Loss of XBP1 Leads to Early-Onset Retinal Neurodegeneration in a Mouse Model of Type I Diabetes


Abstract
:1. Introduction
2. Experimental Section
2.1. Animals
2.2. Electroretinography (ERG)
2.3. Retinal Immunohistochemistry and Morphometry
2.4. Image Analyses
2.5. Statistical Analyses
3. Results
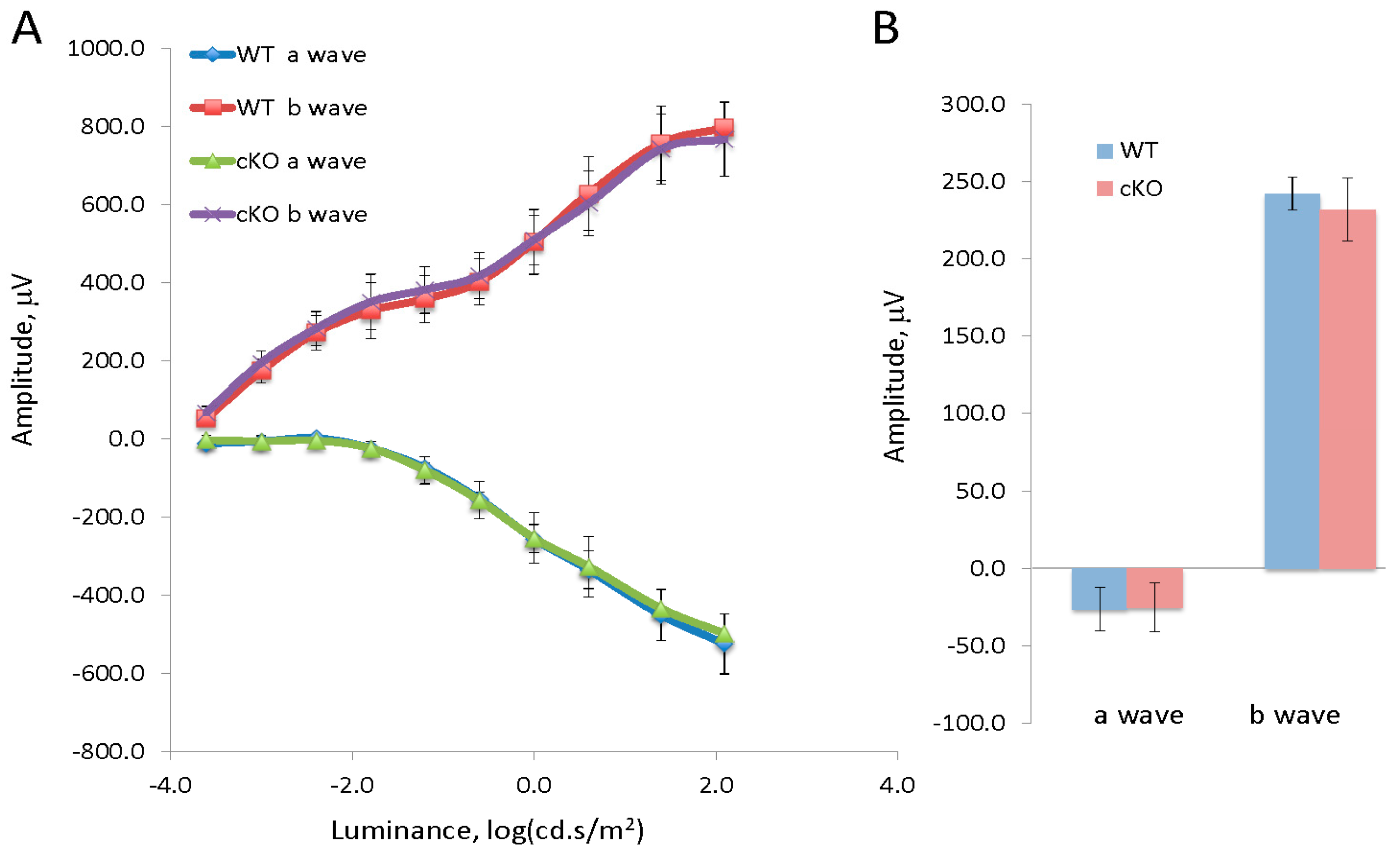
3.1. Conditional Deletion of XBP1 in the Retina Does Not Alter Retinal Function or Morphology at the Onset of Diabetes
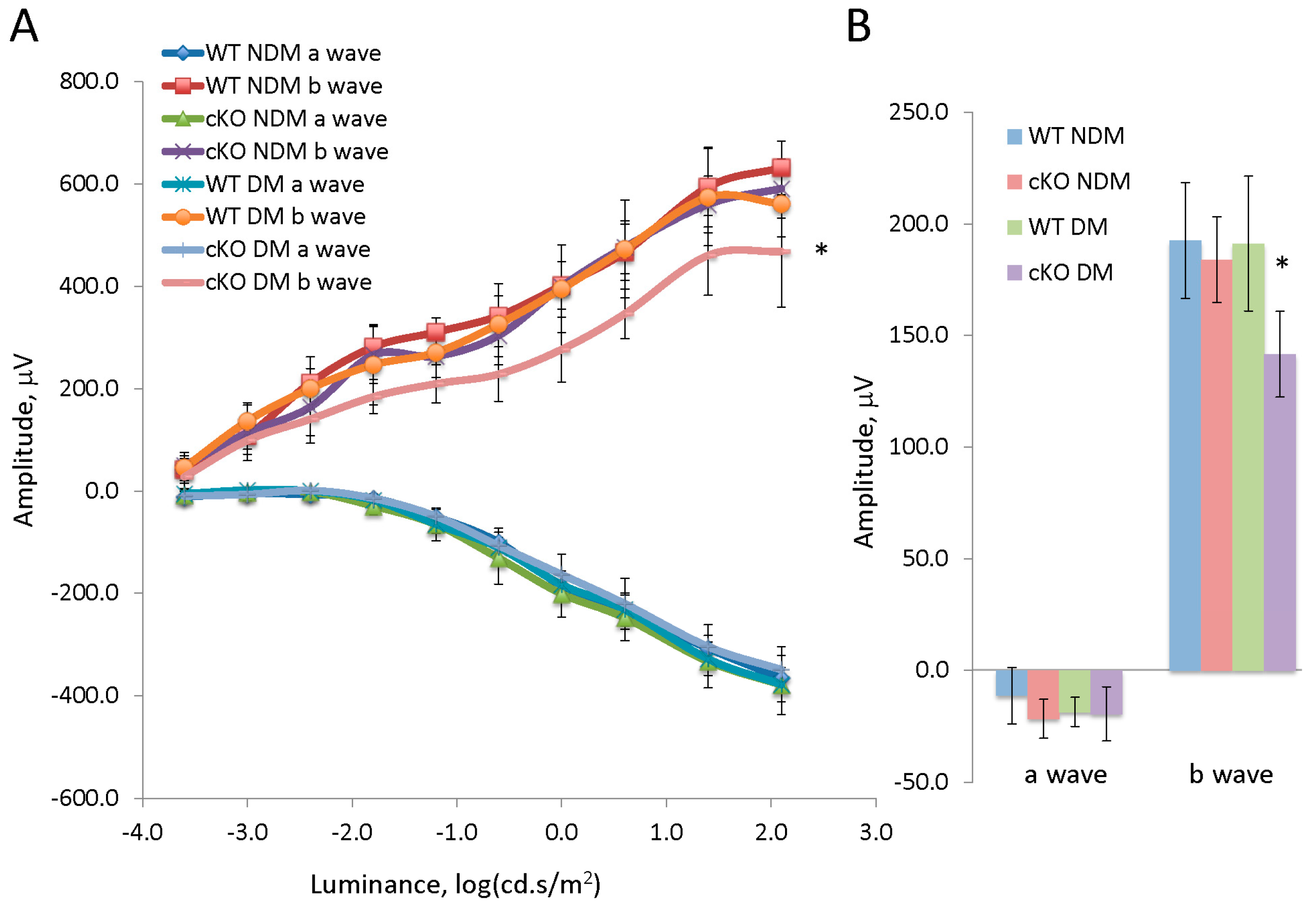
3.2. Conditional Deletion of XBP1 Results in Early Decline of Retinal Function in Diabetic Mice
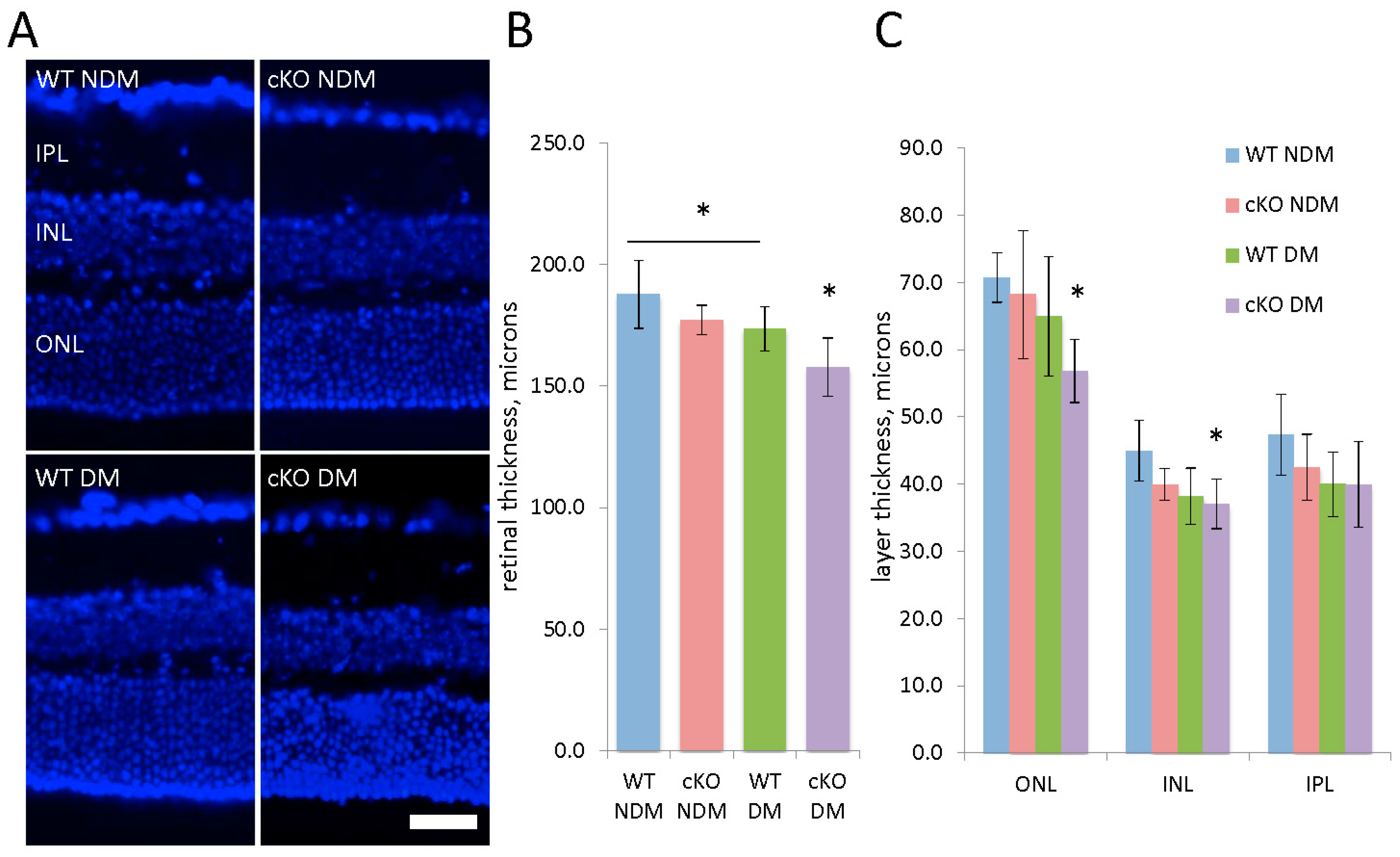
3.3. Conditional Deletion of XBP1 Leads to Retinal Degeneration in Diabetic Mice after 20 Weeks of Hyperglycemia
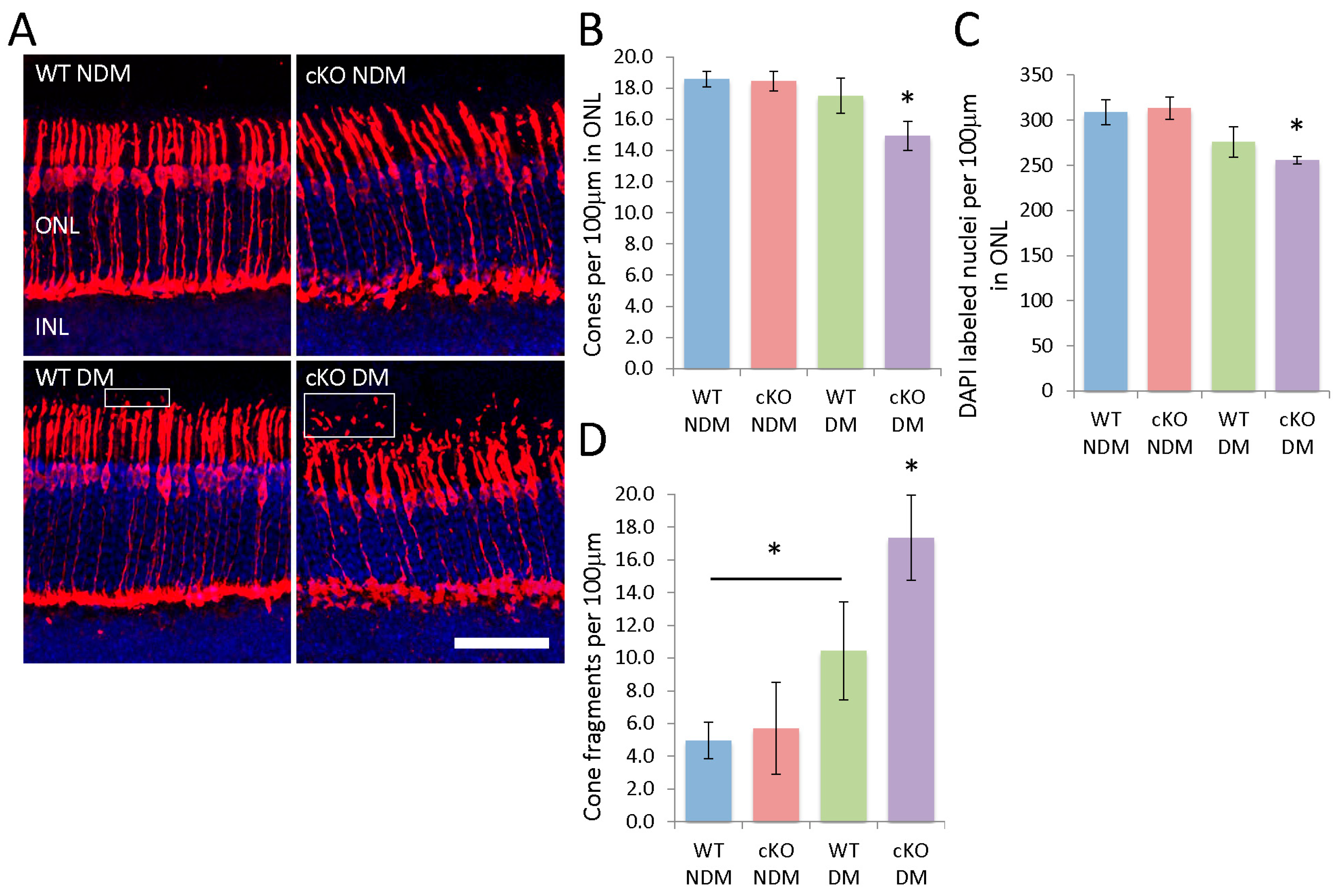
3.4. Loss of Rod and Cone Photoreceptors and Outer Segment Disorganization in Diabetic XBP1 cKO Retinas
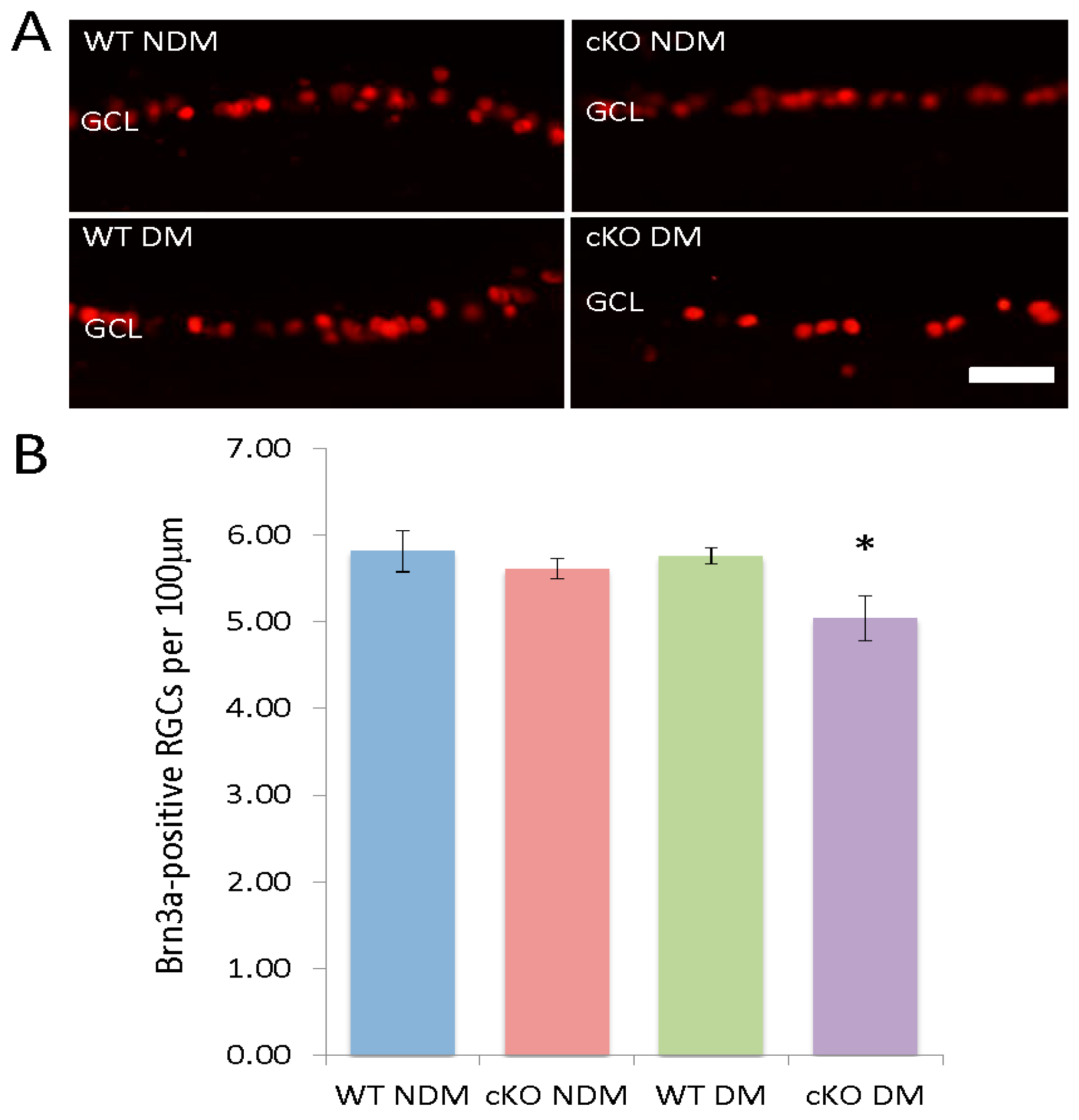
3.5. Loss of RGCs in Diabetic XBP1 cKO Mice
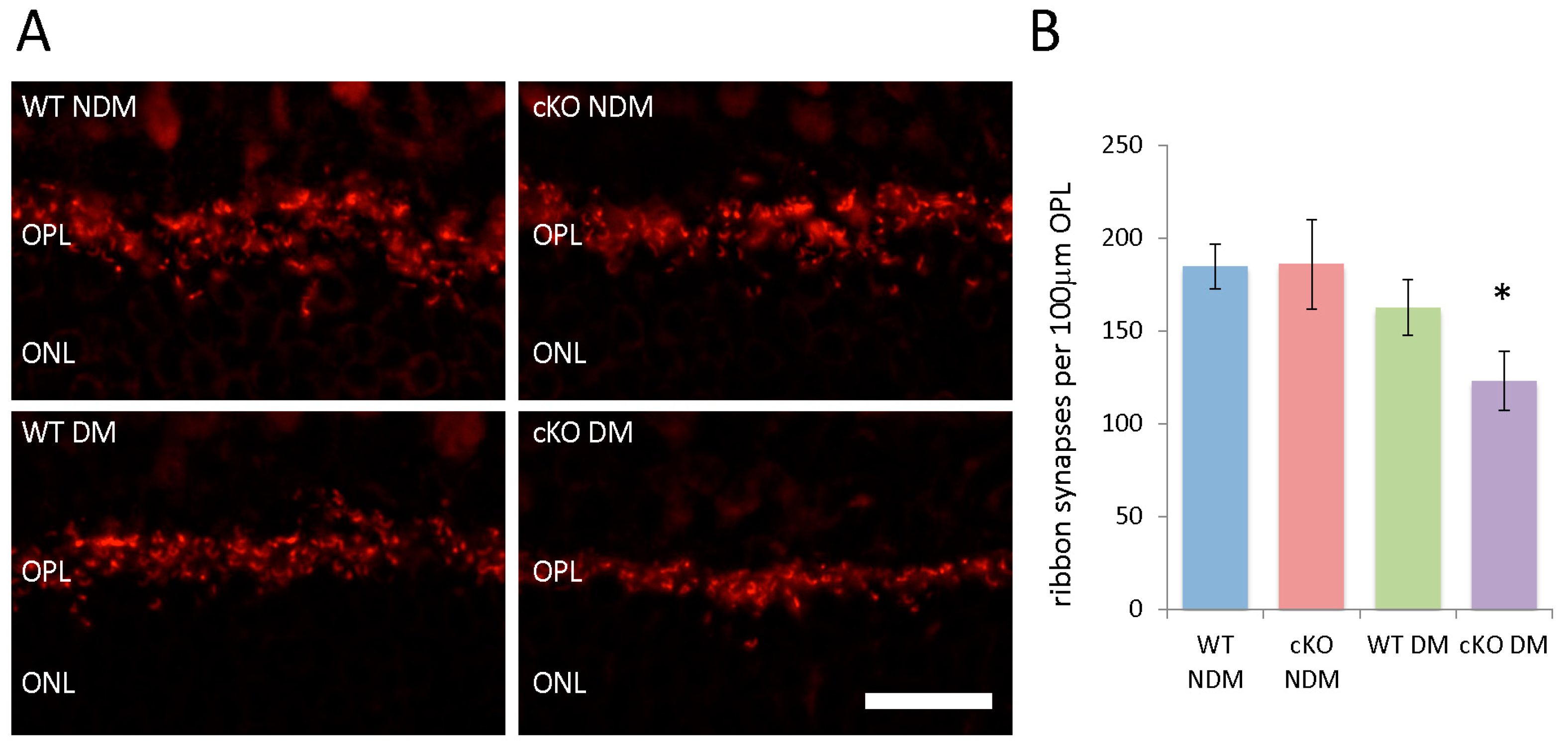
3.6. Fewer Ribbon Synapses in the ONL of Diabetic XBP1 cKO Mice
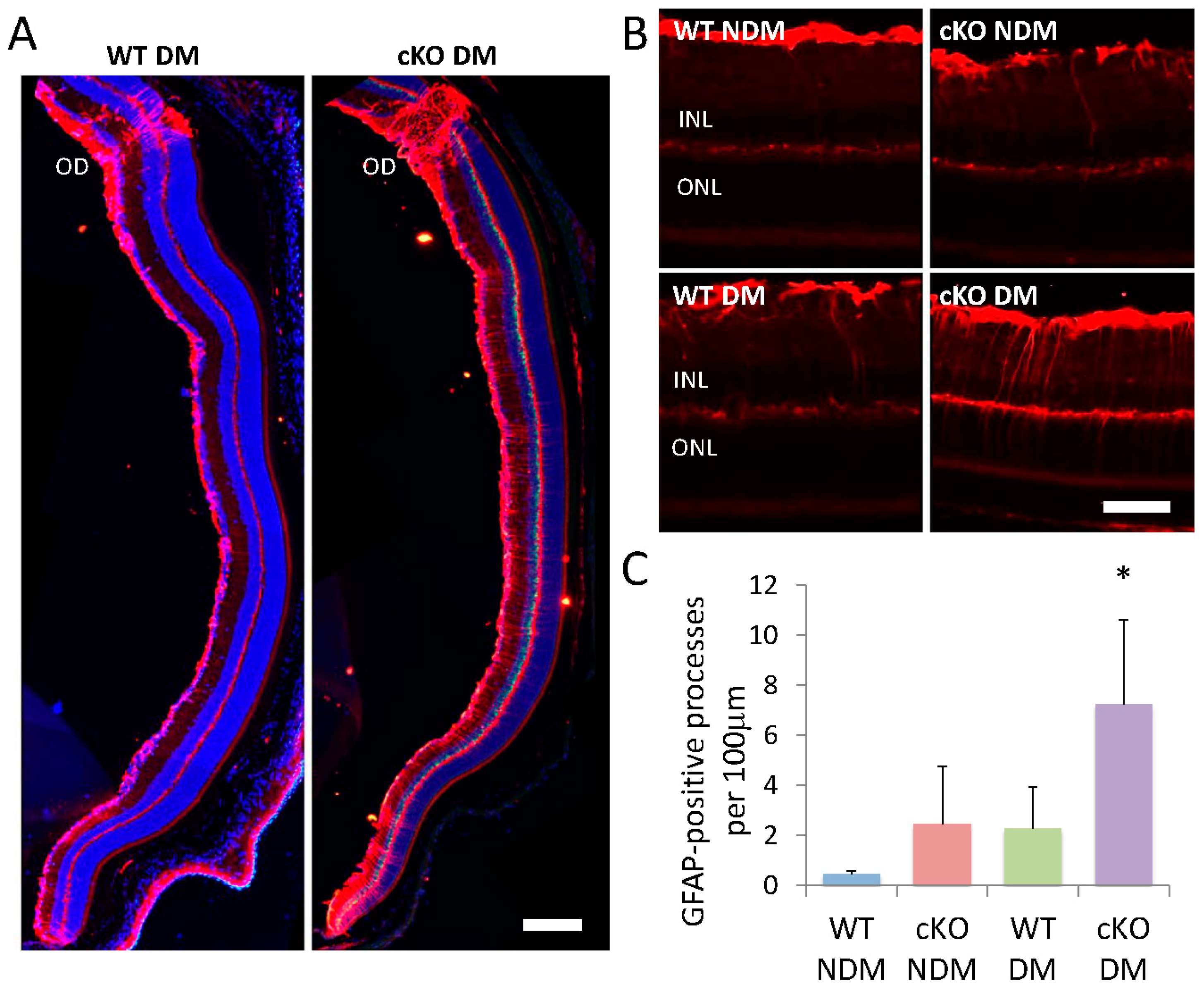
3.7. GFAP Elevation in Müller Cells
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Barber, A.J. Diabetic retinopathy: Recent advances towards understanding neurodegeneration and vision loss. Sci. China. Life Sci. 2015, 58, 541–549. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Wang, J.J.; Zhang, S.X. The unfolded protein response and diabetic retinopathy. J. Diabetes Res. 2014, 2014, 160140. [Google Scholar] [CrossRef] [PubMed]
