UPR: An Upstream Signal to EMT Induction in Cancer

Abstract
:
1. Introduction
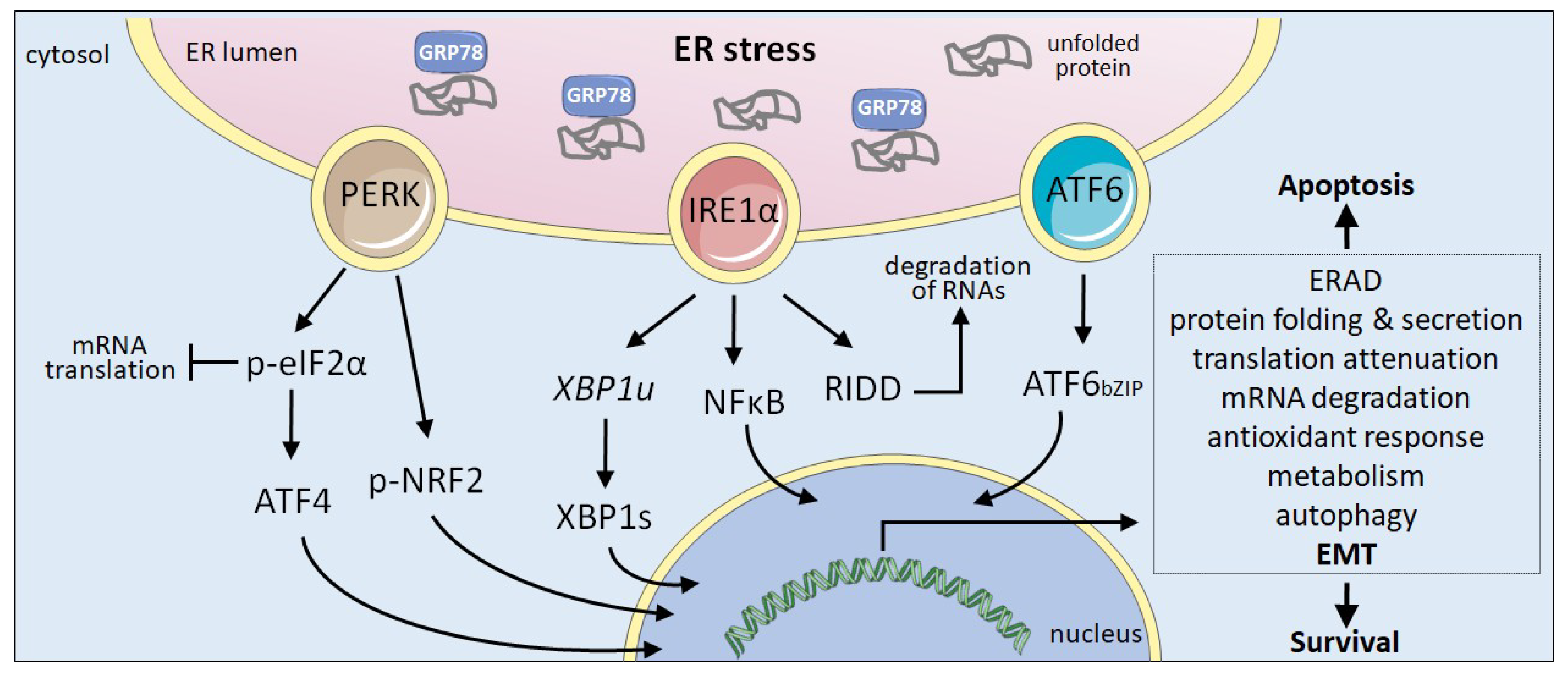
2. UPR Signalling Components
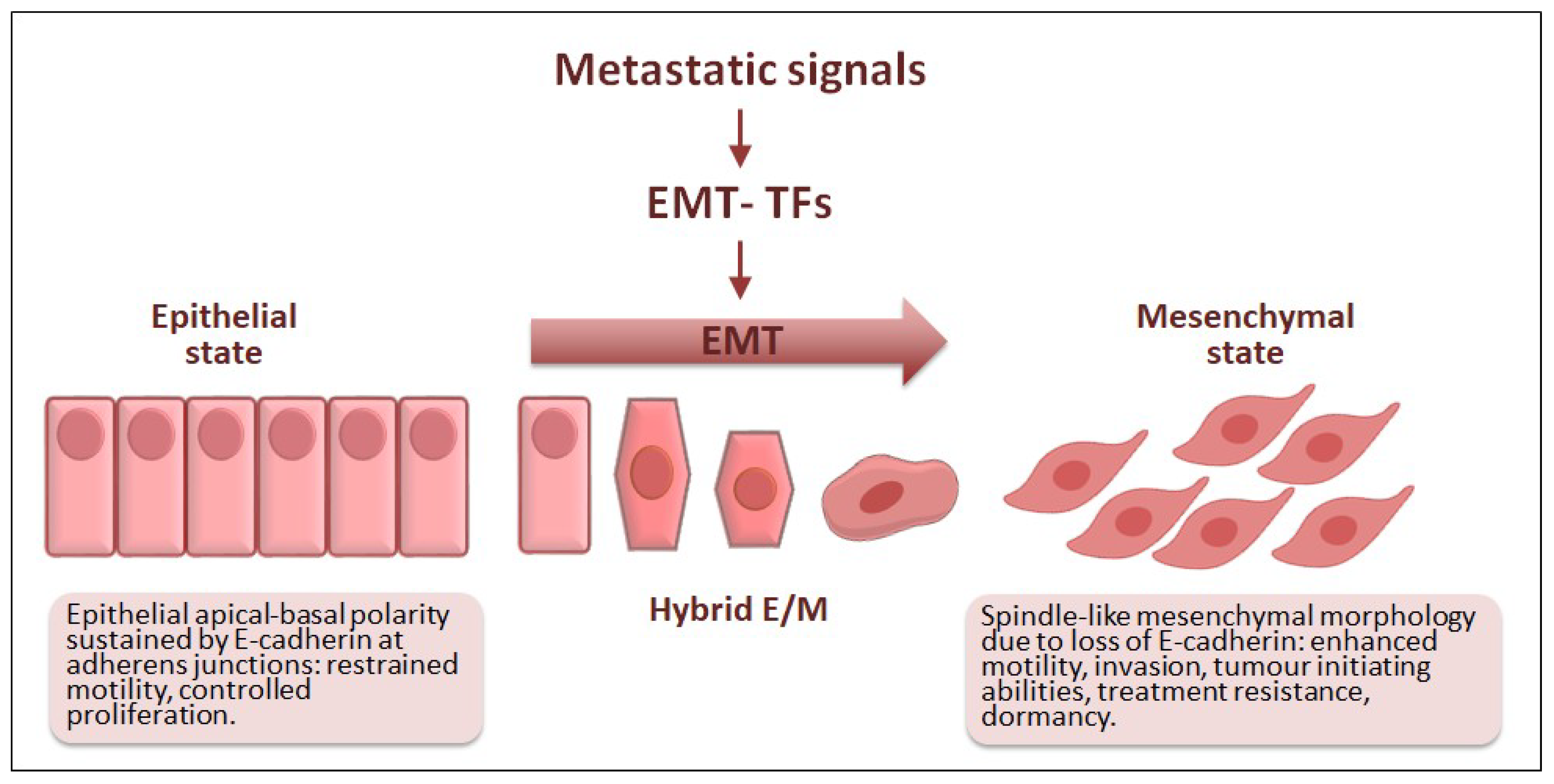
3. EMT: A Brief Update
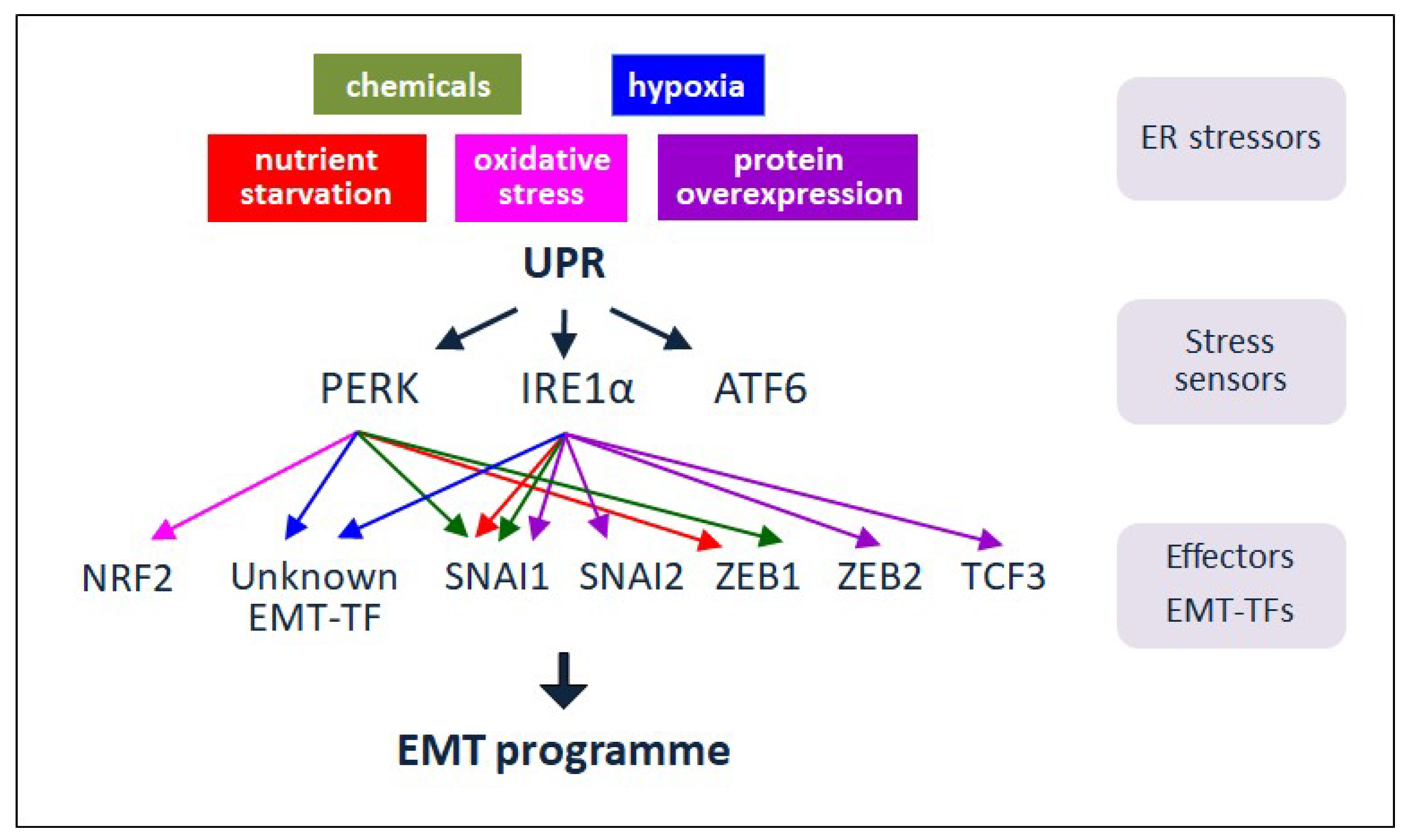
4. ER Stressors, UPR and EMT
4.1. Cell Extrinsic Stressors
4.1.1. Chemicals
4.1.2. Hypoxia
4.1.3. Nutrient Starvation
4.2. Cell Intrinsic Stressors
4.2.1. Oncogenes
4.2.2. Oxidative Stress
4.2.3. Alterations in UPR Signalling Components
4.2.4. Protein Overexpression
5. UPR and EMT Footprint in Human Tumours
6. UPR and Therapeutic Opportunities
7. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Meusser, B.; Hirsch, C.; Jarosch, E.; Sommer, T. ERAD: The long road to destruction. Nat. Cell Biol. 2005, 7, 766–772. [Google Scholar] [CrossRef]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Ron, D.; Walter, P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol. 2007, 8, 519–529. [Google Scholar] [CrossRef]
- Koumenis, C.; Naczki, C.; Koritzinsky, M.; Rastani, S.; Diehl, A.; Sonenberg, N.; Koromilas, A.; Wouters, B.G. Regulation of protein synthesis by hypoxia via activation of the endoplasmic reticulum kinase PERK and phosphorylation of the translation initiation factor eIF2alpha. Mol. Cell. Biol. 2002, 22, 7405–7416. [Google Scholar] [CrossRef]
- Blais, J.D.; Filipenko, V.; Bi, M.; Harding, H.P.; Ron, D.; Koumenis, C.; Wouters, B.G.; Bell, J.C. Activating transcription factor 4 is translationally regulated by hypoxic stress. Mol. Cell. Biol. 2004, 24, 7469–7482. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Diehl, J.A. PERK-dependent activation of Nrf2 contributes to redox homeostasis and cell survival following endoplasmic reticulum stress. J. Biol. Chem. 2004, 279, 20108–20117. [Google Scholar] [CrossRef] [PubMed]
- Cullinan, S.B.; Zhang, D.; Hannink, M.; Arvisais, E.; Kaufman, R.J.; Diehl, J.A. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 2003, 23, 7198–7209. [Google Scholar] [CrossRef]
- Del Vecchio, C.A.; Feng, Y.; Sokol, E.S.; Tillman, E.J.; Sanduja, S.; Reinhardt, F.; Gupta, P.B. De-differentiation confers multidrug resistance via noncanonical PERK-Nrf2 signaling. PLoS Biol. 2014, 12, e1001945. [Google Scholar] [CrossRef]
- Lee, A.H.; Iwakoshi, N.N.; Glimcher, L.H. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol. Cell. Biol. 2003, 23, 7448–7459. [Google Scholar] [CrossRef]
- Acosta-Alvear, D.; Zhou, Y.; Blais, A.; Tsikitis, M.; Lents, N.H.; Arias, C.; Lennon, C.J.; Kluger, Y.; Dynlacht, B.D. XBP1 controls diverse cell type- and condition-specific transcriptional regulatory networks. Mol. Cell 2007, 27, 53–66. [Google Scholar] [CrossRef]
- Hetz, C.; Martinon, F.; Rodriguez, D.; Glimcher, L.H. The unfolded protein response: Integrating stress signals through the stress sensor IRE1alpha. Physiol. Rev. 2011, 91, 1219–1243. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Oakes, S.A. Proteostasis control by the unfolded protein response. Nat. Cell Biol. 2015, 17, 829–838. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, R.; Wang, L.; Wang, E.S.; Perera, B.G.; Igbaria, A.; Morita, S.; Prado, K.; Thamsen, M.; Caswell, D.; Macias, H.; et al. Allosteric inhibition of the IRE1alpha RNase preserves cell viability and function during endoplasmic reticulum stress. Cell 2014, 158, 534–548. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.B.; Koong, A.C.; Niwa, M. Ire1 has distinct catalytic mechanisms for XBP1/HAC1 splicing and RIDD. Cell Rep. 2014, 9, 850–858. [Google Scholar] [CrossRef]
