Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Strategies for In Vivo Administration: Part-II

 ,
,  ,
,  ,
,  , and
, and
Abstract
:1. Introduction
2. Diabetes Mellitus Animal Models
3. Diabetes Mellitus Type 1 Animal Models
3.1. Chemical Induction of Diabetes Mellitus Type 1
3.1.1. Streptozotocin (STZ)-Induced Models
3.1.2. Alloxan-Induced Models
3.2. Spontaneous or Autoimmune of Diabetes Mellitus Type 1
3.2.1. Non-Obese Diabetic Mice
3.2.2. Biobreeding Rats
3.2.3. LEW.1AR1/-Iddm Rats
3.3. Genetically Induced Diabetes Mellitus Type 1
AKITA Mice
3.4. Virally Induced Diabetes Mellitus Type 1
3.5. Non-Rodent Models of Diabetes Mellitus Type 1
3.5.1. Pancreatomy
3.5.2. Chemical Ablation of β-Cells in Large Animals
4. Diabetes Mellitus Type 2 Animal Models
4.1. Obese Monogenic Models of Diabetes Mellitus Type 2
4.1.1. Lepob/ob Mice
4.1.2. Leprdb/db Mice
4.1.3. Zucker Diabetic Fatty (ZDF) Rats
4.2. Obese Polygenic Models of Diabetes Mellitus Type 2
4.2.1. KK Mice Models
4.2.2. Otsuka Long-Evans Tokushima Fat (OLEFT) Rat
4.2.3. New Zealand Obese (NZO) Mice
4.2.4. TallyHo/Jng Mice
4.2.5. NoncNZO10/LtJ Mice
4.3. Induced Obesity Models of Diabetes Mellitus Type 2
4.3.1. High Fat Feeding Models
4.3.2. Dessert Gerbil
4.3.3. Nile Grass Rats
5. Administration Routes
5.1. Enteral Administration
5.2. Intravenous Administration
5.3. Intraosseous Administration
5.4. Dermal Administration
5.5. Muscle Administration
5.6. Epidural and Intrathecal Administration
5.7. Intraperitoneal Administration
5.8. Intranasal, Intratracheal, and Inhalational Administration
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Beverley, B.; Eschwège, E. The diagnosis and classification of diabetes and impaired glucose tolerance. In Textbook of Diabetes; Pickup, J.C., Williams, G., Eds.; John Wiley & Sons: Hoboken, NJ, USA, 2003; pp. 2.1–2.11. [Google Scholar]
- WHO. World Health Organization: Definition, Diagnosis and Classification of Diabetes Mellitus and its Complications. Part 1: Diagnosis and Classification of Diabetes Mellitus; Report No. WHO/NCD/NCS/99.2; WHO: Geneva, Switzerland, 1999. [Google Scholar]
- Forbes, J.M.; Cooper, M.E. Mechanisms of Diabetic Complications. Physiol. Rev. 2013, 93, 137–188. [Google Scholar] [PubMed]
- Fangueiro, J.F.; Silva, A.M.; Garcia, M.L.; Souto, E.B. Current nanotechnology approaches for the treatment and management of diabetic retinopathy. Eur. J. Pharm. Biopharm. 2015, 95, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Souto, S.B.; Souto, E.B.; Braga, D.C.; Medina, J.L. Prevention and current onset delay approaches of type 2 diabetes mellitus (T2DM). Eur. J. Clin. Pharmacol. 2011, 67, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davies, J.L.; Kawaguchi, Y.; Bennett, S.T.; Copeman, J.B.; Cordell, H.J.; Pritchard, L.E.; Reed, P.W.; Gough, S.C.L.; Jenkins, S.C.; Palmer, S.M.; et al. A genome-wide search for human type 1 diabetes susceptibility genes. Nature 1994, 371, 130–136. [Google Scholar] [CrossRef]
- Wagner, D.H.J. Overlooked Mechanisms in Type 1 Diabetes Etiology: How Unique Costimulatory Molecules Contribute to Diabetogenesis. Front. Endocrinol. 2017, 8, 208. [Google Scholar] [Green Version]
- Fangueiro, J.F.; Andreani, T.; Egea, M.A.; Garcia, M.L.; Souto, S.B.; Silva, A.M.; Souto, E.B. Design of cationic lipid nanoparticles for ocular delivery: Development, characterization and cytotoxicity. Int. J. Pharm. 2014, 461, 64–73. [Google Scholar] [PubMed]
