Choe, E.Y.; Choi, Y.; Kim, H.M.; Wang, H.J.; Lee, H.; Kim, C.H.; Kang, E.S. Discovery of DiPeptidyl Peptidase-4 Gene Variants and the Associations with Efficacy of Vildagliptin in Patients with Type 2 Diabetes—A Pilot Study. J. Diabetes Metab. 2013, 4, S13. [Google Scholar]
- Waget, A.; Cabou, C.; Masseboeuf, M.; Cattan, P.; Armanet, M.; Karaca, M.; Castel, J.; Garret, C.; Payros, G.; Maida, A.; et al. Physiological and pharmacological mechanisms through which the DPP-4 inhibitor sitagliptin regulates glycemia in mice. Endocrinology 2011, 152, 3018–3029. [Google Scholar] [CrossRef] [PubMed]
- Pearson, E.R.; Donnelly, L.A.; Kimber, C.; Whitley, A.; Doney, A.S.; McCarthy, M.I.; Hattersley, A.T.; Morris, A.D.; Palmer, C.N. Variation in TCF7L2 influences therapeutic response to sulfonylureas: A GoDARTs study. Diabetes 2007, 56, 2178–2182. [Google Scholar] [CrossRef] [PubMed]
- Lyssenko, V.; Lupi, R.; Marchetti, P.; Del Guerra, S.; Orho-Melander, M.; Almgren, P.; Sjögren, M.; Ling, C.; Eriksson, K.F.; Lethagen, A.L.; et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J. Clin. Investig. 2007, 117, 2155–2163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilgaard, K.; Jensen, C.B.; Schou, J.H.; Lyssenko, V.; Wegner, L.; Brøns, C.; Vilsbøll, T.; Hansen, T.; Madsbad, S.; Holst, J.J.; et al. The T allele of rs7903146 TCF7L2 is associated with impaired insulinotropic action of incretin hormones, reduced 24 h profiles of plasma insulin and glucagon, and increased hepatic glucose production in young healthy men. Diabetologia 2009, 52, 1298–1307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shu, L.; Matveyenko, A.V.; Kerr-Conte, J.; Cho, J.H.; McIntosh, C.H.; Maedler, K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum. Mol. Genet. 2009, 18, 2388–2399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cusi, K. Role of obesity and lipotoxicity in the development of nonalcoholic steatohepatitis: Pathophysiology and clinical implications. Gastroenterology 2012, 142, 711–725. [Google Scholar] [CrossRef] [PubMed]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef]
- Kumashiro, N.; Yoshimura, T.; Cantley, J.L.; Majumdar, S.K.; Guebre-Egziabher, F.; Kursawe, R.; Vatner, D.F.; Fat, I.; Kahn, M.; Erion, D.M.; et al. Role of patatin-like phospholipase domain-containing 3 on lipid-induced hepatic steatosis and insulin resistance in rats. Hepatology 2013, 57, 1763–1772. [Google Scholar] [CrossRef] [PubMed]
- Omori, S.; Tanaka, Y.; Takahashi, A.; Hirose, H.; Kashiwagi, A.; Kaku, K.; Kawamori, R.; Nakamura, Y.; Maeda, S. Association of CDKAL1, IGF2BP2, CDKN2A/B, HHEX, SLC30A8, and KCNJ11 with susceptibility to type 2 diabetes in a Japanese population. Diabetes 2008, 57, 791–795. [Google Scholar] [CrossRef]
- Takeuchi, F.; Serizawa, M.; Yamamoto, K. Confirmation of multiple risk Loci and genetic impacts by a genome-wide association study of type 2 diabetes in the Japanese population. Diabetes 2009, 58, 1690–1699. [Google Scholar] [CrossRef]
- Miki, T.; Seino, S. Roles of KATP channels as metabolic sensors in acute metabolic changes. J. Mol. Cell. Cardiol. 2005, 38, 917–925. [Google Scholar] [CrossRef] [PubMed]
- Gloyn, A.L.; Weedon, M.N.; Owen, K.R.; Turner, M.J.; Knight, B.A.; Hitman, G.; Walker, M.; Levy, J.C.; Sampson, M.; Halford, S.; et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes 2003, 52, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Senokuchi, T.; Lu, M.; Takemoto, M.; Karim, M.F.; Go, C.; Sato, Y.; Hatta, M.; Yoshizawa, T.; Araki, E.; Miyazaki, J. Voltage-gated K+ channel KCNQ1 regulates insulin secretion in MIN6 β-cell line. Biochem. Biophys. Res. Commun. 2011, 407, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Wang, F.; Lu, H.; Ren, X.; Zou, J. Chromanol 293B, an inhibitor of KCNQ1 channels, enhances glucose-stimulated insulin secretion and increases glucagon-like peptide-1 level in mice. Islets 2014, 6, e962386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yasuda, K.; Miyake, K.; Horikawa, Y.; Hara, K.; Osawa, H.; Furuta, H.; Hirota, Y.; Mori, H.; Jonsson, A.; Sato, Y.; et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nat. Genet. 2008, 40, 1092–1097. [Google Scholar] [CrossRef] [PubMed]
- Unoki, H.; Takahashi, A.; Kawaguchi, T.; Hara, K.; Horikoshi, M.; Andersen, G.; Ng, D.P.; Holmkvist, J.; Borch-Johnsen, K.; Jørgensen, T.; et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nat. Genet. 2008, 40, 1098–1102. [Google Scholar] [CrossRef] [PubMed]
- Müssig, K.; Staiger, H.; Machicao, F.; Kirchhoff, K.; Guthoff, M.; Schäfer, S.A.; Kantartzis, K.; Silbernagel, G.; Stefan, N.; Holst, J.; et al. Association of Type 2 diabetes candidate polymorphisms in KCNQ1 with incretin and insulin secretion. Diabetes 2009, 58, 1715–1720. [Google Scholar] [CrossRef] [PubMed]
- Morris, A.P. Fine mapping of type 2 diabetes susceptibility loci. Curr. Diab. Rep. 2014, 14, 549. [Google Scholar] [CrossRef] [PubMed]
- Ferdaoussi, M.; Bergeron, V.; Zarrouki, B.; Kolic, J.; Cantley, J.; Fielitz, J.; Olson, E.N.; Prentki, M.; Biden, T.; MacDonald, P.E.; et al. G protein-coupled receptor (GPR)40-dependent potentiation of insulin secretion in mouse islets is mediated by protein kinase D1. Diabetologia 2012, 55, 2682–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nauck, M.A.; Heimesaat, M.M.; Orskov, C.; Holst, J.J.; Ebert, R.; Creutzfeldt, W. Preserved incretin activity of glucagon-like peptide 1 [7-36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type-2 diabetes mellitus. J. Clin. Investig. 1993, 91, 301–307. [Google Scholar] [CrossRef]