- Han, D.; Lerner, A.G.; Vande Walle, L.; Upton, J.P.; Xu, W.; Hagen, A.; Backes, B.J.; Oakes, S.A.; Papa, F.R. IRE1alpha kinase activation modes control alternate endoribonuclease outputs to determine divergent cell fates. Cell 2009, 138, 562–575. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef] [PubMed]
- Haze, K.; Yoshida, H.; Yanagi, H.; Yura, T.; Mori, K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol. Biol. Cell 1999, 10, 3787–3799. [Google Scholar] [CrossRef]
- Adachi, Y.; Yamamoto, K.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. ATF6 is a transcription factor specializing in the regulation of quality control proteins in the endoplasmic reticulum. Cell Struct. Funct. 2008, 33, 75–89. [Google Scholar] [CrossRef]
- Yoshida, H.; Haze, K.; Yanagi, H.; Yura, T.; Mori, K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J. Biol. Chem. 1998, 273, 33741–33749. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Sato, T.; Matsui, T.; Sato, M.; Okada, T.; Yoshida, H.; Harada, A.; Mori, K. Transcriptional induction of mammalian ER quality control proteins is mediated by single or combined action of ATF6alpha and XBP1. Dev. Cell 2007, 13, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Shoulders, M.D.; Ryno, L.M.; Genereux, J.C.; Moresco, J.J.; Tu, P.G.; Wu, C.; Yates, J.R., 3rd; Su, A.I.; Kelly, J.W.; Wiseman, R.L. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013, 3, 1279–1292. [Google Scholar] [CrossRef]
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Hay, E.D. Organization and Fine Structure of Epithelium and Mesenchyme in the Developing Chick Embryo; Fleischmajer, R., Billingham, R.E., Eds.; Williams & Wilkins Co.: Baltimore, MD, USA, 1968; pp. 31–35. [Google Scholar]
- Nieto, M.A. The ins and outs of the epithelial to mesenchymal transition in health and disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 347–376. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell Stem Cell 2019, 24, 65–78. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- Nieto, M.A.; Cano, A. The epithelial-mesenchymal transition under control: Global programs to regulate epithelial plasticity. Semin. Cancer Biol. 2012, 22, 361–368. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Birchmeier, W.; Behrens, J. Cadherin expression in carcinomas: Role in the formation of cell junctions and the prevention of invasiveness. Biochim. Biophys. Acta 1994, 1198, 11–26. [Google Scholar] [CrossRef]
- De Craene, B.; Berx, G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. [Google Scholar] [CrossRef]
- Ocaña, O.H.; Corcoles, R.; Fabra, A.; Moreno-Bueno, G.; Acloque, H.; Vega, S.; Barrallo-Gimeno, A.; Cano, A.; Nieto, M.A. Metastatic colonization requires the repression of the epithelial-mesenchymal transition inducer Prrx1. Cancer Cell 2012, 22, 709–724. [Google Scholar] [CrossRef]
- Stankic, M.; Pavlovic, S.; Chin, Y.; Brogi, E.; Padua, D.; Norton, L.; Massague, J.; Benezra, R. TGF-beta-Id1 signaling opposes Twist1 and promotes metastatic colonization via a mesenchymal-to-epithelial transition. Cell Rep. 2013, 5, 1228–1242. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef]
- Pastushenko, I.; Blanpain, C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019, 29, 212–226. [Google Scholar] [CrossRef] [PubMed]
- Santamaria, P.G.; Moreno-Bueno, G.; Portillo, F.; Cano, A. EMT: Present and future in clinical oncology. Mol. Oncol. 2017, 11, 718–738. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [Green Version]
- Morel, A.P.; Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Puisieux, A. Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS ONE 2008, 3, e2888. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef]