- Severino, P.; Andreani, T.; Chaud, M.; Benites, C.; Pinho, S.; Souto, E. Essential Oils as Active Ingredients of Lipid Nanocarriers for Chemotherapeutic Use. Curr. Pharm. Biotechnol. 2015, 16, 365–370. [Google Scholar]
- Souto, S.B.; Baptista, P.V.; Braga, D.C.; Carvalho, D. Ovarian Leydig cell tumor in a post-menopausal patient with severe hyperandrogenism. Arq. Bras. Endocrinol. Metabol. 2014, 58, 68–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Souto, S.B.; Fernandes, H.; Matos, M.J.; Braga, D.C.; Pereira, J.; Carvalho, D. Importance of (99)mTc-sestaMIBI thyroid scan in a case of amiodarone-induced thyrotoxicosis. Arq. Bras. Endocrinol. Metabol. 2011, 55, 486–489. [Google Scholar]
- Silva, A.M.; Rosario, L.M.; Santos, R.M. Background Ca2+ influx mediated by a dihydropyridine- and voltage-insensitive channel in pancreatic beta-cells. Modulation by Ni2+, diphenylamine-2-carboxylate, and glucose metabolism. J. Biol. Chem. 1994, 269, 17095–17103. [Google Scholar]
- Barbosa, R.M.; Silva, A.M.; Tome, A.R.; Stamford, J.A.; Santos, R.M.; Rosario, L.M. Control of pulsatile 5-HT/insulin secretion from single mouse pancreatic islets by intracellular calcium dynamics. J. Physiol. 1998, 510, 135–143. [Google Scholar] [PubMed] [Green Version]
- Silva, A.M.; Liu-Gentry, J.; Dickey, A.S.; Barnett, D.W.; Misler, S. alpha-Latrotoxin increases spontaneous and depolarization-evoked exocytosis from pancreatic islet beta-cells. J. Physiol. 2005, 565, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Rodrigues, R.J.; Tome, A.R.; Cunha, R.A.; Misler, S.; Rosario, L.M. Electrophysiological and immunocytochemical evidence for P2X purinergic receptors in pancreatic beta cells. Pancreas 2008, 36, 279–283. [Google Scholar] [CrossRef] [PubMed]
- King, A.J. The use of animal models in diabetes research. Br. J. Pharmacol. 2012, 166, 877–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Yan, L.-J. Streptozotocin-induced type 1 diabetes in rodents as a model for studying mitochondrial mechanisms of diabetic β cell glucotoxicity. Diabetes Metab. Syndr. Obes. Targets Ther. 2015, 8, 181–188. [Google Scholar]
- Furman, B.L. Streptozotocin-Induced Diabetic Models in Mice and Rats. Curr. Protoc. Pharmacol. 2015, 70, 5. [Google Scholar] [PubMed]
- Rohilla, A.; Ali, S. Alloxan Induced Diabetes: Mechanisms and Effects. Int. J. Res. Pharm. Biomed. Sci. 2012, 3, 819–823. [Google Scholar]
- Etuk, E.U. Animals models for studying diabetes mellitus Department of Pharmacology. Agric. Biol. J. North Am. 2010, 1, 130–134. [Google Scholar]
- Pearson, J.A.; Wong, F.S.; Wen, L. The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 2016, 66, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, M.A.; Leiter, E.H. The NOD mouse model of type 1 diabetes: As good as it gets? Nat. Med. 1999, 5, 601–604. [Google Scholar] [CrossRef]
- Kleinert, M.; Clemmensen, C.; Hofmann, S.M.; Moore, M.C.; Renner, S.; Woods, S.C.; Huypens, P.; Beckers, J.; De Angelis, M.H.; Schürmann, A.; et al. Animal models of obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2018, 14, 140–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medina, A.; Parween, S.; Ullsten, S.; Vishnu, N.; Siu, Y.T.; Quach, M. Early deficits in insulin secretion, beta cell mass and islet blood perfusion precede onset of autoimmune type 1 diabetes in BioBreeding rats. Diabetologia 2018, 61, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, K.; Ramarao, P. Animal models in type 2 diabetes research: An overview K. Indian J. Med. Res. 2012, 136, 451–472. [Google Scholar]
