Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations

 , , , and add
Show full author list
, , , and add
Show full author list
Abstract
:1. Introduction
2. Experimental Section
2.1. Subjects
2.2. Genetic Studies
2.3. Statistical Analysis
3. Results
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tattersall, R.B. Mild familial diabetes with dominant inheritance. QJM Int. J. Med. 1974, 43, 339–357. [Google Scholar]
- Ledermann, H.M. Maturity-onset diabetes of the young (MODY) at least ten times more common in Europe than previously assumed? Diabetologia 1995, 38, 1482. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.M.; Hicks, S.; Shepherd, M.H.; Colclough, K.; Hattersley, A.T.; Ellard, S. Maturity-onset diabetes of the young (MODY): How many cases are we missing? Diabetologia 2010, 53, 2504–2508. [Google Scholar] [CrossRef] [PubMed]
- Hattersley, A.T.; Patel, K.A. Precision diabetes: Learning from monogenic diabetes. Diabetologia 2017, 60, 769–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Firdous, P.; Nissar, K.; Ali, S.; Ganai, B.A.; Shabir, U.; Hassan, T.; Masoodi, S.R. Genetic Testing of Maturity-Onset Diabetes of the Young Current Status and Future Perspectives. Front. Endocrinol. 2018, 9, 253. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, J.W.; Pollin, T.I. Undiagnosed MODY: Time for Action. Curr. Diabetes Rep. 2015, 15, 110. [Google Scholar] [CrossRef]
- Lemos, M.C.; Regateiro, F.J. N-acetyltransferase genotypes in the Portuguese population. Pharmacogenetics 1998, 8, 561–564. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The human genomic variant search engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Gragnoli, C.; Cockburn, B.N.; Chiaramonte, F.; Gorini, A.; Marietti, G.; Marozzi, G.; Signorini, A.M. Early-onset Type II diabetes mellitus in Italian families due to mutations in the genes encoding hepatic nuclear factor 1 alpha and glucokinase. Diabetologia 2001, 44, 1326–1329. [Google Scholar] [CrossRef] [Green Version]
- Massa, O.; Meschi, F.; Cuesta-Munoz, A.; Caumo, A.; Cerutti, F.; Toni, S.; Cherubini, V.; Guazzarotti, L.; Sulli, N.; Matschinsky, F.M.; et al. High prevalence of glucokinase mutations in Italian children with MODY. Influence on glucose tolerance, first-phase insulin response, insulin sensitivity and BMI. Diabetologia 2001, 44, 898–905. [Google Scholar]
- Froguel, P.; Zouali, H.; Vionnet, N.; Velho, G.; Vaxillaire, M.; Sun, F.; Lesage, S.; Stoffel, M.; Takeda, J.; Passa, P.; et al. Familial hyperglycemia due to mutations in glucokinase. Definition of a subtype of diabetes mellitus. N. Engl. J. Med. 1993, 328, 697–702. [Google Scholar] [CrossRef]