- Avril, T.; Vauleon, E.; Chevet, E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis 2017, 6, e373. [Google Scholar] [CrossRef] [Green Version]
- Papaioannou, A.; Chevet, E. Driving Cancer Tumorigenesis and Metastasis Through UPR Signaling. Curr. Top. Microbiol. Immunol. 2018, 414, 159–192. [Google Scholar]
- Clarke, H.J.; Chambers, J.E.; Liniker, E.; Marciniak, S.J. Endoplasmic reticulum stress in malignancy. Cancer Cell 2014, 25, 563–573. [Google Scholar] [CrossRef]
- Chevet, E.; Hetz, C.; Samali, A. Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov. 2015, 5, 586–597. [Google Scholar] [CrossRef]
- Dejeans, N.; Barroso, K.; Fernandez-Zapico, M.E.; Samali, A.; Chevet, E. Novel roles of the unfolded protein response in the control of tumor development and aggressiveness. Semin. Cancer Biol. 2015, 33, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Urra, H.; Dufey, E.; Avril, T.; Chevet, E.; Hetz, C. Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2016, 2, 252–262. [Google Scholar] [CrossRef] [Green Version]
- Cubillos-Ruiz, J.R.; Bettigole, S.E.; Glimcher, L.H. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell 2017, 168, 692–706. [Google Scholar] [CrossRef] [Green Version]
- Obacz, J.; Avril, T.; Le Reste, P.J.; Urra, H.; Quillien, V.; Hetz, C.; Chevet, E. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Sci. Signal. 2017, 10, eaal2323. [Google Scholar] [CrossRef]
- Feng, Y.X.; Sokol, E.S.; Del Vecchio, C.A.; Sanduja, S.; Claessen, J.H.; Proia, T.A.; Jin, D.X.; Reinhardt, F.; Ploegh, H.L.; Wang, Q.; et al. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discov. 2014, 4, 702–715. [Google Scholar] [CrossRef] [PubMed]
- Zeindl-Eberhart, E.; Brandl, L.; Liebmann, S.; Ormanns, S.; Scheel, S.K.; Brabletz, T.; Kirchner, T.; Jung, A. Epithelial-mesenchymal transition induces endoplasmic-reticulum-stress response in human colorectal tumor cells. PLoS ONE 2014, 9, e87386. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, T.J.; Fregno, I.; Fumagalli, F.; Rinaldi, A.; Bertoni, F.; Boersema, P.J.; Picotti, P.; Molinari, M. Chemical stresses fail to mimic the unfolded protein response resulting from luminal load with unfolded polypeptides. J. Biol. Chem. 2018, 293, 5600–5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mujtaba, T.; Dou, Q.P. Advances in the understanding of mechanisms and therapeutic use of bortezomib. Discov. Med. 2011, 12, 471–480. [Google Scholar]
- Jiang, D.; Lynch, C.; Medeiros, B.C.; Liedtke, M.; Bam, R.; Tam, A.B.; Yang, Z.; Alagappan, M.; Abidi, P.; Le, Q.T.; et al. Identification of Doxorubicin as an Inhibitor of the IRE1alpha-XBP1 Axis of the Unfolded Protein Response. Sci. Rep. 2016, 6, 33353. [Google Scholar] [CrossRef]
- Logue, S.E.; McGrath, E.P.; Cleary, P.; Greene, S.; Mnich, K.; Almanza, A.; Chevet, E.; Dwyer, R.M.; Oommen, A.; Legembre, P.; et al. Inhibition of IRE1 RNase activity modulates the tumor cell secretome and enhances response to chemotherapy. Nat. Commun. 2018, 9, 3267. [Google Scholar] [CrossRef]
- Nguyen, H.G.; Conn, C.S.; Kye, Y.; Xue, L.; Forester, C.M.; Cowan, J.E.; Hsieh, A.C.; Cunningham, J.T.; Truillet, C.; Tameire, F.; et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci. Transl. Med. 2018, 10, eaar2036. [Google Scholar] [CrossRef]
- Tanjore, H.; Cheng, D.S.; Degryse, A.L.; Zoz, D.F.; Abdolrasulnia, R.; Lawson, W.E.; Blackwell, T.S. Alveolar epithelial cells undergo epithelial-to-mesenchymal transition in response to endoplasmic reticulum stress. J. Biol. Chem. 2011, 286, 30972–30980. [Google Scholar] [CrossRef]