- Oshitari, T.; Hata, N.; Yamamoto, S. Endoplasmic reticulum stress and diabetic retinopathy. Vasc. Health Risk Manag. 2008, 4, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.X.; Sanders, E.; Fliesler, S.J.; Wang, J.J. Endoplasmic reticulum stress and the unfolded protein responses in retinal degeneration. Exp. Eye Res. 2014, 125C, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Li, H.; Yasumura, D.; Cohen, H.R.; Zhang, C.; Panning, B.; Shokat, K.M.; Lavail, M.M.; Walter, P. IRE1 signaling affects cell fate during the unfolded protein response. Science 2007, 318, 944–949. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, T.; Falkowski, M.; Park, J.W.; Keegan, S.; Elliott, M.; Wang, J.J.; Zhang, S.X. Loss of XBP1 accelerates age-related decline in retinal function and neurodegeneration. Mol. Neurodegener. 2018, 13, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valensi, P.; Giroux, C.; Seeboth-Ghalayini, B.; Attali, J.R. Diabetic peripheral neuropathy: Effects of age, duration of diabetes, glycemic control, and vascular factors. J. Diabetes Its Complicat. 1997, 11, 27–34. [Google Scholar] [CrossRef]
- Munshi, M.N. Cognitive Dysfunction in Older Adults with Diabetes: What a Clinician Needs to Know. Diabetes Care 2017, 40, 461–467. [Google Scholar] [CrossRef]
- Kalyani, R.R.; Golden, S.H.; Cefalu, W.T. Diabetes and Aging: Unique Considerations and Goals of Care. Diabetes Care 2017, 40, 440–443. [Google Scholar] [CrossRef] [Green Version]
- Kirkman, M.S.; Briscoe, V.J.; Clark, N.; Florez, H.; Haas, L.B.; Halter, J.B.; Huang, E.S.; Korytkowski, M.T.; Munshi, M.N.; Odegard, P.S.; et al. Diabetes in older adults. Diabetes Care 2012, 35, 2650–2664. [Google Scholar] [CrossRef]