- Katagiri, H.; Asano, T.; Ishihara, H.; Inukai, K.; Anai, M.; Miyazaki, J.; Tsukuda, K.; Kikuchi, M.; Yazaki, Y.; Oka, Y. Nonsense mutation of glucokinase gene in late-onset non-insulin-dependent diabetes mellitus. Lancet 1992, 340, 1316–1317. [Google Scholar] [CrossRef]
- Wheeler, B.J.; Patterson, N.; Love, D.R.; Prosser, D.; Tomlinson, P.; Taylor, B.J.; Manning, P. Frequency and genetic spectrum of maturity-onset diabetes of the young (MODY) in southern New Zealand. J. Diabetes Metab. Disord. 2013, 12, 46. [Google Scholar] [CrossRef] [Green Version]
- Weinert, L.S.; Silveiro, S.P.; Giuffrida, F.M.; Cunha, V.T.; Bulcao, C.; Calliari, L.E.; Della Manna, T.; Kunii, I.S.; Dotto, R.P.; Dias-da-Silva, M.R.; et al. Three unreported glucokinase (GCK) missense mutations detected in the screening of thirty-two Brazilian kindreds for GCK and HNF1A-MODY. Diabetes Res. Clin. Pract. 2014, 106, e44–e48. [Google Scholar] [CrossRef]
- Velho, G.; Blanche, H.; Vaxillaire, M.; Bellanne-Chantelot, C.; Pardini, V.C.; Timsit, J.; Passa, P.; Deschamps, I.; Robert, J.J.; Weber, I.T.; et al. Identification of 14 new glucokinase mutations and description of the clinical profile of 42 MODY-2 families. Diabetologia 1997, 40, 217–224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osbak, K.K.; Colclough, K.; Saint-Martin, C.; Beer, N.L.; Bellanne-Chantelot, C.; Ellard, S.; Gloyn, A.L. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum. Mutat. 2009, 30, 1512–1526. [Google Scholar] [CrossRef] [PubMed]
- Vaxillaire, M.; Rouard, M.; Yamagata, K.; Oda, N.; Kaisaki, P.J.; Boriraj, V.V.; Chevre, J.C.; Boccio, V.; Cox, R.D.; Lathrop, G.M.; et al. Identification of nine novel mutations in the hepatocyte nuclear factor 1 alpha gene associated with maturity-onset diabetes of the young (MODY3). Hum. Mol. Genet. 1997, 6, 583–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chevre, J.C.; Hani, E.H.; Boutin, P.; Vaxillaire, M.; Blanche, H.; Vionnet, N.; Pardini, V.C.; Timsit, J.; Larger, E.; Charpentier, G.; et al. Mutation screening in 18 Caucasian families suggest the existence of other MODY genes. Diabetologia 1998, 41, 1017–1023. [Google Scholar] [CrossRef] [Green Version]
- Bellanne-Chantelot, C.; Carette, C.; Riveline, J.P.; Valero, R.; Gautier, J.F.; Larger, E.; Reznik, Y.; Ducluzeau, P.H.; Sola, A.; Hartemann-Heurtier, A.; et al. The type and the position of HNF1A mutation modulate age at diagnosis of diabetes in patients with maturity-onset diabetes of the young (MODY)-3. Diabetes 2008, 57, 503–508. [Google Scholar] [CrossRef] [Green Version]