- O’Brien, P.D.; Sakowski, S.A.; Feldman, E.L. Mouse Models of Diabetic Neuropathy. ILAR J. 2014, 54, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Guberski, D.L. Diabetes-Prone and Diabetes-Resistant BB Rats: Animal Models of Spontaneous and Virally Induced Diabetes Mellitus, Lymphocytic Thyroiditis, and Collagen-Induced Arthritis. ILAR J. 2013, 35, 29–37. [Google Scholar] [CrossRef]
- Rossetti, L.; Shulman, G.I.; Zawalich, W.; DeFronzo, R.A. Effect of chronic hyperglycemia on in vivo insulin secretion in partially pancreatectomized rats. J. Clin. Investig. 1987, 80, 1037–1044. [Google Scholar] [CrossRef]
- Bracke, A.; Domanska, G.; Bracke, K.; Harzsch, S.; Brandt, J.V.D.; Broeker, B.; Halbach, O.V.B.U. Obesity alters mobility and adult neurogenesis, but not hippocampal dependent learning in ob/ob mice. In bioRxiv; Cold Spring Harbor Laboratory: Cold Spring Harbor, NY, USA, 2019; p. 537720. [Google Scholar]
- Guimbal, S.; Couffinhal, T.; Hollier, P.; Chapouly, C.; Caradu, C.; Gadeau, A.; Renault, M. Leptin receptor deficient female mice as a mouse model of heart failure with preserve ejection fraction. Arch. Cardiovasc. Dis. Suppl. 2019, 11, 226–227. [Google Scholar] [CrossRef]
- Peterson, R.G.; Shaw, W.N.; Neel, M.-A.; Little, L.A.; Eichberg, J. Zucker Diabetic Fatty Rat as a Model for Non-insulin-dependent Diabetes Mellitus. ILAR J. 1990, 32, 16–19. [Google Scholar] [CrossRef] [Green Version]
- John, C.; Grune, J.; Ott, C.; Nowotny, K.; Deubel, S.; Kühne, A.; Schubert, C.; Kintscher, U.; Regitz-Zagrosek, V.; Grune, T. Sex Differences in Cardiac Mitochondria in the New Zealand Obese Mouse. Front. Endocrinol. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Pomp, D. Genetic Dissection of Obesity in Polygenic Animal Models. Behav. Genet. 1997, 27, 285–306. [Google Scholar] [CrossRef]
- Rees, D.A.; Alcolado, J.C. Animal models of diabetes mellitus. Diabet. Med. 2005, 22, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Roland, B.; Jürgen, S.; Cornelius, B.L. High-fat Diets: Modeling the Metabolic Disorders of Human Obesity in Rodents. Obesity 2012, 15, 798–808. [Google Scholar]
- Reuter, T.Y. Diet-induced models for obesity and type 2 diabetes. Drug Discov. Today Dis. Model. 2007, 4, 3–8. [Google Scholar] [CrossRef]
- Wall, R.; Shani, M. Are animal models as good as we think? Theriogenology 2008, 69, 2–9. [Google Scholar] [PubMed] [Green Version]
- Von Herrath, M.; Nepom, G.T. Animal models of human type 1 diabetes. Nat. Immunol. 2009, 10, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Graham, M.L.; Schuurman, H.-J. Validity of animal models of type 1 diabetes, and strategies to enhance their utility in translational research. Eur. J. Pharmacol. 2015, 759, 221–230. [Google Scholar]
- Wu, K.K.; Huan, Y. Streptozotocin-Induced Diabetic Models in Mice and Rats. Current Protocols in Pharmacology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Schnedl, W.J.; Ferber, S.; Johnson, J.H.; Newgard, C.B. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 1994, 43, 1326–1333. [Google Scholar] [CrossRef] [PubMed]