- Stratton, I.M.; Kohner, E.M.; Aldington, S.J.; Turner, R.C.; Holman, R.R.; Manley, S.E.; Matthews, D.R. UKPDS 50: Risk factors for incidence and progression of retinopathy in Type II diabetes over 6 years from diagnosis. Diabetologia 2001, 44, 156–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolesnikov, A.V.; Fan, J.; Crouch, R.K.; Kefalov, V.J. Age-related deterioration of rod vision in mice. J. Neurosci. Off. J. Soc. Neurosci. 2010, 30, 11222–11231. [Google Scholar] [CrossRef] [PubMed]
- Sergeys, J.; Etienne, I.; Van Hove, I.; Lefevere, E.; Stalmans, I.; Feyen, J.H.M.; Moons, L.; Van Bergen, T. Longitudinal In Vivo Characterization of the Streptozotocin-Induced Diabetic Mouse Model: Focus on Early Inner Retinal Responses. Investig. Ophthalmol. Vis. Sci. 2019, 60, 807–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C.; Lee, A.H.; Gonzalez-Romero, D.; Thielen, P.; Castilla, J.; Soto, C.; Glimcher, L.H. Unfolded protein response transcription factor XBP-1 does not influence prion replication or pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 757–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rowan, S.; Cepko, C.L. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev. Biol. 2004, 271, 388–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jeon, C.J.; Strettoi, E.; Masland, R.H. The major cell populations of the mouse retina. J. Neurosci. Off. J. Soc. Neurosci. 1998, 18, 8936–8946. [Google Scholar] [CrossRef]
- Barber, A.J.; Antonetti, D.A.; Gardner, T.W. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Investig. Ophthalmol. Vis. Sci. 2000, 41, 3561–3568. [Google Scholar]