- Zhong, Q.; Zhou, B.; Ann, D.K.; Minoo, P.; Liu, Y.; Banfalvi, A.; Krishnaveni, M.S.; Dubourd, M.; Demaio, L.; Willis, B.C.; et al. Role of endoplasmic reticulum stress in epithelial-mesenchymal transition of alveolar epithelial cells: Effects of misfolded surfactant protein. Am. J. Respir. Cell Mol. Biol. 2011, 45, 498–509. [Google Scholar] [CrossRef]
- Mo, X.T.; Zhou, W.C.; Cui, W.H.; Li, D.L.; Li, L.C.; Xu, L.; Zhao, P.; Gao, J. Inositol-requiring protein 1—X-box-binding protein 1 pathway promotes epithelial-mesenchymal transition via mediating snail expression in pulmonary fibrosis. Int. J. Biochem. Cell Biol. 2015, 65, 230–238. [Google Scholar] [CrossRef]
- Shah, P.P.; Dupre, T.V.; Siskind, L.J.; Beverly, L.J. Common cytotoxic chemotherapeutics induce epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2017, 8, 22625–22639. [Google Scholar] [CrossRef] [Green Version]
- Rankin, E.B.; Giaccia, A.J. Hypoxic control of metastasis. Science 2016, 352, 175–180. [Google Scholar] [CrossRef] [Green Version]
- Mujcic, H.; Nagelkerke, A.; Rouschop, K.M.; Chung, S.; Chaudary, N.; Span, P.N.; Clarke, B.; Milosevic, M.; Sykes, J.; Hill, R.P.; et al. Hypoxic activation of the PERK/eIF2alpha arm of the unfolded protein response promotes metastasis through induction of LAMP3. Clin. Cancer Res. 2013, 19, 6126–6137. [Google Scholar] [CrossRef]
- Romero-Ramirez, L.; Cao, H.; Nelson, D.; Hammond, E.; Lee, A.H.; Yoshida, H.; Mori, K.; Glimcher, L.H.; Denko, N.C.; Giaccia, A.J.; et al. XBP1 is essential for survival under hypoxic conditions and is required for tumor growth. Cancer Res. 2004, 64, 5943–5947. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1alpha pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef]
- Madden, E.; Logue, S.E.; Healy, S.J.; Manie, S.; Samali, A. The role of the unfolded protein response in cancer progression: From oncogenesis to chemoresistance. Biol. Cell 2019, 111, 1–17. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2010, 120, 127–141. [Google Scholar] [CrossRef]
- Ghosh, R.; Lipson, K.L.; Sargent, K.E.; Mercurio, A.M.; Hunt, J.S.; Ron, D.; Urano, F. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 2010, 5, e9575. [Google Scholar] [CrossRef]
- Blais, J.D.; Addison, C.L.; Edge, R.; Falls, T.; Zhao, H.; Wary, K.; Koumenis, C.; Harding, H.P.; Ron, D.; Holcik, M.; et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol. Cell. Biol. 2006, 26, 9517–9532. [Google Scholar] [CrossRef]
- Wang, Y.; Alam, G.N.; Ning, Y.; Visioli, F.; Dong, Z.; Nor, J.E.; Polverini, P.J. The unfolded protein response induces the angiogenic switch in human tumor cells through the PERK/ATF4 pathway. Cancer Res. 2012, 72, 5396–5406. [Google Scholar] [CrossRef]
- Shen, X.; Xue, Y.; Si, Y.; Wang, Q.; Wang, Z.; Yuan, J.; Zhang, X. The unfolded protein response potentiates epithelial-to-mesenchymal transition (EMT) of gastric cancer cells under severe hypoxic conditions. Med. Oncol. 2015, 32, 447. [Google Scholar] [CrossRef]
- Sarrio, D.; Rodriguez-Pinilla, S.M.; Hardisson, D.; Cano, A.; Moreno-Bueno, G.; Palacios, J. Epithelial-mesenchymal transition in breast cancer relates to the basal-like phenotype. Cancer Res. 2008, 68, 989–997. [Google Scholar] [CrossRef]
- Gwinn, D.M.; Lee, A.G.; Briones-Martin-Del-Campo, M.; Conn, C.S.; Simpson, D.R.; Scott, A.I.; Le, A.; Cowan, T.M.; Ruggero, D.; Sweet-Cordero, E.A. Oncogenic KRAS Regulates Amino Acid Homeostasis and Asparagine Biosynthesis via ATF4 and Alters Sensitivity to L-Asparaginase. Cancer Cell 2018, 33, 91–107.e6. [Google Scholar] [CrossRef]