- Pieper, A.A.; Brat, D.J.; Krug, D.K.; Watkins, C.C.; Gupta, A.; Blackshaw, S.; Verma, A.; Wang, Z.-Q.; Snyder, S.H. Poly(ADP-ribose) polymerase-deficient mice are protected from streptozotocin-induced diabetes. Proc. Natl. Acad. Sci. USA 1999, 96, 3059–3064. [Google Scholar] [CrossRef] [Green Version]
- Deeds, M.C.; Anderson, J.M.; Armstrong, A.S.; Gastineau, D.A.; Hiddinga, H.J.; Jahangir, A.; Eberhardt, N.L.; Kudva, Y.C. Single Dose Streptozotocin Induced Diabetes: Considerations for Study Design in Islet Transplantation Models. Lab. Anim. 2011, 45, 131–140. [Google Scholar] [CrossRef]
- Lenzen, S. The mechanisms of alloxan- and streptozotocin-induced diabetes. Diabetologia 2008, 51, 216–226. [Google Scholar] [CrossRef]
- Szkudelski, T. The mechanism of alloxan and streptozotocin action in B cells of the rat pancreas. Physiol. Res. 2001, 50, 537–546. [Google Scholar] [PubMed]
- Ighodaro, O.M.; Adeosun, A.M.; Akinloye, O.A. Alloxan-induced diabetes, a common model for evaluating the glycemic-control potential of therapeutic compounds and plants extracts in experimental studies. Medicina 2017, 53, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Willcox, A.; Richardson, S.J.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Analysis of islet inflammation in human type 1 diabetes. Clin. Exp. Immunol. 2009, 155, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Azushima, K.; Gurley, S.B.; Coffman, T.M. Modelling diabetic nephropathy in mice. Nat. Rev. Nephrol. 2017, 14, 48–56. [Google Scholar] [CrossRef]
- Arndt, T.; Jörns, A.; Wedekind, D. Changes in immune cell frequencies in primary and secondary lymphatic organs of LEW.1AR1-iddm rats, a model of human type 1 diabetes compared to other MHC congenic LEW inbred strains. Immunol. Res. 2018, 66, 462–470. [Google Scholar]
- Todd, J.A. Intolerable secretion and diabetes in tolerant transgenic mice, revisited. Nat. Genet. 2016, 48, 476–477. [Google Scholar] [CrossRef]
- Dhuria, R.S.; Singh, G.; Kaur, A.; Kaur, R.; Kaur, T. Current status and patent prospective of animal models in diabetic research. Adv. Biomed. Res. 2015, 4, 117. [Google Scholar]
- Ramos-Lobo, A.M.; Donato, J. The role of leptin in health and disease. Temperature 2017, 4, 258–291. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.-W.; Sun, G.-D.; Sun, J.; Liu, S.-J.; Wang, J.; Xu, X.-H.; Miao, L.-N. Spontaneous Type 2 Diabetic Rodent Models. J. Diabetes Res. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Kulkarni, S.; Sharda, S.; Watve, M. Bi-stability in type 2 diabetes mellitus multi-organ signalling network. PLoS ONE 2017, 12, e0181536. [Google Scholar] [CrossRef]
- Guilbaud, A.; Howsam, M.; Niquet-Léridon, C.; Delguste, F.; Boulanger, E.; Tessier, F.J. The LepR db/db mice model for studying glycation in the context of diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3103. [Google Scholar] [CrossRef] [PubMed]
- King, A.; Bowe, J. Animal models for diabetes: Understanding the pathogenesis and finding new treatments. Biochem. Pharmacol. 2016, 99, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Clee, S.M.; Attie, A.D. The Genetic Landscape of Type 2 Diabetes in Mice. Endocr. Rev. 2007, 28, 48–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomino, Y. Lessons From the KK-Ay Mouse, a Spontaneous Animal Model for the Treatment of Human Type 2 Diabetic Nephropathy. Nephro-Urology Mon. 2012, 4, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Bi, S.; Moran, T.H. Obesity in the Otsuka Long Evans Tokushima Fatty Rat: Mechanisms and Discoveries. Front. Nutr. 2016, 3, 1–5. [Google Scholar]