- Zheng, L.; Du, Y.; Miller, C.; Gubitosi-Klug, R.; Kern, T.; Ball, S.; Berkowitz, B. Critical role of inducible nitric oxide synthase in degeneration of retinal capillaries in mice with streptozotocin-induced diabetes. Diabetologia 2007, 50, 1987–1996. [Google Scholar] [CrossRef] [Green Version]
- Samuels, I.S.; Lee, C.-A.; Petrash, J.M.; Peachey, N.S.; Kern, T.S. Exclusion of aldose reductase as a mediator of ERG deficits in a mouse model of diabetic eye disease. Vis. Neurosci. 2012, 29, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Bogdanov, P.; Corraliza, L.; Villena, J.A.; Carvalho, A.R.; Garcia-Arumí, J.; Ramos, D.; Ruberte, J.; Simó, R.; Hernández, C. The db/db Mouse: A Useful Model for the Study of Diabetic Retinal Neurodegeneration. PLoS ONE 2014, 9, e97302. [Google Scholar] [CrossRef]
- Rajagopal, R.; Bligard, G.W.; Zhang, S.; Yin, L.; Lukasiewicz, P.; Semenkovich, C.F. Functional Deficits Precede Structural Lesions in Mice With High-Fat Diet-Induced Diabetic Retinopathy. Diabetes 2016, 65, 1072–1084. [Google Scholar] [CrossRef] [PubMed]
- Akimov, N.P.; Rentería, R.C. Spatial frequency threshold and contrast sensitivity of an optomotor behavior are impaired in the Ins2Akita mouse model of diabetes. Behav. Brain Res. 2012, 226, 601–605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hombrebueno, J.R.; Chen, M.; Penalva, R.G.; Xu, H. Loss of synaptic connectivity, particularly in second order neurons is a key feature of diabetic retinal neuropathy in the Ins2Akita mouse. PLoS ONE 2014, 9, e97970. [Google Scholar] [CrossRef] [PubMed]