- Eng, J.; Kleinman, W.A.; Singh, L.; Singh, G.; Raufman, J.P. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom. Further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J. Biol. Chem. 1992, 267, 7402–7405. [Google Scholar] [PubMed]
- Greig, N.H.; Holloway, H.W.; De Ore, K.A.; Jani, D.; Wang, Y.; Zhou, J.; Garant, M.J.; Egan, J.M. Once daily injection of exendin-4 to diabetic mice achieves long-term beneficial effects on blood glucose concentrations. Diabetologia 1999, 42, 45–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, A.A.; Gedulin, B.R.; Bhavsar, S.; Bodkin, N.; Jodka, C.; Hansen, B.; Denaro, M. Glucose-lowering and insulin-sensitizing actions of exendin-4: Studies in obese diabetic (ob/ob, db/db) mice, diabetic fatty Zucker rats, and diabetic rhesus monkeys (Macaca mulatta). Diabetes 1999, 48, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Stoffers, D.A.; Habener, J.F.; Bonner-Weir, S. Exendin-4 stimulates both beta-cell replication and neogenesis, resulting in increased beta-cell mass and improved glucose tolerance in diabetic rats. Diabetes 1999, 48, 2270–2276. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.M.; Stanley, S.A.; Davis, R.; Brynes, A.E.; Frost, G.S.; Seal, L.J.; Ghatei, M.A.; Bloom, S.R. Exendin-4 reduces fasting and postprandial glucose and decreases energy intake in healthy volunteers. Am. J. Physiol. Endocrinol. Metab. 2001, 281, E155–E161. [Google Scholar] [CrossRef]
- Egan, J.M.; Clocquet, A.R.; Elahi, D. The insulinotropic effect of acute exendin-4 administered to humans: Comparison of nondiabetic state to type 2 diabetes. J. Clin. Endocrinol. Metab. 2002, 87, 1282–1290. [Google Scholar] [CrossRef]
- Kolterman, O.G.; Buse, J.B.; Fineman, M.S.; Gaines, E.; Heintz, S.; Bicsak, T.A.; Taylor, K.; Kim, D.; Aisporna, M.; Wang, Y.; et al. Synthetic exendin-4 (exenatide) significantly reduces postprandial and fasting plasma glucose in subjects with type 2 diabetes. J. Clin. Endocrinol. Metab. 2003, 88, 3082–3089. [Google Scholar] [CrossRef]
- Fineman, M.S.; Bicsak, T.A.; Shen, L.Z.; Taylor, K.; Gaines, E.; Varns, A.; Kim, D.; Baron, A.D. Effect on glycemic control of exenatide (synthetic exendin-4) additive to existing metformin and/or sulfonylurea treatment in patients with type 2 diabetes. Diabetes Care 2003, 26, 2370–2377. [Google Scholar] [CrossRef]
- Degn, K.B.; Brock, B.; Juhl, C.B.; Djurhuus, C.B.; Grubert, J.; Kim, D.; Han, J.; Taylor, K.; Fineman, M.; Schmitz, O. Effect of intravenous infusion of exenatide (synthetic exendin-4) on glucose-dependent insulin secretion and counterregulation during hypoglycemia. Diabetes 2004, 53, 2397–2403. [Google Scholar] [CrossRef]
- Buse, J.B.; Henry, R.R.; Han, J.; Kim, D.D.; Fineman, M.S.; Baron, A.D.; Exenatide-113 Clinical Study Group. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in sulfonylurea-treated patients with type 2 diabetes. Diabetes Care 2004, 27, 2628–2635. [Google Scholar] [CrossRef]
- Kendall, D.M.; Riddle, M.C.; Rosenstock, J.; Zhuang, D.; Kim, D.D.; Fineman, M.S.; Baron, A.D. Effects of exenatide (exendin-4) on glycemic control over 30 weeks in patients with type 2 diabetes treated with metformin and a sulfonylurea. Diabetes Care 2005, 28, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Ratner, R.E.; Han, J.; Kim, D.D.; Fineman, M.S.; Baron, A.D. Effects of exenatide (exendin-4) on glycemic control and weight over 30 weeks in metformin-treated patients with type 2 diabetes. Diabetes Care 2005, 28, 1092–1100. [Google Scholar] [CrossRef] [PubMed]
- Exenatide (Byetta) for type 2 diabetes. Med. Lett. Drugs Ther. 2005, 47, 45–46.
- Tran, K.L.; Park, Y.I.; Pandya, S.; Muliyil, N.J.; Jensen, B.D.; Huynh, K.; Nguyen, Q.T. Overview of Glucagon-Like Peptide-1 Receptor Agonists for the Treatment of Patients with Type 2 Diabetes. Am. Health Drug Benefits 2017, 10, 178–188. [Google Scholar]
- Kapitza, C.; Nosek, L.; Jensen, L.; Hartvig, H.; Jensen, C.B.; Flint, A. Semaglutide, a once-weekly human GLP-1 analog, does not reduce the bioavailability of the combined oral contraceptive, ethinylestradiol/levonorgestrel. J. Clin. Pharmacol. 2015, 55, 497–504. [Google Scholar] [CrossRef]
- Sharma, D.; Verma, S.; Vaidya, S.; Kalia, K.; Tiwari, V. Recent updates on GLP-1 agonists: Current advancements & challenges. Biomed. Pharmacother. 2018, 108, 952–962. [Google Scholar] [PubMed]
- Perfetti, R. Combining basal insulin analogs with glucagon-like peptide-1 mimetics. Diabetes Technol. Ther. 2011, 13, 873–881. [Google Scholar] [CrossRef]
- Raccah, D.; Lin, J.; Wang, E.; Germé, M.; Perfetti, R.; Bonadonna, R.C.; de Pablos-Velasco, P.; Roussel, R.; Rosenstock, J. Once-daily prandial lixisenatide versus once-daily rapid-acting insulin in patients with type 2 diabetes mellitus insufficiently controlled with basal insulin: Analysis of data from five randomized, controlled trials. J. Diabetes Complicat. 2014, 28, 40–44. [Google Scholar] [CrossRef]
- Mathieu, C.; Rodbard, H.W.; Cariou, B.; Handelsman, Y.; Philis-Tsimikas, A.; Ocampo Francisco, A.M.; Rana, A.; Zinman, B.; BEGIN: VICTOZA ADD-ON (NN1250-3948) study group. A comparison of adding liraglutide versus a single daily dose of insulin aspart to insulin degludec in subjects with type 2 diabetes (BEGIN: VICTOZA ADD-ON). Diabetes Obes. Metab. 2014, 16, 636–644. [Google Scholar] [CrossRef] [Green Version]