- Kim, J.H.; Stewart, T.P.; Soltani-Bejnood, M.; Wang, L.; Fortuna, J.M. Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J. Endocrinol. 2006, 191, 437–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leiter, E.H.; Reifsnyder, P.C. Section I: Genetic Factors in Type 2 Diabetes—In Search of New Links. Diabetes. 2004, 53, 4–11. [Google Scholar] [CrossRef]
- Hirata, T.; Yoshitomi, T.; Inoue, M.; Iigo, Y.; Matsumoto, K.; Kubota, K.; Shinagawa, A. Pathological and gene expression analysis of a polygenic diabetes model, NONcNZO10/LtJ mice. Gene 2017, 629, 52–58. [Google Scholar] [CrossRef]
- Winzell, M.S.; Ahrén, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53, S215–S219. [Google Scholar] [CrossRef]
- Shafrir, E.; Ziv, E.; Kalman, R. Nutritionally induced diabetes in desert rodents as models of type 2 diabetes: Acomys cahirinus (spiny mice) and Psammomys obesus (desert gerbil). ILAR J. 2006, 47, 212–224. [Google Scholar] [CrossRef]
- Shafrir, E.; Ziv, E.; Mosthaf, L. Nutritionally Induced Insulin Resistance and Receptor Defect Leading to beta-Cell Failure in Animal Models. Ann. N. Y. Acad. Sci. 1999, 892, 223–246. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Melhorn, M.I.; Zandi, S.; Frimmel, S.; Tayyari, F.; Hisatomi, T.; Almulki, L.; Pronczuk, A.; Hayes, K.C.; Hafezi-Moghadam, A. An animal model of spontaneous metabolic syndrome: Nile grass rat. FASEB J. 2010, 24, 2443–2453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, L.; Panchal, S.K. Rodent models for metabolic syndrome research. J. Biomed. Biotechnol. 2011, 2011, 351982. [Google Scholar]
- Turner, P.V.; Brabb, T.; Pekow, C.; Vasbinder, M.A. Administration of Substances to Laboratory Animals: Routes of Administration and Factors to Consider. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 600–613. [Google Scholar]
- Sánchez-López, E.; Egea, M.; Cano, A.; Espina, M.; Calpena, A.; Ettcheto, M.; Camins, A.; Souto, E.; Silva, A.; García, M.; et al. PEGylated PLGA nanospheres optimized by design of experiments for ocular administration of dexibuprofen—In vitro, ex vivo and in vivo characterization. Colloids Surfaces B Biointerfaces 2016, 145, 241–250. [Google Scholar] [CrossRef]
- Fangueiro, J.F.; Calpena, A.C.; Clares, B.; Andreani, T.; Egea, M.A.; Veiga, F.J.; Garcia, M.L.; Silva, A.M.; Souto, E.B. Biopharmaceutical evaluation of epigallocatechin gallate-loaded cationic lipid nanoparticles (EGCG-LNs): In vivo, in vitro and ex vivo studies. Int. J. Pharm. 2016, 502, 161–169. [Google Scholar] [CrossRef]
- Faustino-Rocha, A.I.; Gama, A.; Oliveira, P.A.; Vanderperren, K.; Saunders, J.H.; Pires, M.J.; Ferreira, R.; Ginja, M. Modulation of mammary tumor vascularization by mast cells: Ultrasonographic and histopathological approaches. Life Sci. 2017, 176, 35–41. [Google Scholar] [CrossRef]
- Nogueira, A.; Vala, H.; Vasconcelos-Nóbrega, C.; Faustino-Rocha, A.I.; Pires, C.A.; Colaço, A.; Oliveira, P.A.; Pires, M.J. Long-term treatment with chaethomellic acid A reduces glomerulosclerosis and arteriolosclerosis in a rat model of chronic kidney disease. Biomed. Pharmacother. 2017, 96, 489–496. [Google Scholar] [CrossRef] [Green Version]