- Wang, Z.V.; Deng, Y.; Gao, N.; Pedrozo, Z.; Li, D.L.; Morales, C.R.; Criollo, A.; Luo, X.; Tan, W.; Jiang, N.; et al. Spliced X-box binding protein 1 couples the unfolded protein response to hexosamine biosynthetic pathway. Cell 2014, 156, 1179–1192. [Google Scholar] [CrossRef] [PubMed]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef]
- Falletta, P.; Sanchez-Del-Campo, L.; Chauhan, J.; Effern, M.; Kenyon, A.; Kershaw, C.J.; Siddaway, R.; Lisle, R.; Freter, R.; Daniels, M.J.; et al. Translation reprogramming is an evolutionarily conserved driver of phenotypic plasticity and therapeutic resistance in melanoma. Genes Dev. 2017, 31, 18–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caramel, J.; Papadogeorgakis, E.; Hill, L.; Browne, G.J.; Richard, G.; Wierinckx, A.; Saldanha, G.; Osborne, J.; Hutchinson, P.; Tse, G.; et al. A switch in the expression of embryonic EMT-inducers drives the development of malignant melanoma. Cancer Cell 2013, 24, 466–480. [Google Scholar] [CrossRef] [PubMed]
- Bobak, Y.; Kurlishchuk, Y.; Vynnytska-Myronovska, B.; Grydzuk, O.; Shuvayeva, G.; Redowicz, M.J.; Kunz-Schughart, L.A.; Stasyk, O. Arginine deprivation induces endoplasmic reticulum stress in human solid cancer cells. Int. J. Biochem. Cell Biol. 2016, 70, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Sun, L.; Qin, X.; Liu, T.; Zhang, S.; Liu, Y.; Li, S.; Guo, K. HSF1 promotes the inhibition of EMT-associated migration by low glucose via directly regulating Snail1 expression in HCC cells. Discov. Med. 2016, 22, 87–96. [Google Scholar]
- Garcia-Jimenez, C.; Goding, C.R. Starvation and Pseudo-Starvation as Drivers of Cancer Metastasis through Translation Reprogramming. Cell Metab. 2019, 29, 254–267. [Google Scholar] [CrossRef]
- Hart, L.S.; Cunningham, J.T.; Datta, T.; Dey, S.; Tameire, F.; Lehman, S.L.; Qiu, B.; Zhang, H.; Cerniglia, G.; Bi, M.; et al. ER stress-mediated autophagy promotes Myc-dependent transformation and tumor growth. J. Clin. Investig. 2012, 122, 4621–4634. [Google Scholar] [CrossRef]
- Croft, A.; Tay, K.H.; Boyd, S.C.; Guo, S.T.; Jiang, C.C.; Lai, F.; Tseng, H.Y.; Jin, L.; Rizos, H.; Hersey, P.; et al. Oncogenic activation of MEK/ERK primes melanoma cells for adaptation to endoplasmic reticulum stress. J. Investig. Dermatol. 2014, 134, 488–497. [Google Scholar] [CrossRef]
- Wang, M.; Kaufman, R.J. The impact of the endoplasmic reticulum protein-folding environment on cancer development. Nat. Rev. Cancer 2014, 14, 581–597. [Google Scholar] [CrossRef]
- Blazanin, N.; Son, J.; Craig-Lucas, A.B.; John, C.L.; Breech, K.J.; Podolsky, M.A.; Glick, A.B. ER stress and distinct outputs of the IRE1alpha RNase control proliferation and senescence in response to oncogenic Ras. Proc. Natl. Acad. Sci. USA 2017, 114, 9900–9905. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, D.R.; Sukhdeo, K.; Protopopova, M.; Sinha, R.; Enos, M.; Carrasco, D.E.; Zheng, M.; Mani, M.; Henderson, J.; Pinkus, G.S.; et al. The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 2007, 11, 349–360. [Google Scholar] [CrossRef]
- Fujimoto, T.; Onda, M.; Nagai, H.; Nagahata, T.; Ogawa, K.; Emi, M. Upregulation and overexpression of human X-box binding protein 1 (hXBP-1) gene in primary breast cancers. Breast Cancer 2003, 10, 301–306. [Google Scholar] [CrossRef]
- Fujimoto, T.; Yoshimatsu, K.; Watanabe, K.; Yokomizo, H.; Otani, T.; Matsumoto, A.; Osawa, G.; Onda, M.; Ogawa, K. Overexpression of human X-box binding protein 1 (XBP-1) in colorectal adenomas and adenocarcinomas. Anticancer Res. 2007, 27, 127–131. [Google Scholar]