- Rosenstock, J.; Guerci, B.; Hanefeld, M.; Gentile, S.; Aronson, R.; Tinahones, F.J.; Roy-Duval, C.; Souhami, E.; Wardecki, M.; Ye, J.; et al. GetGoal Duo-2 Trial Investigators. Prandial Options to Advance Basal Insulin Glargine Therapy: Testing Lixisenatide Plus Basal Insulin Versus Insulin Glulisine Either as Basal-Plus or Basal-Bolus in Type 2 Diabetes: The GetGoal Duo-2 Trial. Diabetes Care 2016, 39, 1318–1328. [Google Scholar] [CrossRef]
- Dempsey, M.; Mocarski, M.; Langer, J.; Hunt, B. Ideglira is Associated with Improved Short-Term Clinical Outcomes and Cost Savings Compared with Insulin Glargine U100 Plus Insulin Aspart in the U.S. Endocr. Pract. 2018, 24, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Htike, Z.Z.; Zaccardi, F.; Papamargaritis, D.; Webb, D.R.; Khunti, K.; Davies, M.J. Efficacy and safety of glucagon-like peptide-1 receptor agonists in type 2 diabetes: A systematic review and mixed-treatment comparison analysis. Diabetes Obes. Metab. 2017, 19, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Y.; Quan, X.; Yang, Z.; Zeng, X.; Ji, L.; Sun, F.; Zhan, S. Efficacy and Acceptability of Glycemic Control of Glucagon-Like Peptide-1 Receptor Agonists among Type 2 Diabetes: A Systematic Review and Metwork Meta-Analysis. PLoS ONE 2016, 11, e0154206. [Google Scholar] [CrossRef] [PubMed]
- Bethel, M.A.; Patel, R.A.; Merrill, P.; Lokhnygina, Y.; Buse, J.B.; Mentz, R.J.; Pagidipati, N.J.; Chan, J.C.; Gustavson, S.M.; Iqbal, N.; et al. Cardiovascular outcomes with glucagon-like peptide-1 receptor agonists in patients with type 2 diabetes: A meta-analysis. Lancet Diabetes Endocrinol. 2018, 6, 105–113. [Google Scholar] [CrossRef]
- Tokuyama, Y.; Matsui, K.; Egashira, T.; Nozaki, O.; Ishizuka, T.; Kanatsuka, A. Five missense mutations in glucagon-like peptide 1 receptor gene in Japanese population. Diabetes Res. Clin. Pract. 2004, 66, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Beinborn, M.; Worrall, C.I.; McBride, E.W.; Kopin, A.S. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul. Pept. 2005, 130, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Sathananthan, A.; Man, C.D.; Micheletto, F.; Zinsmeister, A.R.; Camilleri, M.; Giesler, P.D.; Laugen, J.M.; Toffolo, G.; Rizza, R.A.; Cobelli, C.; et al. Common genetic variation in GLP1R and insulin secretion in response to exogenous GLP-1 in nondiabetic subjects: A pilot study. Diabetes Care 2010, 33, 2074–2076. [Google Scholar] [CrossRef] [PubMed]
- Koole, C.; Wootten, D.; Simms, J.; Valant, C.; Miller, L.J.; Christopoulos, A.; Sexton, P.M. Polymorphism and ligand dependent changes in human glucagon-like peptide-1 receptor (GLP-1R) function: Allosteric rescue of loss of function mutation. Mol. Pharmacol. 2011, 80, 486–497. [Google Scholar] [CrossRef] [PubMed]
- Koole, C.; Wootten, D.; Simms, J.; Miller, L.J.; Christopoulos, A.; Sexton, P.M. Differential impact of amino acid substitutions on critical residues of the human glucagon-like peptide-1 receptor involved in peptide activity and small-molecule allostery. J. Pharmacol. Exp. Ther. 2015, 353, 52–63. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Lee, Y.S.; Huang, Y.Y.; Hsieh, S.H.; Chen, Z.S.; Tsai, C.N. Polymorphisms of GLP-1 Receptor Gene and Response to GLP-1 Analogue in Patients with Poorly Controlled Type 2 Diabetes. J. Diabetes Res. 2015, 2015, 176949. [Google Scholar] [CrossRef] [PubMed]
- De Luis, D.A.; Diaz Soto, G.; Izaola, O.; Romero, E. Evaluation of weight loss and metabolic changes in diabetic patients treated with liraglutide, effect of RS 6923761 gene variant of glucagon-like peptide 1 receptor. J. Diabetes Complicat. 2015, 29, 595–598. [Google Scholar] [CrossRef] [PubMed]
- Jensterle, M.; Pirš, B.; Goričar, K.; Dolžan, V.; Janež, A. Genetic variability in GLP-1 receptor is associated with inter-individual differences in weight lowering potential of liraglutide in obese women with PCOS: A pilot study. Eur. J. Clin. Pharmacol. 2015, 71, 817–824. [Google Scholar] [CrossRef]
- Chedid, V.; Vijayvargiya, P.; Carlson, P.; Van Malderen, K.; Acosta, A.; Zinsmeister, A.; Camilleri, M. Allelic variant in the glucagon-like peptide 1 receptor gene associated with greater effect of liraglutide and exenatide on gastric emptying: A pilot pharmacogenetics study. Neurogastroenterol. Motil. 2018, 30, e13313. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.J.; Lundkvist, P.; Kamble, P.G.; Lau, J.; Martins, J.G.; Sjöström, C.D.; Schnecke, V.; Walentinsson, A.; Johnsson, E.; Eriksson, J.W. A Randomized Controlled Trial of Dapagliflozin Plus Once-Weekly Exenatide Versus Placebo in Individuals with Obesity and Without Diabetes: Metabolic Effects and Markers Associated with Bodyweight Loss. Diabetes Ther. 2018, 9, 1511–1532. [Google Scholar] [CrossRef] [PubMed]
- De Luis, D.A.; Ovalle, H.F.; Soto, G.D.; Izaola, O.; de la Fuente, B.; Romero, E. Role of genetic variation in the cannabinoid receptor gene (CNR1) (G1359A polymorphism) on weight loss and cardiovascular risk factors after liraglutide treatment in obese patients with diabetes mellitus type 2. J. Investig. Med. 2014, 62, 324–327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.M.; Xu, W.; Yan, X.M.; Li, M.X.Y.; Liang, H.; Weng, J.P. Association between SORCS1 rs1416406 and therapeutic effect of exenatide. Zhonghua Yi Xue Za Zhi 2017, 97, 1415–1419. [Google Scholar] [PubMed]
- Ferreira, M.C.; da Silva, M.E.R.; Fukui, R.T.; do Carmo Arruda-Marques, M.; Azhar, S.; Dos Santos, R.F. Effect of TCF7L2 polymorphism on pancreatic hormones after exenatide in type 2 diabetes. Diabetol. Metab. Syndr. 2019, 11, 10. [Google Scholar] [CrossRef] [PubMed]