- Nebendahl, K. Chapter 24—Routes of Administration A2—Krinke, Georg J. The Laboratory Rat; Academic Press: London, UK, 2000; pp. 463–483. [Google Scholar]
- Lax, E.R.; Militzer, K.; Trauschel, A. A simple method for oral administration of drugs in solid form to fully conscious rats. Lab. Anim. 1983, 17, 50–54. [Google Scholar] [CrossRef] [Green Version]
- Mesejo, A.; Montejo-González, J.C.; Vaquerizo-Alonso, C.; Lobo-Tamer, G.; Zabarte-Martinez, M.; Herrero-Meseguer, J.I.; Acosta-Escribano, J.; Blesa-Malpica, A.; Martinez-Lozano, F. Diabetes-specific enteral nutrition formula in hyperglycemic, mechanically ventilated, critically ill patients: A prospective, open-label, blind-randomized, multicenter study. Crit. Care 2015, 19, 75. [Google Scholar] [CrossRef]
- Diehl, K.-H.; Hull, R.; Morton, D.; Pfister, R.; Rabemampianina, Y.; Smith, D.; Vidal, J.-M.; Van De Vorstenbosch, C. A good practice guide to the administration of substances and removal of blood, including routes and volumes. J. Appl. Toxicol. 2001, 21, 15–23. [Google Scholar] [PubMed]
- Umpierrez, G.; Korytkowski, M. Diabetic emergencies — ketoacidosis, hyperglycaemic hyperosmolar state and hypoglycaemia. Nat. Rev. Endocrinol. 2016, 12, 222–232. [Google Scholar] [CrossRef] [PubMed]
- Elliott, A.; Dubé, P.-A.; Cossette-Côté, A.; Patakfalvi, L.; Villeneuve, E.; Morris, M. Intraosseous administration of antidotes—A systematic review. Clin. Toxicol. 2017, 55, 1025–1054. [Google Scholar] [CrossRef] [PubMed]
- Andreani, T.; Macedo, A.S.; Ferreira, S.F.; Silva, A.M.; Rosmaninho, A.; Souto, E.B. Topical Targeting Therapies for Sexually Transmitted Diseases. Curr. Nanosci. 2012, 8, 486–490. [Google Scholar]
- Rini, C.J.; McVey, E.; Sutter, D.; Keith, S.; Kurth, H.-J.; Nosek, L.; Kapitza, C.; Rebrin, K.; Hirsch, L.; Pettis, R.J. Intradermal insulin infusion achieves faster insulin action than subcutaneous infusion for 3-day wear. Drug Deliv. Transl. Res. 2015, 5, 332–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Ji, S.; Wu, H.; Tian, S.; Zhang, Y.; Wang, L.; Fang, H.; Luo, P.; Wang, X.; Hu, X.; et al. Topical administration of cryopreserved living micronized amnion accelerates wound healing in diabetic mice by modulating local microenvironment. Biomaterials 2017, 113, 56–67. [Google Scholar] [PubMed]
- Demyanenko, I.A.; Zakharova, V.V.; Ilyinskaya, O.P.; Vasilieva, T.V.; Fedorov, A.V.; Manskikh, V.N.; Zinovkin, R.A.; Pletjushkina, O.Y.; Chernyak, B.V.; Skulachev, V.P.; et al. Mitochondria-Targeted Antioxidant SkQ1 Improves Dermal Wound Healing in Genetically Diabetic Mice. Oxidative Med. Cell. Longev. 2017, 2017, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, S. Routes of Administration. In The Laboratory Mouse; Elsevier: Amsterdam, The Netherlands, 2004; pp. 527–541. [Google Scholar]
- Turner, P.V.; Pekow, C.; Vasbinder, M.A.; Brabb, T. Administration of Substances to Laboratory Animals: Equipment Considerations, Vehicle Selection, and Solute Preparation. J. Am. Assoc. Lab. Anim. Sci. 2011, 50, 614–627. [Google Scholar]
- Wu, S.C.; Pollak, R.; Frykberg, R.G.; Karnoub, M.; Fischkoff, S.A.; Chitkara, D.; Zhou, W.; Jankovic, V. Safety and efficacy of intramuscular human placenta-derived mesenchymal stromal-like cells (cenplacel [PDA-002]) in patients who have a diabetic foot ulcer with peripheral arterial disease. Int. Wound J. 2017, 14, 823–829. [Google Scholar] [CrossRef] [Green Version]