- VanGuilder, H.D.; Brucklacher, R.M.; Patel, K.; Ellis, R.W.; Freeman, W.M.; Barber, A.J. Diabetes downregulates presynaptic proteins and reduces basal synapsin I phosphorylation in rat retina. Eur. J. Neurosci. 2008, 28, 1–11. [Google Scholar] [CrossRef] [PubMed]
- D’Cruz, T.S.; Weibley, B.N.; Kimball, S.R.; Barber, A.J. Post-translational processing of synaptophysin in the rat retina is disrupted by diabetes. PLoS ONE 2012, 7, e44711. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, J.J.; Yu, Q.; Wang, M.; Zhang, S.X. Endoplasmic Reticulum Stress is implicated in Retinal Inflammation and Diabetic Retinopathy. FEBS Lett. 2009, 583, 1521–1527. [Google Scholar] [CrossRef]
- Zhong, Y.; Wang, J.J.; Zhang, S.X. Intermittent but not constant high glucose induces ER stress and inflammation in human retinal pericytes. Adv. Exp. Med. Biol. 2012, 723, 285–292. [Google Scholar]
- Zhong, Y.; Li, J.; Chen, Y.; Wang, J.J.; Ratan, R.; Zhang, S.X. Activation of Endoplasmic Reticulum Stress by Hyperglycemia Is Essential for Muller Cell-Derived Inflammatory Cytokine Production in Diabetes. Diabetes 2012, 61, 492–504. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, J.J.; Li, J.; Hosoya, K.I.; Ratan, R.; Townes, T.; Zhang, S.X. Activating transcription factor 4 mediates hyperglycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia 2012, 55, 2533–2545. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Jing, G.; Wang, J.; Sheibani, N.; Zhang, S. ATF4 is a novel regulator of MCP-1 in microvascular endothelial cells. J. Inflamm. 2015, 12, 31. [Google Scholar] [CrossRef]