- Škrha, J.; Šoupal, J.; Škrha, J., Jr.; Prázný, M. Glucose variability, HbA1c and microvascular complications. Rev. Endocr. Metab. Disord. 2016, 17, 103–110. [Google Scholar]
- Felder, C.C.; Glass, M. Cannabinoid receptors and their endogenous agonists. Ann. Rev. Pharmacol. Toxicol. 1998, 38, 179–200. [Google Scholar] [CrossRef]
- Aberle, J.; Fedderwitz, I.; Klages, N.; George, E.; Beil, F.U. Genetic variation in two proteins of the endocannabinoid system and their influence on body mass index and metabolism under low fat diet. Horm. Metab. Res. 2007, 39, 395–397. [Google Scholar] [CrossRef] [PubMed]
- De Luis, D.A.; González Sagrado, M.; Aller, R.; Conde, R.; Izaola, O.; de la Fuente, B.; Primo, D. Roles of G1359A polymorphism of the cannabinoid receptor gene (CNR1) on weight loss and adipocytokines after a hypocaloric diet. Nutr. Hosp. 2011, 26, 317–322. [Google Scholar] [PubMed]
- Antonio de Luis, D.; Sagrado, M.G.; Aller, R.; Conde, R.; Izaola, O.; de la Fuente, B.; Primo, D. Role of G1359A polymorphism of the cannabinoid receptor gene on weight loss and adipocytokines levels after two different hypocaloric diets. J. Nutr. Biochem. 2012, 23. [Google Scholar] [CrossRef] [PubMed]
- Clee, S.M.; Yandell, B.S.; Schueler, K.M.; Rabaglia, M.E.; Richards, O.C.; Raines, S.M.; Kabara, E.A.; Klass, D.M.; Mui, E.T.; Stapleton, D.S.; et al. Positional cloning of Sorcs1, a type 2 diabetes quantitative trait locus. Nat. Genet. 2006, 38, 688–693. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, M.O.; Lehman, D.M.; Taylor, K.D.; Guo, X.; Cui, J.; Quiñones, M.J.; Clee, S.M.; Yandell, B.S.; Blangero, J.; Hsueh, W.A.; et al. SORCS1: A novel human type 2 diabetes susceptibility gene suggested by the mouse. Diabetes 2007, 56, 1922–1929. [Google Scholar] [CrossRef] [PubMed]
- Florez, J.C.; Manning, A.K.; Dupuis, J.; McAteer, J.; Irenze, K.; Gianniny, L.; Mirel, D.B.; Fox, C.S.; Cupples, L.A.; Meigs, J.B. A 100K genome-wide association scan for diabetes and related traits in the Framingham Heart Study: Replication and integration with other genome-wide datasets. Diabetes 2007, 56, 3063–3074. [Google Scholar] [CrossRef]
- Schäfer, S.A.; Müssig, K.; Staiger, H.; Machicao, F.; Stefan, N.; Gallwitz, B.; Häring, H.U.; Fritsche, A. A common genetic variant in WFS1 determines impaired glucagon-like peptide-1-induced insulin secretion. Diabetologia 2009, 52, 1075–1082. [Google Scholar] [CrossRef] [Green Version]
- Sandhu, M.S.; Weedon, M.N.; Fawcett, K.A.; Wasson, J.; Debenham, S.L.; Daly, A.; Lango, H.; Frayling, T.M.; Neumann, R.J.; Sherva, R.; et al. Common variants in WFS1 confer risk of type 2 diabetes. Nat. Genet. 2007, 39, 951–953. [Google Scholar] [CrossRef]
- Pfutzner, A.; Kunt, T.; Hohberg, C.; Mondok, A.; Pahler, S.; Konrad, T.; Lubben, G.; Forst, T. Fasting intact proinsulin is a highly specific predictor of insulin resistance in type 2 diabetes. Diabetes Care 2004, 27, 682–687. [Google Scholar] [CrossRef]
- Strawbridge, R.J.; Dupuis, J.; Prokopenko, I.; Barker, A.; Ahlqvist, E.; Rybin, D.; Petrie, J.R.; Travers, M.E.; Bouatia-Naji, N.; Dimas, A.S.; et al. Genome-wide association identifies nine common variants associated with fasting proinsulin levels and provides new insights into the pathophysiology of type 2 diabetes. Diabetes 2011, 60, 2624–2634. [Google Scholar] [CrossRef]
- Shannon, J.A.; Fisher, S. The renal tubular reabsorption of glucose in the normal dog. Am. J. Physiol. 1938, 122, 765–774. [Google Scholar] [CrossRef]
- Vick, H.; Diedrich, D.F.; Baumann, K. Reevaluation of renal tubular glucose transport inhibition by phlorizin analogs. Am. J. Physiol. 1973, 224, 552–557. [Google Scholar] [CrossRef] [PubMed]
- Turner, R.J.; Moran, A. Heterogeneity of sodium-dependent D-glucose transport sites along the proximal tubule: Evidence from vesicle studies. Am. J. Physiol. 1982, 242, F406–F414. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Coady, M.J.; Ikeda, T.S.; Wright, E.M. Expression cloning and cDNA sequencing of the Na+/glucose co-transporter. Nature 1987, 330, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Kanai, Y.; Lee, W.S.; You, G.; Brown, D.; Hediger, M.A. The human kidney low affinity Na+/glucose cotransporter SGLT2. Delineation of the major renal reabsorptive mechanism for D-glucose. J. Clin. Investig. 1994, 93, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.M.; Loo, D.D.; Hirayama, B.A. Biology of human sodium glucose transporters. Physiol. Rev. 2011, 91, 733–794. [Google Scholar] [CrossRef] [PubMed]
- Vallon, V.; Platt, K.A.; Cunard, R.; Schroth, J.; Whaley, J.; Thomson, S.C.; Koepsell, H.; Rieg, T. SGLT2 mediates glucose reabsorption in the early proximal tubule. J. Am. Soc. Nephrol. 2011, 22, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Gorboulev, V.; Schürmann, A.; Vallon, V.; Kipp, H.; Jaschke, A.; Klessen, D.; Friedrich, A.; Scherneck, S.; Rieg, T.; Cunard, R.; et al. Na+-D-glucose cotransporter SGLT1 is pivotal for intestinal glucose absorption and glucose-dependent incretin secretion. Diabetes 2012, 61, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Rieg, T.; Masuda, T.; Gerasimova, M.; Mayoux, E.; Platt, K.; Powell, D.R.; Thomson, S.C.; Koepsell, H.; Vallon, V. Increase in SGLT1-mediated transport explains renal glucose reabsorption during genetic and pharmacological SGLT2 inhibition in euglycemia. Am. J. Physiol. Renal Physiol. 2014, 306, F188–F193. [Google Scholar] [CrossRef]