- Dickinson, B.C.; Chang, C.J. Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat. Chem. Biol. 2011, 7, 504–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trachootham, D.; Alexandre, J.; Huang, P. Targeting cancer cells by ROS-mediated mechanisms: A radical therapeutic approach? Nat. Rev. Drug Discov. 2009, 8, 579–591. [Google Scholar] [CrossRef]
- Marciniak, S.J.; Yun, C.Y.; Oyadomari, S.; Novoa, I.; Zhang, Y.; Jungreis, R.; Nagata, K.; Harding, H.P.; Ron, D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004, 18, 3066–3077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutomi, G.; Tamura, Y.; Tanaka, T.; Kajiwara, T.; Kukita, K.; Ohmura, T.; Shima, H.; Takamaru, T.; Satomi, F.; Suzuki, Y.; et al. Human endoplasmic reticulum oxidoreductin 1-alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci. 2013, 104, 1091–1096. [Google Scholar] [CrossRef]
- Malhotra, J.D.; Kaufman, R.J. Endoplasmic reticulum stress and oxidative stress: A vicious cycle or a double-edged sword? Antioxid. Redox Signal. 2007, 9, 2277–2293. [Google Scholar] [CrossRef]
- Avivar-Valderas, A.; Salas, E.; Bobrovnikova-Marjon, E.; Diehl, J.A.; Nagi, C.; Debnath, J.; Aguirre-Ghiso, J.A. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Mol. Cell. Biol. 2011, 31, 3616–3629. [Google Scholar] [CrossRef]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. ATF4-dependent induction of heme oxygenase 1 prevents anoikis and promotes metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [Green Version]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef]
- Liu, F.; Gomez Garcia, A.M.; Meyskens, F.L., Jr. NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase-2 and epithelial-mesenchymal transition in melanoma cells. J. Investig. Dermatol. 2012, 132, 2033–2041. [Google Scholar] [CrossRef] [PubMed]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and thioredoxin antioxidant pathways synergize to drive cancer initiation and progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Bobrovnikova-Marjon, E.; Grigoriadou, C.; Pytel, D.; Zhang, F.; Ye, J.; Koumenis, C.; Cavener, D.; Diehl, J.A. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene 2010, 29, 3881–3895. [Google Scholar] [CrossRef] [Green Version]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef] [PubMed]
- Arfmann-Knubel, S.; Struck, B.; Genrich, G.; Helm, O.; Sipos, B.; Sebens, S.; Schafer, H. The Crosstalk between Nrf2 and TGF-beta1 in the Epithelial-Mesenchymal Transition of Pancreatic Duct Epithelial Cells. PLoS ONE 2015, 10, e0132978. [Google Scholar] [CrossRef]
- Shen, H.; Yang, Y.; Xia, S.; Rao, B.; Zhang, J.; Wang, J. Blockage of Nrf2 suppresses the migration and invasion of esophageal squamous cell carcinoma cells in hypoxic microenvironment. Dis. Esophagus 2014, 27, 685–692. [Google Scholar] [CrossRef]
- Hetz, C.; Chevet, E.; Harding, H.P. Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov. 2013, 12, 703–719. [Google Scholar] [CrossRef]
- Ma, Y.; Hendershot, L.M. The role of the unfolded protein response in tumour development: Friend or foe? Nat. Rev. Cancer 2004, 4, 966–977. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.M.; Tabbara, S.O.; Jacobs, L.K.; Manning, F.C.; Tsangaris, T.N.; Schwartz, A.M.; Kennedy, K.A.; Patierno, S.R. Overexpression of the glucose-regulated stress gene GRP78 in malignant but not benign human breast lesions. Breast Cancer Res. Treat. 2000, 59, 15–26. [Google Scholar] [CrossRef]
- Sakatani, T.; Maemura, K.; Hiyama, N.; Amano, Y.; Watanabe, K.; Kage, H.; Fukayama, M.; Nakajima, J.; Yatomi, Y.; Nagase, T.; et al. High expression of IRE1 in lung adenocarcinoma is associated with a lower rate of recurrence. Jpn. J. Clin. Oncol. 2017, 47, 543–550. [Google Scholar] [CrossRef] [Green Version]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef]