- De Barros, G.A.M.; Marques, M.E.A.; Ganem, E.M. The effects of intrathecal administration of betamethasone over the dogs’ spinal cord and meninges. Acta Cir. Bras. 2007, 22, 361–365. [Google Scholar] [CrossRef]
- Pentel, P.R.; Jentzen, J.; Sievert, J. Myocardial necrosis due to intraperitoneal administration of phenylpropanolamine in rats. Fundam. Appl. Toxicol. 1987, 9, 167–172. [Google Scholar] [PubMed]
- Palleria, C.; Leo, A.; Andreozzi, F.; Citraro, R.; Iannone, M.; Spiga, R.; Sesti, G.; Constanti, A.; De Sarro, G.; Arturi, F.; et al. Liraglutide prevents cognitive decline in a rat model of streptozotocin-induced diabetes independently from its peripheral metabolic effects. Behav. Brain Res. 2017, 321, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.Y.; Wang, J.; Zhao, Y.; Shen, L.; Jiang, X.; Xie, Z.G. Anti-diabetic effects of Panax notoginseng saponins and its major anti-hyperglycemic components. J. Ethnopharmacol. 2010, 130, 231–236. [Google Scholar] [PubMed]
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal Administration of CNS Therapeutics to Awake Mice. J. Vis. Exp. 2013, 74, e4440. [Google Scholar] [CrossRef] [PubMed]
- Illum, L. Nasal drug delivery: New developments and strategies. Drug Discov. Today 2002, 7, 1184–1189. [Google Scholar] [CrossRef]



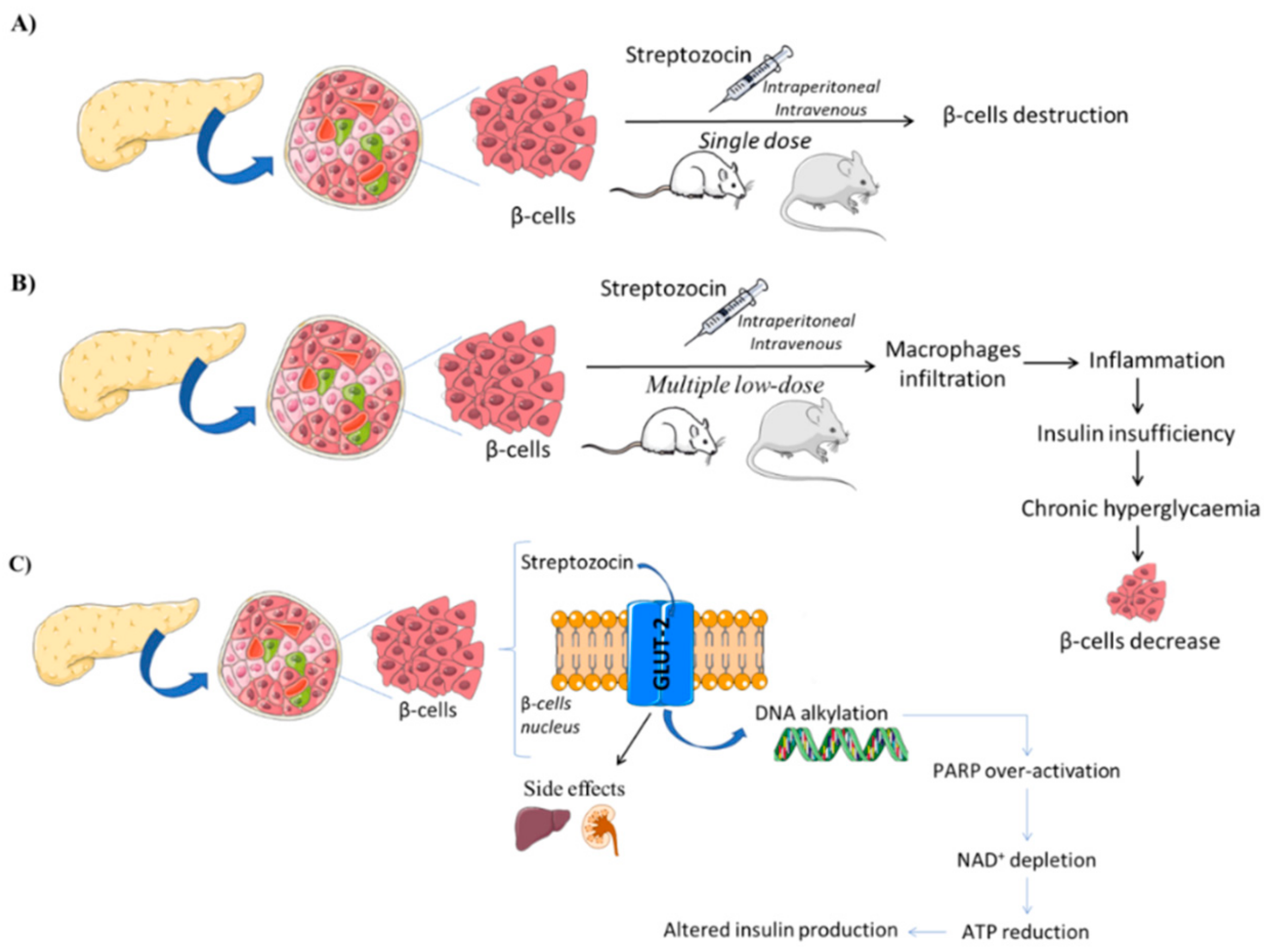{kind=link}
{kind=link}
| Induction Mechanism | Model | Main Features | Possible Uses | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|---|
| Chemical induction | High single-dose streptozotocin (STZ) * | Simple model of hyperglycemia | Testing drugs (new insulin formulations) or therapies (transplantation) |
|
| [17,18] |
| ||||||
| Multiple low dose streptozotocin (STZ) * | Model of induced insulitis | Treatments that may prevent β-cell death |
|
| ||
|
| |||||
| Alloxan * | Simple model of hyperglycemia | Transplantation models |
|
| [19,20] | |
|
| |||||
|
| |||||
|
| |||||
| Spontaneous autoimmune | Non-obese diabetic (NOD) mice(Spontaneous autoimmune model of choice) | β-cell destruction due to an autoimmune process | Understanding genetics of T1DM |
|
| [16,21,22] |
| Biobreeding (BB) rats | Understanding mechanism of T1DM |
|
| [23,24] | ||
| LEW.1AR1/-iidm rats | Treatments that may prevent β-cell death and/or manipulate autoimmune process | |||||
| ||||||
| Genetically induced | AKITA mice * | β-cell destruction due to ER stress. Insulin dependent. | New formulations of insulin Transplantation models Treatments to prevent ER stress |
| [25,26] | |
| Virally-induced | Coxsackie B virus | β-cell destruction induced by viral infection of b-cells | Establish potential role of viruses in the development of T1DM |
|
| [27] |
| Encephalomyocarditis virus | ||||||
| Kilham rat virus | ||||||
| LCMV under insulin promoter | ||||||
| ||||||
| Non-rodent models | Pancreatectomy | Hyperglycemia induction in pigs, dogs and primates | Treatments that may prevent β-cell death Transplantation models |
|
| [28] |
| ||||||
| Chemical ablation of β-cells in large animals |
|
| [28] | |||
|
| Induction Mechanism | Model | Main Features | Possible Uses | Advantages | Disadvantages | Ref. |
|---|---|---|---|---|---|---|