- Choi, C.I. Sodium-Glucose Cotranspoter 2 (SGLT2) Inhibitors from Natural Products: Discovery of Next-Generation Antihyperglycemic Agents. Molecules 2016, 21, 1136. [Google Scholar] [CrossRef] [PubMed]
- Blaschek, W. Natural Products as Lead Compounds for Sodium Glucose Cotransporter (SGLT) Inhibitors. Planta Med. 2017, 83, 985–993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rieg, T.; Vallon, V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia 2018, 61, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Ehrenkranz, R.R.L.; Lewis, N.G.; Kahn, C.R.; Roth, J. Phlorizin: A review. Diabetes Metab. Res. Rev. 2005, 21, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Chasis, H.; Jolliffe, N.; Smith, H.W. The action of phlorizin on the excretion of glucose, xylose, sucrose, creatinine and urea by man. J. Clin. Investig. 1933, 12, 1083–1090. [Google Scholar] [CrossRef]
- Rossetti, L.; Smith, D.; Shulman, G.I.; Papachristou, D.; DeFronzo, R.A. Correction of hyperglycemia with phlorizin normalizes tissue sensitivity to insulin in diabetic rats. J. Clin. Investig. 1987, 79, 1510–1515. [Google Scholar] [CrossRef] [PubMed]
- Dimitrakoudis, D.; Vranic, M.; Klip, A. Effects of hyperglycemia on glucose transporters of the muscle: Use of the renal glucose reabsorption inhibitor phlorizin to control glycemia. J. Am. Soc. Nephrol. 1992, 3, 1078–1091. [Google Scholar] [PubMed]
- Jonas, J.C.; Sharma, A.; Hasenkamp, W.; Ilkova, H.; Patane, G.; Laybutt, R.; Bonner-Weir, S.; Weir, G.C. Chronic hyperglycemia triggers loss of pancreatic β cell differentiation in an animal model of diabetes. J. Biol. Chem. 1999, 274, 14112–14121. [Google Scholar] [CrossRef] [PubMed]
- Katsuno, K.; Fujimori, Y.; Takemura, Y.; Hiratochi, M.; Itoh, F.; Komatsu, Y.; Fujikura, H.; Isaji, M. Sergliflozin, a novel selective inhibitor of low-affinity sodium glucose cotransporter (SGLT2), validates the critical role of SGLT2 in renal glucose reabsorption and modulates plasma glucose level. J. Pharmacol. Exp. Ther. 2007, 320, 323–330. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Ghani, M.A.; Norton, L.; DeFronzo, R.A. Role of sodium-glucose cotransporter 2 (SGLT 2) inhibitors in the treatment of type 2 diabetes. Endocr. Rev. 2011, 32, 515–531. [Google Scholar] [CrossRef]
- Meng, W.; Ellsworth, B.A.; Nirschl, A.A.; McCann, P.J.; Patel, M.; Girotra, R.N.; Wu, G.; Sher, P.M.; Morrison, E.P.; Biller, S.A.; et al. Discovery of dapagliflozin: A potent, selective renal sodium-dependent glucose cotransporter 2 (SGLT2) inhibitor for the treatment of type 2 diabetes. J. Med. Chem. 2008, 51, 1145–1149. [Google Scholar] [CrossRef]
- Han, S.; Hagan, D.L.; Taylor, J.R.; Xin, L.; Meng, W.; Biller, S.A.; Wetterau, J.R.; Washburn, W.N.; Whaley, J.M. Dapagliflozin, a selective SGLT2 inhibitor, improves glucose homeostasis in normal and diabetic rats. Diabetes 2008, 57, 1723–1729. [Google Scholar] [CrossRef]
- Komoroski, B.; Vachharajani, N.; Feng, Y.; Li, L.; Kornhauser, D.; Pfister, M. Dapagliflozin, a novel, selective SGLT2 inhibitor, improved glycemic control over 2 weeks in patients with type 2 diabetes mellitus. Clin. Pharmacol. Ther. 2009, 85, 513–519. [Google Scholar] [CrossRef] [PubMed]
- List, J.F.; Woo, V.; Morales, E.; Tang, W.; Fiedorek, F.T. Sodium-glucose cotransport inhibition with dapagliflozin in type 2 diabetes. Diabetes Care 2009, 32, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Wilding, J.P.; Norwood, P.; T’joen, C.; Bastien, A.; List, J.F.; Fiedorek, F.T. A study of dapagliflozin in patients with type 2 diabetes receiving high doses of insulin plus insulin sensitizers: Applicability of a novel insulin-independent treatment. Diabetes Care 2009, 32, 1656–1662. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Ramos, S.J.; Salsali, A.; Tang, W.; List, J.F. Dapagliflozin monotherapy in type 2 diabetic patients with inadequate glycemic control by diet and exercise: A randomized, double-blind, placebo-controlled, phase III trial. Diabetes Care 2010, 33, 2217–2224. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J.; Gross, J.L.; Pieters, A.; Bastien, A.; List, J.F. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with metformin: A randomised, double-blind, placebo-controlled trial. Lancet 2010, 375, 2223–2233. [Google Scholar] [CrossRef]
- Strojek, K.; Yoon, K.H.; Hruba, V.; Elze, M.; Langkilde, A.M.; Parikh, S. Effect of dapagliflozin in patients with type 2 diabetes who have inadequate glycaemic control with glimepiride: A randomized, 24-week, double-blind, placebo-controlled trial. Diabetes Obes. Metab. 2011, 13, 928–938. [Google Scholar] [CrossRef] [PubMed]
- Nauck, M.A.; Del Prato, S.; Meier, J.J.; Durán-García, S.; Rohwedder, K.; Elze, M.; Parikh, S.J. Dapagliflozin versus glipizide as add-on therapy in patients with type 2 diabetes who have inadequate glycemic control with metformin: A randomized, 52-week, double-blind, active controlled noninferiority trial. Diabetes Care 2011, 34, 2015–2022. [Google Scholar] [CrossRef]
- Rosenstock, J.; Vico, M.; Wei, L.; Salsali, A.; List, J.F. Effects of dapagliflozin, an SGLT2 inhibitor, on HbA1c, body weight, and hypoglycemia risk in patients with type 2 diabetes inadequately controlled on pioglitazone monotherapy. Diabetes Care 2012, 35, 1473–1478. [Google Scholar] [CrossRef]
- Aylsworth, A.; Dean, Z.; VanNorman, C.; Nkemdirim Okere, A. Dapagliflozin for the Treatment of Type 2 Diabetes Mellitus. Ann. Pharmacother. 2014, 48, 1202–1208. [Google Scholar] [CrossRef]