- Lhomond, S.; Avril, T.; Dejeans, N.; Voutetakis, K.; Doultsinos, D.; McMahon, M.; Pineau, R.; Obacz, J.; Papadodima, O.; Jouan, F.; et al. Dual IRE1 RNase functions dictate glioblastoma development. EMBO Mol. Med. 2018, 10, e7929. [Google Scholar] [CrossRef] [Green Version]
- Sheshadri, N.; Catanzaro, J.M.; Bott, A.J.; Sun, Y.; Ullman, E.; Chen, E.I.; Pan, J.A.; Wu, S.; Crawford, H.C.; Zhang, J.; et al. SCCA1/SERPINB3 promotes oncogenesis and epithelial-mesenchymal transition via the unfolded protein response and IL6 signaling. Cancer Res. 2014, 74, 6318–6329. [Google Scholar] [CrossRef]
- Cuevas, E.P.; Eraso, P.; Mazon, M.J.; Santos, V.; Moreno-Bueno, G.; Cano, A.; Portillo, F. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signalling pathway. Sci. Rep. 2017, 7, 44988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cano, A.; Santamaria, P.G.; Moreno-Bueno, G. LOXL2 in epithelial cell plasticity and tumor progression. Future Oncol. 2012, 8, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Peinado, H.; Moreno-Bueno, G.; Hardisson, D.; Perez-Gomez, E.; Santos, V.; Mendiola, M.; de Diego, J.I.; Nistal, M.; Quintanilla, M.; Portillo, F.; et al. Lysyl oxidase-like 2 as a new poor prognosis marker of squamous cell carcinomas. Cancer Res. 2008, 68, 4541–4550. [Google Scholar] [CrossRef]
- Moreno-Bueno, G.; Salvador, F.; Martin, A.; Floristan, A.; Cuevas, E.P.; Santos, V.; Montes, A.; Morales, S.; Castilla, M.A.; Rojo-Sebastian, A.; et al. Lysyl oxidase-like 2 (LOXL2), a new regulator of cell polarity required for metastatic dissemination of basal-like breast carcinomas. EMBO Mol. Med. 2011, 3, 528–544. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.X.; Jin, D.X.; Sokol, E.S.; Reinhardt, F.; Miller, D.H.; Gupta, P.B. Cancer-specific PERK signaling drives invasion and metastasis through CREB3L1. Nat. Commun. 2017, 8, 1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Jin, Z.; Chen, N.Z.; Lu, M.; Liu, C.B.; Hu, W.L.; Zheng, C.G. Activation of IRE1alpha-XBP1 pathway induces cell proliferation and invasion in colorectal carcinoma. Biochem. Biophys. Res. Commun. 2016, 470, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Du, R.; Gao, C.; Kang, J.; Wen, J.; Sun, T. The role of XBP1s in the metastasis and prognosis of hepatocellular carcinoma. Biochem. Biophys. Res. Commun. 2018, 500, 530–537. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.P.; Beverly, L.J. Regulation of VCP/p97 demonstrates the critical balance between cell death and epithelial-mesenchymal transition (EMT) downstream of ER stress. Oncotarget 2015, 6, 17725–17737. [Google Scholar] [CrossRef] [Green Version]
- Auf, G.; Jabouille, A.; Guerit, S.; Pineau, R.; Delugin, M.; Bouchecareilh, M.; Magnin, N.; Favereaux, A.; Maitre, M.; Gaiser, T.; et al. Inositol-requiring enzyme 1alpha is a key regulator of angiogenesis and invasion in malignant glioma. Proc. Natl. Acad. Sci. USA 2010, 107, 15553–15558. [Google Scholar] [CrossRef]
- Niu, Z.; Wang, M.; Zhou, L.; Yao, L.; Liao, Q.; Zhao, Y. Elevated GRP78 expression is associated with poor prognosis in patients with pancreatic cancer. Sci. Rep. 2015, 5, 16067. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Chen, X.; Chen, B.; Chen, B.; Song, W.; Sun, D.; Zhao, Y. Activating transcription factor 4 promotes esophageal squamous cell carcinoma invasion and metastasis in mice and is associated with poor prognosis in human patients. PLoS ONE 2014, 9, e103882. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Hu, S.; Wang, W.; Li, Y.; Zhi, W.; Lu, S.; Shi, Q.; Wang, Y.; Yang, Y. Matrine inhibits prostate cancer via activation of the unfolded protein response/endoplasmic reticulum stress signaling and reversal of epithelial to mesenchymal transition. Mol. Med. Rep. 2018, 18, 945–957. [Google Scholar] [CrossRef]