- Zhu, S.; Liu, H.; Sha, H.; Qi, L.; Gao, D.S.; Zhang, W. PERK and XBP1 differentially regulate CXCL10 and CCL2 production. Exp. Eye Res. 2017, 155, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Wang, J.J.; Zhang, S.X. Preconditioning with endoplasmic reticulum stress mitigates retinal endothelial inflammation via activation of X-box binding protein 1. J. Biol. Chem. 2011, 286, 4912–4921. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Chen, C.; McLaughlin, T.; Wang, Y.; Le, Y.Z.; Wang, J.J.; Zhang, S.X. Loss of X-box binding protein 1 in Muller cells augments retinal inflammation in a mouse model of diabetes. Diabetologia 2019, 62, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Li, J.; Wang, J.J.; Chen, C.; Tran, J.-T.A.; Saadi, A.; Yu, Q.; Le, Y.-z.; Mandal, M.N.A.; Anderson, R.E.; et al. X-Box Binding Protein 1 Is Essential for the Anti-Oxidant Defense and Cell Survival in the Retinal Pigment Epithelium. PLoS ONE 2012, 7, e38616. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Cano, M.; Wang, J.J.; Li, J.; Huang, C.; Yu, Q.; Herbert, T.P.; Handa, J.T.; Zhang, S.X. Role of unfolded protein response dysregulation in oxidative injury of retinal pigment epithelial cells. Antioxid. Redox Signal. 2014, 20, 2091–2106. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, J.J.; Ma, J.H.; Jin, C.; Yu, Q.; Zhang, S.X. Activation of the UPR Protects Against Cigarette Smoke-induced RPE Apoptosis through Up-Regulation of Nrf2. J. Biol. Chem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.H.; Wang, J.J.; Li, J.; Pfeffer, B.A.; Zhong, Y.; Zhang, S.X. The Role of IRE-XBP1 Pathway in Regulation of Retinal Pigment Epithelium Tight JunctionsXBP1 Regulates the RPE Tight Junctions. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5244–5252. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, J.M.; Hernandez, C.; Weber, S.R.; Zhao, Y.; Dunklebarger, M.; Tiberti, N.; Laremore, T.; Simo-Servat, O.; Garcia-Ramirez, M.; Barber, A.J.; et al. Proteomic Analysis of Early Diabetic Retinopathy Reveals Mediators of Neurodegenerative Brain Diseases. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2264–2274. [Google Scholar] [CrossRef]
- Kumar, S.; Zhuo, L. Longitudinal in vivo imaging of retinal gliosis in a diabetic mouse model. Exp. Eye Res. 2010, 91, 530–536. [Google Scholar] [CrossRef]
- Attwell, D.; Buchan, A.M.; Charpak, S.; Lauritzen, M.; Macvicar, B.A.; Newman, E.A. Glial and neuronal control of brain blood flow. Nature 2010, 468, 232–243. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Xu, X.; Elliott, M.H.; Zhu, M.; Le, Y.Z. Muller cell-derived VEGF is essential for diabetes-induced retinal inflammation and vascular leakage. Diabetes 2010, 59, 2297–2305. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Dong, S.; Zhu, M.; Sherry, D.M.; Wang, C.; You, Z.; Haigh, J.J.; Le, Y.-Z. Müller Glia Are a Major Cellular Source of Survival Signals for Retinal Neurons in Diabetes. Diabetes 2015, 64, 3554–3563. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, E.; Hwang, G.S.; Pan, Z.H. Characterization of transgenic mouse lines expressing Cre recombinase in the retina. Neuroscience 2010, 165, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Dilutions | Catalog No. | Company |
|---|---|---|---|
| anti-Ribeye, B-domain | 1:800 | 192 003 | Synaptic Systems |