- Ansary, T.M.; Nakano, D.; Nishiyama, A. Diuretic Effects of Sodium Glucose Cotransporter 2 Inhibitors and Their Influence on the Renin-Angiotensin System. Int. J. Mol. Sci. 2019, 20, 629. [Google Scholar] [CrossRef]
- DeFronzo, R.A.; Davidson, J.A.; Del Prato, S. The role of the kidneys in glucose homeostasis: A new path towards normalizing glycaemia. Diabetes Obes. Metab. 2012, 14, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Gallo, L.A.; Wright, E.M.; Vallon, V. Probing SGLT2 as a therapeutic target for diabetes: Basic physiology and consequences. Diab. Vasc. Dis. Res. 2015, 12, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Fitchett, D.; Zinman, B.; Wanner, C.; Lachin, J.M.; Hantel, S.; Salsali, A.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Inzucchi, S.E.; et al. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: Results of the EMPA-REG OUTCOME® trial. Eur. Heart J. 2016, 37, 1526–1534. [Google Scholar] [CrossRef] [PubMed]
- Wanner, C.; Inzucchi, S.E.; Lachin, J.M.; Fitchett, D.; von Eynatten, M.; Mattheus, M.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Zinman, B.; et al. Empagliflozin and Progression of Kidney Disease in Type 2 Diabetes. N. Engl. J. Med. 2016, 375, 323–334. [Google Scholar] [CrossRef]
- Neal, B.; Perkovic, V.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Erondu, N.; Shaw, W.; Law, G.; Desai, M.; Matthews, D.R.; et al. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Jardine, M.J.; Mahaffey, K.W.; Neal, B.; Agarwal, R.; Bakris, G.L.; Brenner, B.M.; Bull, S.; Cannon, C.P.; Charytan, D.M.; de Zeeuw, D.; et al. The Canagliflozin and Renal Endpoints in Diabetes with Established Nephropathy Clinical Evaluation (CREDENCE) Study Rationale, Design, and Baseline Characteristics. Am. J. Nephrol. 2017, 46, 462–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rådholm, K.; Figtree, G.; Perkovic, V.; Solomon, S.D.; Mahaffey, K.W.; de Zeeuw, D.; Fulcher, G.; Barrett, T.D.; Shaw, W.; Desai, M.; et al. Canagliflozin and Heart Failure in Type 2 Diabetes Mellitus. Circulation 2018, 138, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francke, S.; Mamidi, R.N.; Solanki, B.; Scheers, E.; Jadwin, A.; Favis, R.; Devineni, D. In vitro metabolism of canagliflozin in human liver, kidney, intestine microsomes, and recombinant uridine diphosphate glucuronosyltransferases (UGT) and the effect of genetic variability of UGT enzymes on the pharmacokinetics of canagliflozin in humans. J. Clin. Pharmacol. 2015, 55, 1061–1072. [Google Scholar] [CrossRef]
- Hoeben, E.; De Winter, W.; Neyens, M.; Devineni, D.; Vermeulen, A.; Dunne, A. Population Pharmacokinetic Modeling of Canagliflozin in Healthy Volunteers and Patients with Type 2 Diabetes Mellitus. Clin. Pharmacokinet. 2016, 55, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Zimdahl, H.; Haupt, A.; Brendel, M.; Bour, L.; Machicao, F.; Salsali, A.; Broedl, U.C.; Woerle, H.J.; Häring, H.U.; Staiger, H. Influence of common polymorphisms in the SLC5A2 gene on metabolic traits in subjects at increased risk of diabetes and on response to empagliflozin treatment in patients with diabetes. Pharmacogenet. Genom. 2017, 27, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, J.W.; Lundkvist, P.; Jansson, P.A.; Johansson, L.; Kvarnström, M.; Moris, L.; Miliotis, T.; Forsberg, G.B.; Risérus, U.; Lind, L.; Oscarsson, J. Effects of dapagliflozin and n-3 carboxylic acids on non-alcoholic fatty liver disease in people with type 2 diabetes: A double-blind randomised placebo-controlled study. Diabetologia 2018, 61, 1923–1934. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Tugnait, M.; Zhu, T.; Kimoto, E.; Miao, Z.; Mascitti, V.; Yang, X.; Tan, B.; Walsky, R.L.; Chupka, J.; et al. Preclinical species and human disposition of PF-04971729, a selective inhibitor of the sodium-dependent glucose cotransporter 2 and clinical candidate for the treatment of type 2 diabetes mellitus. Drug. Metab. Dispos. 2011, 39, 1609–1619. [Google Scholar] [CrossRef]
- Kasichayanula, S.; Liu, X.; Griffen, S.C.; Lacreta, F.P.; Boulton, D.W. Effects of rifampin and mefenamic acid on the pharmacokinetics and pharmacodynamics of dapagliflozin. Diabetes Obes. Metab. 2013, 15, 280–283. [Google Scholar] [CrossRef]
- Miyata, A.; Hasegawa, M.; Hachiuma, K.; Mori, H.; Horiuchi, N.; Mizuno-Yasuhira, A.; Chino, Y.; Jingu, S.; Sakai, S.; Samukawa, Y.; et al. Metabolite profiling and enzyme reaction phenotyping of luseogliflozin, a sodium-glucose cotransporter 2 inhibitor, in humans. Xenobiotica 2017, 47, 332–345. [Google Scholar] [CrossRef]
- Stingl, J.C.; Bartels, H.; Viviani, R.; Lehmann, M.L.; Brockmöller, J. Relevance of UDP-glucuronosyltransferase polymorphisms for drug dosing: A quantitative systematic review. Pharmacol. Ther. 2014, 141, 92–116. [Google Scholar] [CrossRef]
- Hu, D.G.; Mackenzie, P.I.; McKinnon, R.A.; Meech, R. Genetic polymorphisms of human UDP-glucuronosyltransferase (UGT) genes and cancer risk. Drug Metab. Rev. 2016, 48, 47–69. [Google Scholar] [CrossRef]
- Calado, J.; Loeffler, J.; Sakallioglu, O.; Gok, F.; Lhotta, K.; Barata, J.; Rueff, J. Familial renal glucosuria: SLC5A2 mutation analysis and evidence of salt-wasting. Kidney Int. 2006, 69, 852–855. [Google Scholar] [CrossRef] [Green Version]
- Santer, R.; Calado, J. Familial renal glucosuria and SGLT2: From a mendelian trait to a therapeutic target. Clin. J. Am. Soc. Nephrol. 2010, 5, 133–141. [Google Scholar] [CrossRef]