| Obese monogenic models | Lepob/ob mice (mutated leptin gene) | Obesity-induced hyperglycemia, with hyperphagic, obese, hyperinsulinaemic and hyperglycemic animals | Treatments to improve insulin resistance |
|
| [29] |
| ||||||
| ||||||
| Leprdb/db mice (mutated leptin receptor gene) | Treatments to improve β-cell function |
| [30] | |||
| Zucker Diabetic Fatty (ZDF) Rats (mutated leptin receptor gene) |
|
| [31] | |||
| ||||||
| Obese polygenic models | KK mice | Obesity-induced hyperglycemia | Treatments to improve insulin resistance | |||
| Otsuka Long-Evans Tokushima Fat (OLEFT) rat | Treatments to improve β-cell function |
| [32,33] | |||
| New Zealand Obese (NZO) mice | Some models show diabetic complications |
| ||||
| TallyHo/Jng mice |
|
| ||||
| NoncNZO10/LtJ mice |
| |||||
| ||||||
| Induced obesity models | High fat feeding (mice or rats) | Obesity-induced hyperglycemia | Treatments to improve insulin resistance |
|
| [31,33] |
| Desert gerbil | Treatments to improve β-cell function |
|
| |||
| Nile grass rat | Treatments to prevent diet-induced obesity |
| ||||
| Non-obese models | Goto-Kakizaki (GK) rat | Hyperglycemia induced by insufficient β-cell function or mass | Treatments to improve β-cell function |
|
| [31,33] |
| Treatments to improve β-cell survival | ||||||
| Genetically induced models of β-cell dysfunction | Human islet amyloid polypeptide-expressing (hIAPP) mice | Amyloid deposition in islets | Treatments to prevent amyloid deposition |
|
| [31,33] |
| Treatments to improve β-cell function | ||||||
| β-cell destruction due to ER stress | Treatments to prevent ER stress |
| ||||
| Treatments to improve β-cell survival | ||||||
| Non-rodent models | Cat models | Amyloid deposition in islets | Treatments to improve β-cell function |
|
| [31,33,34,35,36,37] |
| β-cell destruction | Treatments to prevent diet-induced obesity | |||||
| Old-world non-human primates |
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vieira, R.; Souto, S.B.; Sánchez-López, E.; López Machado, A.; Severino, P.; Jose, S.; Santini, A.; Silva, A.M.; Fortuna, A.; García, M.L.; et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Strategies for In Vivo Administration: Part-II. J. Clin. Med. 2019, 8, 1332. https://doi.org/10.3390/jcm8091332
Vieira R, Souto SB, Sánchez-López E, López Machado A, Severino P, Jose S, Santini A, Silva AM, Fortuna A, García ML, et al. Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Strategies for In Vivo Administration: Part-II. Journal of Clinical Medicine. 2019; 8(9):1332. https://doi.org/10.3390/jcm8091332
Chicago/Turabian StyleVieira, Raquel, Selma B. Souto, Elena Sánchez-López, Ana López Machado, Patricia Severino, Sajan Jose, Antonello Santini, Amelia M. Silva, Ana Fortuna, Maria Luisa García, and et al. 2019. "Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Strategies for In Vivo Administration: Part-II" Journal of Clinical Medicine 8, no. 9: 1332. https://doi.org/10.3390/jcm8091332
APA StyleVieira, R., Souto, S. B., Sánchez-López, E., López Machado, A., Severino, P., Jose, S., Santini, A., Silva, A. M., Fortuna, A., García, M. L., & Souto, E. B. (2019). Sugar-Lowering Drugs for Type 2 Diabetes Mellitus and Metabolic Syndrome—Strategies for In Vivo Administration: Part-II. Journal of Clinical Medicine, 8(9), 1332. https://doi.org/10.3390/jcm8091332