| anti-Cone Arrestin | 1:1000 | AB15282 | Millipore |
| anti-Brn3a | 1:800 | sc-31984 | Santa Cruz Biotechnology |
| anti-GFAP | 1:800 | Z0334 | Dako |
| Alexa Fluor-488 | 1:800 | A11001 | Molecular Probes |
| Alexa Fluor-594 | 1:800 | A11005 | Molecular Probes |
| Alexa Fluor-594 | 1:800 | A11080 | Molecular Probes |
| Texas Red | 1:800 | T6391 | Molecular Probes |
| Body Weight | Blood Glucose | ||
|---|---|---|---|
| n | (g) | (mmol/L) | |
| WT NDM | 8 | 26.6 ± 1.5 | 6.6 ± 0.8 |
| cKO NDM | 8 | 25.9 ± 1.1 | 6.5 ± 0.6 |
| WT DM | 8 | 21.0 ± 1.3 | 23.3 ± 2.1 |
| cKO DM | 8 | 20.8 ± 1.8 | 24.1 ± 2.5 |
| p values | p < 0.01 | p < 0.01 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McLaughlin, T.; Siddiqi, M.; Wang, J.J.; Zhang, S.X. Loss of XBP1 Leads to Early-Onset Retinal Neurodegeneration in a Mouse Model of Type I Diabetes. J. Clin. Med. 2019, 8, 906. https://doi.org/10.3390/jcm8060906
McLaughlin T, Siddiqi M, Wang JJ, Zhang SX. Loss of XBP1 Leads to Early-Onset Retinal Neurodegeneration in a Mouse Model of Type I Diabetes. Journal of Clinical Medicine. 2019; 8(6):906. https://doi.org/10.3390/jcm8060906
Chicago/Turabian StyleMcLaughlin, Todd, Manhal Siddiqi, Joshua J. Wang, and Sarah X. Zhang. 2019. "Loss of XBP1 Leads to Early-Onset Retinal Neurodegeneration in a Mouse Model of Type I Diabetes" Journal of Clinical Medicine 8, no. 6: 906. https://doi.org/10.3390/jcm8060906
APA StyleMcLaughlin, T., Siddiqi, M., Wang, J. J., & Zhang, S. X. (2019). Loss of XBP1 Leads to Early-Onset Retinal Neurodegeneration in a Mouse Model of Type I Diabetes. Journal of Clinical Medicine, 8(6), 906. https://doi.org/10.3390/jcm8060906