- Merino, J.; Udler, M.S.; Leong, A.; Meigs, J.B. A Decade of Genetic and Metabolomic Contributions to Type 2 Diabetes Risk Prediction. Curr. Diab. Rep. 2017, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wessel, J. The Continuing Evolution of Precision Health in Type 2 Diabetes: Achievements and Challenges. Curr. Diab. Rep. 2019, 19, 16. [Google Scholar] [CrossRef] [PubMed]
- Vincent, S.H.; Reed, J.R.; Bergman, A.J.; Elmore, C.S.; Zhu, B.; Xu, S.; Ebel, D.; Larson, P.; Zeng, W.; Chen, L.; et al. Metabolism and excretion of the dipeptidyl peptidase 4 inhibitor [14C]sitagliptin in humans. Drug Metab. Dispos. 2007, 35, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Boulton, D.W.; Barros, A., Jr.; Wang, L.; Cao, K.; Bonacorsi, S.J., Jr.; Iyer, R.A.; Humphreys, W.G.; Christopher, L.J. Characterization of the in vitro and in vivo metabolism and disposition and cytochrome P450 inhibition/induction profile of saxagliptin in human. Drug Metab. Dispos. 2012, 40, 1345–1356. [Google Scholar] [CrossRef] [PubMed]
- Ishiguro, N.; Shimizu, H.; Kishimoto, W.; Ebner, T.; Schaefer, O. Evaluation and prediction of potential drug-drug interactions of linagliptin using in vitro cell culture methods. Drug Metab. Dispos. 2013, 41, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Kasichayanula, S.; Liu, X.; Lacreta, F.; Griffen, S.C.; Boulton, D.W. Clinical pharmacokinetics and pharmacodynamics of dapagliflozin, a selective inhibitor of sodium-glucose co-transporter type 2. Clin. Pharmacokinet. 2014, 53, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Breljak, D.; Onishi, A.; Batz, F.; Patel, R.; Huang, W.; Song, P.; Freeman, B.; Mayoux, E.; Koepsell, H.; et al. Organic anion transporter OAT3 enhances the glucosuric effect of the SGLT2 inhibitor empagliflozin. Am. J. Physiol. Renal Physiol. 2018, 315, F386–F394. [Google Scholar] [CrossRef] [PubMed]
- Garber, A.J.; Abrahamson, M.J.; Barzilay, J.I.; Blonde, L.; Bloomgarden, Z.T.; Bush, M.A.; Dagogo-Jack, S.; DeFronzo, R.A.; Einhorn, D.; Fonseca, V.A.; et al. Consensus Statement by the American Association of Clinical Endocrinologists and American College of Endocrinology on the Comprehensive Type 2 Diabetes Management Algorithm—2018 Executive Summary. Endocr. Pract. 2018, 24, 91–120. [Google Scholar] [CrossRef] [PubMed]
- Yassin, S.A.; Aroda, V.R. Sodium-glucose cotransporter 2 inhibitors combined with dipeptidyl peptidase-4 inhibitors in the management of type 2 diabetes: A review of current clinical evidence and rationale. Drug Des. Devel. Ther. 2017, 11, 923–937. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Gao, X.; Yang, W.; Han, X.; Ji, L. Efficacy and Safety of Initial Combination Therapy in Treatment-Naïve Type 2 Diabetes Patients: A Systematic Review and Meta-analysis. Diabetes Ther. 2018, 9, 1995–2014. [Google Scholar] [CrossRef]
- Milder, T.Y.; Stocker, S.L.; Abdel Shaheed, C.; McGrath-Cadell, L.; Samocha-Bonet, D.; Greenfield, J.R.; Day, R.O. Combination Therapy with an SGLT2 Inhibitor as Initial Treatment for Type 2 Diabetes: A Systematic Review and Meta-Analysis. J. Clin. Med. 2019, 8, 45. [Google Scholar] [CrossRef]


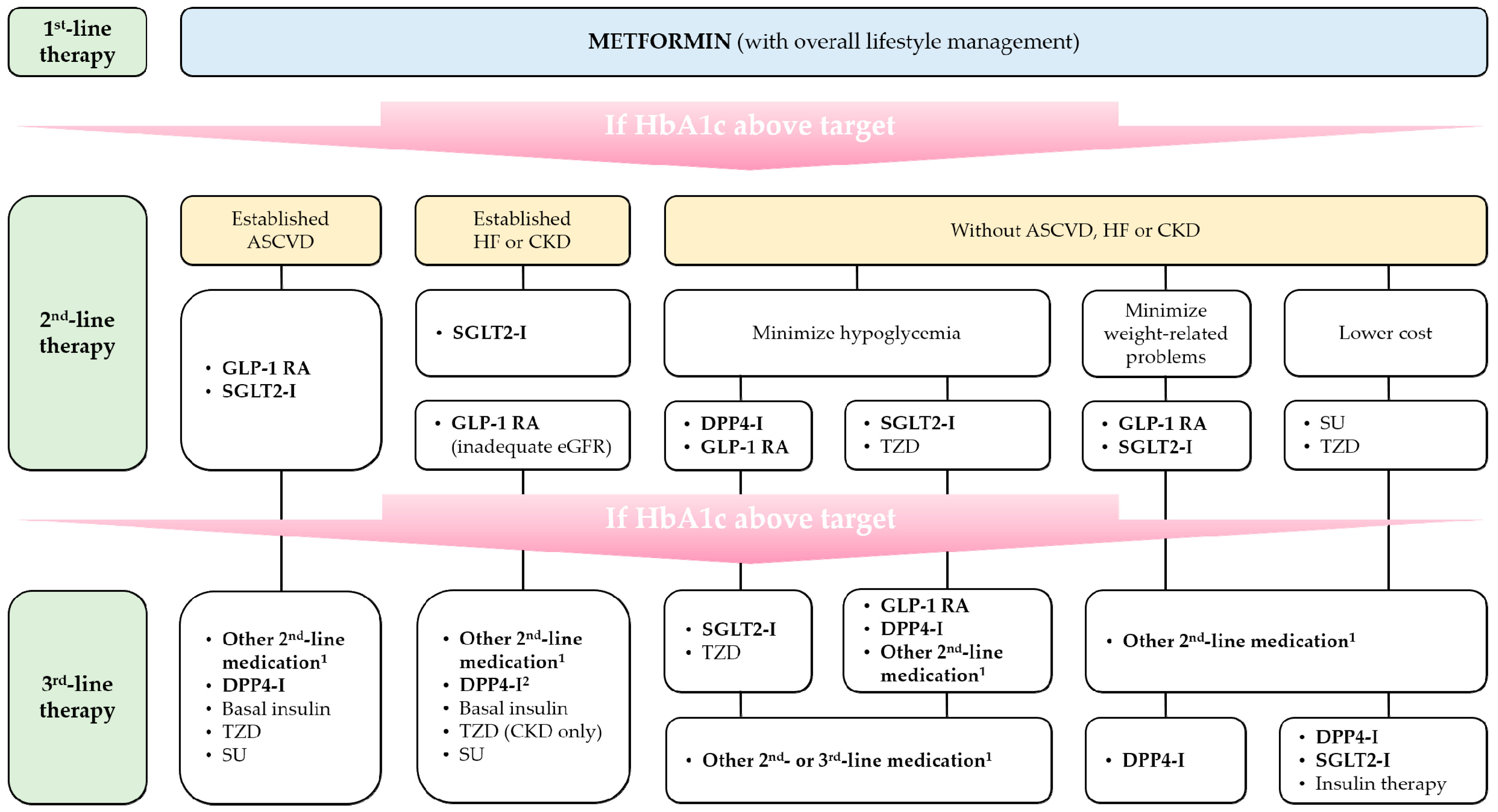{kind=link}
| Drug Class | Examples 1 (Currently Marketed as a Single Active Ingredient) |
|---|---|
| Oral | |
| α-Glucosidase inhibitors | Acarbose, miglitol |
| Biguanides | Metformin |
| Bile acid sequestrants | Colesevelam |
| DPP-4 inhibitors | Alogliptin, linagliptin, saxagliptin, sitagliptin, vildagliptin |
| Dopamine-2 agonists | Bromocriptine |
| Meglitinides | Nateglinide, repaglinide |
| SGLT2 inhibitors | Canagliflozin, dapagliflozin, empagliflozin, ertugliflozin |
| Sulfonylureas | Chlorpropamide, gliclazide, glimepiride, glipizide, glyburide (glibenclamide), tolazamide, tolbutamide |
| Thiazolidinediones | Pioglitazone, rosiglitazone |
| Injectable | |
| Amylin analogs | Pramlintide |
| GLP-1 receptor agonists | Albiglutide, dulaglutide, exenatide, liraglutide, lixisenatide, semaglutide |
| Insulin and its analogs | Insulin aspart, insulin degludec, insulin detemir, insulin glargine, insulin glulisine, insulin human, insulin lispro, human NPH (neutral protamine Hagedorn), human regular |
| Gene | Study Population | Dosage | Genetic Variant(s) | Clinical Outcome(s) | Ref. |
|---|---|---|---|---|---|
| DPP4 | 27 T2D patients with hypertension and 38 healthy controls | Sitagliptin 100 mg/day or a single dose of 200 mg | rs2909451 rs759717 | Increased DPP-4 activity during sitagliptin treatment in rs2909451 TT genotype and rs759717 CC genotype; rs2909451 genotype was only considered a predictive factor for DPP-4 activity | [32] |
| GLP1R | 246 Korean patients with T2D | Various (dosage not provided) | rs3765467 | Higher reduction in HbA1c and higher DPP-4 inhibitor responder proportion observed in GA/AA genotype | [33] |
| 140 white patients treated for T2D in outpatient clinics | Sitagliptin or vildagliptin 100 mg/day | rs6923761 (p.Gly168Ser) | Lower reduction in HbA1c in Ser/Ser genotype | [34] | |
| TCF7L2 | 693 T2D patients from four phase III clinical trials | Linagliptin 5 mg/day | rs7903146 | Lower decreases in HbA1c and 2-h PG levels from baseline in TT genotype | [35] |
| PNPLA3 | 41 patients with T2D and NAFLD | Alogliptin 25 mg/day | rs738409 (p.Ile148Met) | Positive correlation between improvement in HbA1c and changes in liver aminotransferase levels in CG/GG genotype; higher reductions in total cholesterol, hyaluronic acid, and triglyceride in CG/GG genotype | [36] |