- Dekervel, J.; Bulle, A.; Windmolders, P.; Lambrechts, D.; Van Cutsem, E.; Verslype, C.; van Pelt, J. Acriflavine Inhibits Acquired Drug Resistance by Blocking the Epithelial-to-Mesenchymal Transition and the Unfolded Protein Response. Transl. Oncol. 2017, 10, 59–69. [Google Scholar] [CrossRef]
- Dasgupta, A.; Sawant, M.A.; Kavishwar, G.; Lavhale, M.; Sitasawad, S. AECHL-1 targets breast cancer progression via inhibition of metastasis, prevention of EMT and suppression of Cancer Stem Cell characteristics. Sci. Rep. 2016, 6, 38045. [Google Scholar] [CrossRef] [Green Version]
- Jiang, Y.; Jiao, Y.; Liu, Y.; Zhang, M.; Wang, Z.; Li, Y.; Li, T.; Zhao, X.; Wang, D. Sinomenine Hydrochloride Inhibits the Metastasis of Human Glioblastoma Cells by Suppressing the Expression of Matrix Metalloproteinase-2/-9 and Reversing the Endogenous and Exogenous Epithelial-Mesenchymal Transition. Int. J. Mol. Sci. 2018, 19, 844. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.; Mills, T.M.; Shafie, N.H.; Merlot, A.M.; Saleh Moussa, R.; Kalinowski, D.S.; Kovacevic, Z.; Richardson, D.R. Expanding horizons in iron chelation and the treatment of cancer: Role of iron in the regulation of ER stress and the epithelial-mesenchymal transition. Biochim. Biophys. Acta 2014, 1845, 166–181. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Cancer | UPR Activation | EMT Footprint | Source | Prognosis | Reference |
|---|---|---|---|---|---|
| Breast cancer | active PERK (ATF4 target genes) | EMT gene signature | human breast cancer datasets | NA | [49] |
| active PERK (ATF4 target genes) | EMT gene signature | human breast cancer datasets | increased metastasis | [112] | |
| Colon cancer | active PERK (ATF4 target genes) | EMT gene signature | human colon cancer datasets | NA | [49] |
| Colorectal carcinoma | IRE1 | E-cad, N-cad | CRC tumour tissues and CRC cell lines | shorter overall survival | [113] |
| GRP78 | β-catenin | CRC tumour tissues | NA | [50] | |
| Gastric cancer | active PERK (ATF4 target genes) | EMT gene signature | human gastric cancer datasets | NA | [49] |
| Glioblastoma | IRE1/XBP1 axis | VIM, ZEB1, TGFβ2 | human GBM cancer datasets and primary derived GBM cell lines | shorter overall survival, increased tumour aggressiveness | [106] |
| Hepatocellular carcinoma | XBP1 | VIM, E-cad | HCC tumour tissue | increased tumour size, increased metastasis | [114] |
| Lung cancer | active PERK (ATF4 target genes) | EMT gene signature | human cancer datasets | NA | [49] |
| IRE1, PERK | ZEB1, SNAI2, SNAI1 | LAC tumours | NA | [115] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Santamaría, P.G.; Mazón, M.J.; Eraso, P.; Portillo, F. UPR: An Upstream Signal to EMT Induction in Cancer. J. Clin. Med. 2019, 8, 624. https://doi.org/10.3390/jcm8050624
Santamaría PG, Mazón MJ, Eraso P, Portillo F. UPR: An Upstream Signal to EMT Induction in Cancer. Journal of Clinical Medicine. 2019; 8(5):624. https://doi.org/10.3390/jcm8050624
Chicago/Turabian StyleSantamaría, Patricia G., María J. Mazón, Pilar Eraso, and Francisco Portillo. 2019. "UPR: An Upstream Signal to EMT Induction in Cancer" Journal of Clinical Medicine 8, no. 5: 624. https://doi.org/10.3390/jcm8050624
APA StyleSantamaría, P. G., Mazón, M. J., Eraso, P., & Portillo, F. (2019). UPR: An Upstream Signal to EMT Induction in Cancer. Journal of Clinical Medicine, 8(5), 624. https://doi.org/10.3390/jcm8050624