| CDKAL1 | 512 T2D patients receiving DPP-4 inhibitor treatment | Various (dosage not provided) | rs7754840 rs7756992 | Higher reduction in HbA1c in rs7754840 CC genotype and rs7756992 GG genotype | [37] |
| KCNJ11 | 662 subjects with T2D (331 receiving DPP-4 inhibitor and 331 receiving other medications) | Sitagliptin 100 mg/day; vildagliptin 50–200 mg/day; linagliptin 5 mg/day | rs2285676 | Strong association between poor DPP-4 inhibitor efficacy and T-allele (OR = 1.479; 95% CI = 0.753–1.403), as a genetic predictor of DPP-4 inhibitor treatment response | [38] |
| KCNQ1 | 137 European patients from five outpatient clinics treated for T2D | Sitagliptin or vildagliptin 100 mg/day | rs163184 | Less reduction in HbA1c in G-allele carriers | [39] |
| PRKD1 | 88 resistant and 83 sensitive responders to T2D treatment | Various (dosage not provided) | rs57803087 | Strong association with DPP-4 inhibitor response and rs57803087 SNP from GWAS and replication study | [40] |
| Gene | Study Population | Dosage | Genetic Variant(s) | Clinical Outcome(s) | Ref. |
|---|---|---|---|---|---|
| GLP1R | 36 patients with T2D | Exenatide 5 μg twice daily | rs3765467 rs761386 (in complete LD (r2 = 1) with rs5875654) | Decreased (rs3765467 CT/TT) or increased (rs761386 CT/TT) SDPG levels; higher 2-h PG after 75 g OGTT in rs761386 CT/TT genotype | [94] |
| 90 patients with T2D and overweight (BMI > 25 kg/m2) | Liraglutide 1.8 mg/day | rs6923761 | Decreased waist circumference, waist-to-hip ratio and systolic blood pressure in GA/AA genotype, as an independent predictor for weight and fat mass reduction | [95] | |
| 20 strong responder and 37 poor responder obese women with PCOS | Liraglutide 1.2 mg/day | rs10305420 rs6923761 | Higher rs10305420 T-allele in poor responders; higher rs6923761 A-allele in strong responders; the best response to liraglutide observed in combined C-A haplotype (OR = 3.85; 95% CI = 1.24–11.96) | [96] | |
| 20 obese individuals with rapid gastric emptying (exenatide) and 40 obese individuals with normal or rapid gastric emptying (liraglutide) | Exenatide 5 μg twice daily; liraglutide 3 mg/day | rs6923761 | Prolonged gastric emptying half-life in GA/AA genotype (more remarkable in liraglutide treatment); no effect on body weight | [97] | |
| WFS1 | 40 obese patient without T2D | Long-acting exenatide 2 mg/week (plus dapagliflozin 10 mg) | rs10010131 | Higher body weight loss in A-allele carriers | [98] |
| CNR1 | 86 patients with T2D and obesity (BMI > 30 kg/m2) | Liraglutide 1.8 mg/day | rs1049353 (G1359A) | Decreased total cholesterol and LDL-cholesterol in GG genotype; improved HOMA-IR in GA/AA genotype | [99] |
| SORCS1 | 101 newly diagnosed T2D patients from CONFIDENCE study | Exenatide 5 μg twice daily (week 1–4) 10 μg twice daily (week 5–48) | rs1416406 | Higher reduction in proinsulin/insulin ratio in GG genotype | [100] |
| TCF7L2 | 46 patients with T2D completed a 500-kcal mixed-meal test | Exenatide 5 μg twice daily (week 1–4) 10 μg twice daily (week 5–8) | rs7903146 | Higher basal insulin and proinsulin, and reductions in insulin, proinsulin and C-peptide in CT/TT genotype | [101] |
| Gene | Study Population | Dosage | Genetic Variant(s) | Clinical Outcome(s) | Ref. |
|---|---|---|---|---|---|
| UGT1A9 | 134 healthy participants and T2D patients from 7 clinical trials | Canagliflozin 50–300 mg/day | UGT1A9*3 (rs72551330; p.Met33Thr) | Higher dose-normalized steady-state AUC (AUCτ,ss) for canagliflozin and M5, M/P ratio for M5 AUCτ,ss; lower AUCτ,ss for M7, M/P ratio for M7 AUCτ,ss (UGT1A9*3 carriers) | [153] |
| 1616 healthy volunteers and T2D patients from 14 clinical trials | Canagliflozin 50–1600 mg/day | UGT1A9*3 (rs72551330; p.Met33Thr) | Higher median dose-normalized canagliflozin AUC in UGT1A9*3 carriers (ratio = 1.26; 95% CI = 1.08–1.44) | [154] | |
| SLC5A2 | 979 patients from 4 phase III clinical trials | Empagliflozin 10 or 25 mg/day | rs3116650 rs3116149 rs11646054 | Increased systolic blood pressure by rs3116650 A-allele, rs3116149 A-allele and rs11646054 C-allele; decreased FPG levels in rs3116149 AA genotype | [155] |
| PNPLA3 | 20 dapagliflozin alone and 20 dapagliflozin plus omega-3 carboxylic acids treated subjects with T2D and NAFLD | Dapagliflozin 10 mg/day | rs738409 (p.Ile148Met) | Lower reduction in liver PDFF in dapagliflozin alone treatment group; higher reduction in liver PDFF in dapagliflozin plus omega-3 carboxylic acids treatment group (CG/GG genotype) | [156] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Heo, C.U.; Choi, C.-I. Current Progress in Pharmacogenetics of Second-Line Antidiabetic Medications: Towards Precision Medicine for Type 2 Diabetes. J. Clin. Med. 2019, 8, 393. https://doi.org/10.3390/jcm8030393
Heo CU, Choi C-I. Current Progress in Pharmacogenetics of Second-Line Antidiabetic Medications: Towards Precision Medicine for Type 2 Diabetes. Journal of Clinical Medicine. 2019; 8(3):393. https://doi.org/10.3390/jcm8030393
Chicago/Turabian StyleHeo, Chan Uk, and Chang-Ik Choi. 2019. "Current Progress in Pharmacogenetics of Second-Line Antidiabetic Medications: Towards Precision Medicine for Type 2 Diabetes" Journal of Clinical Medicine 8, no. 3: 393. https://doi.org/10.3390/jcm8030393
APA StyleHeo, C. U., & Choi, C. -I. (2019). Current Progress in Pharmacogenetics of Second-Line Antidiabetic Medications: Towards Precision Medicine for Type 2 Diabetes. Journal of Clinical Medicine, 8(3), 393. https://doi.org/10.3390/jcm8030393