- Colclough, K.; Bellanne-Chantelot, C.; Saint-Martin, C.; Flanagan, S.E.; Ellard, S. Mutations in the genes encoding the transcription factors hepatocyte nuclear factor 1 alpha and 4 alpha in maturity-onset diabetes of the young and hyperinsulinemic hypoglycemia. Hum. Mutat. 2013, 34, 669–685. [Google Scholar] [CrossRef]
- Dyle, M.C.; Kolakada, D.; Cortazar, M.A.; Jagannathan, S. How to get away with nonsense: Mechanisms and consequences of escape from nonsense-mediated RNA decay. Wiley Interdiscip. Rev. RNA 2019, e1560. [Google Scholar] [CrossRef]
- Ellard, S.; Lango Allen, H.; de Franco, E.; Flanagan, S.E.; Hysenaj, G.; Colclough, K.; Houghton, J.A.; Shepherd, M.; Hattersley, A.T.; Weedon, M.N.; et al. Improved genetic testing for monogenic diabetes using targeted next-generation sequencing. Diabetologia 2013, 56, 1958–1963. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.H. Maturity-Onset Diabetes of the Young: What Do Clinicians Need to Know? Diabetes Metab. J. 2015, 39, 468–477. [Google Scholar] [CrossRef] [Green Version]
- Ellard, S.; Bellanne-Chantelot, C.; Hattersley, A.T. Best practice guidelines for the molecular genetic diagnosis of maturity-onset diabetes of the young. Diabetologia 2008, 51, 546–553. [Google Scholar] [CrossRef] [Green Version]
- Pezzilli, S.; Ludovico, O.; Biagini, T.; Mercuri, L.; Alberico, F.; Lauricella, E.; Dallali, H.; Capocefalo, D.; Carella, M.; Miccinilli, E.; et al. Insights From Molecular Characterization of Adult Patients of Families With Multigenerational Diabetes. Diabetes 2018, 67, 137–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Annunzio, G.; Marchi, M.; Aloi, C.; Salina, A.; Lugani, F.; Lorini, R. Hyperglycaemia and beta-cell antibodies: Is it always pre-type 1 diabetes? Diabetes Res. Clin. Pract. 2013, 100, e20–e22. [Google Scholar] [CrossRef] [PubMed]
- Shields, B.M.; Shepherd, M.; Hudson, M.; McDonald, T.J.; Colclough, K.; Peters, J.; Knight, B.; Hyde, C.; Ellard, S.; Pearson, E.R.; et al. Population-Based Assessment of a Biomarker-Based Screening Pathway to Aid Diagnosis of Monogenic Diabetes in Young-Onset Patients. Diabetes Care 2017, 40, 1017–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, B.M.; McDonald, T.J.; Ellard, S.; Campbell, M.J.; Hyde, C.; Hattersley, A.T. The development and validation of a clinical prediction model to determine the probability of MODY in patients with young-onset diabetes. Diabetologia 2012, 55, 1265–1272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Misra, S.; Owen, K.R. Genetics of Monogenic Diabetes: Present Clinical Challenges. Curr. Diabetes Rep. 2018, 18, 141. [Google Scholar] [CrossRef] [Green Version]
- Naylor, R. Economics of Genetic Testing for Diabetes. Curr. Diabetes Rep. 2019, 19, 23. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
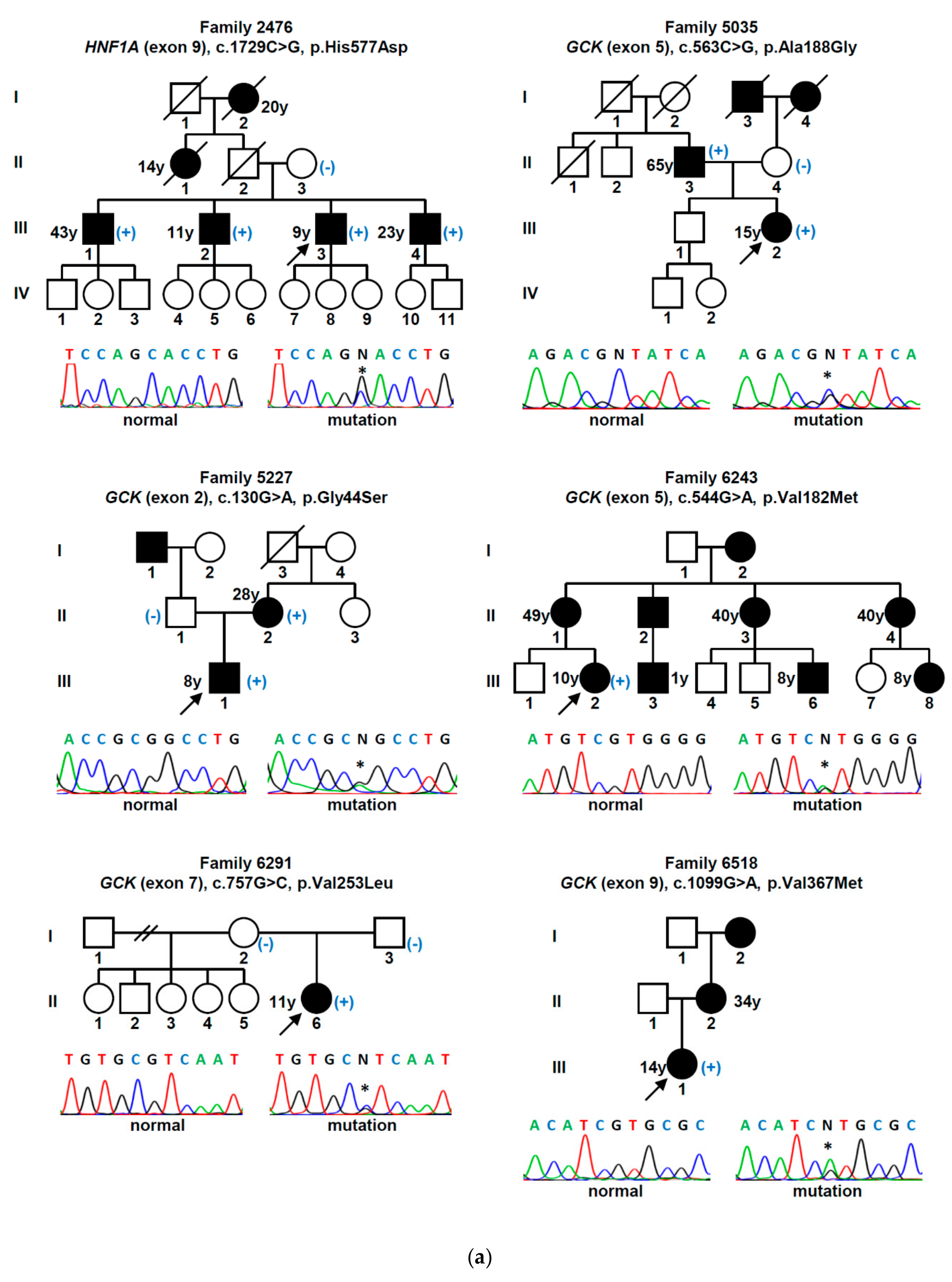
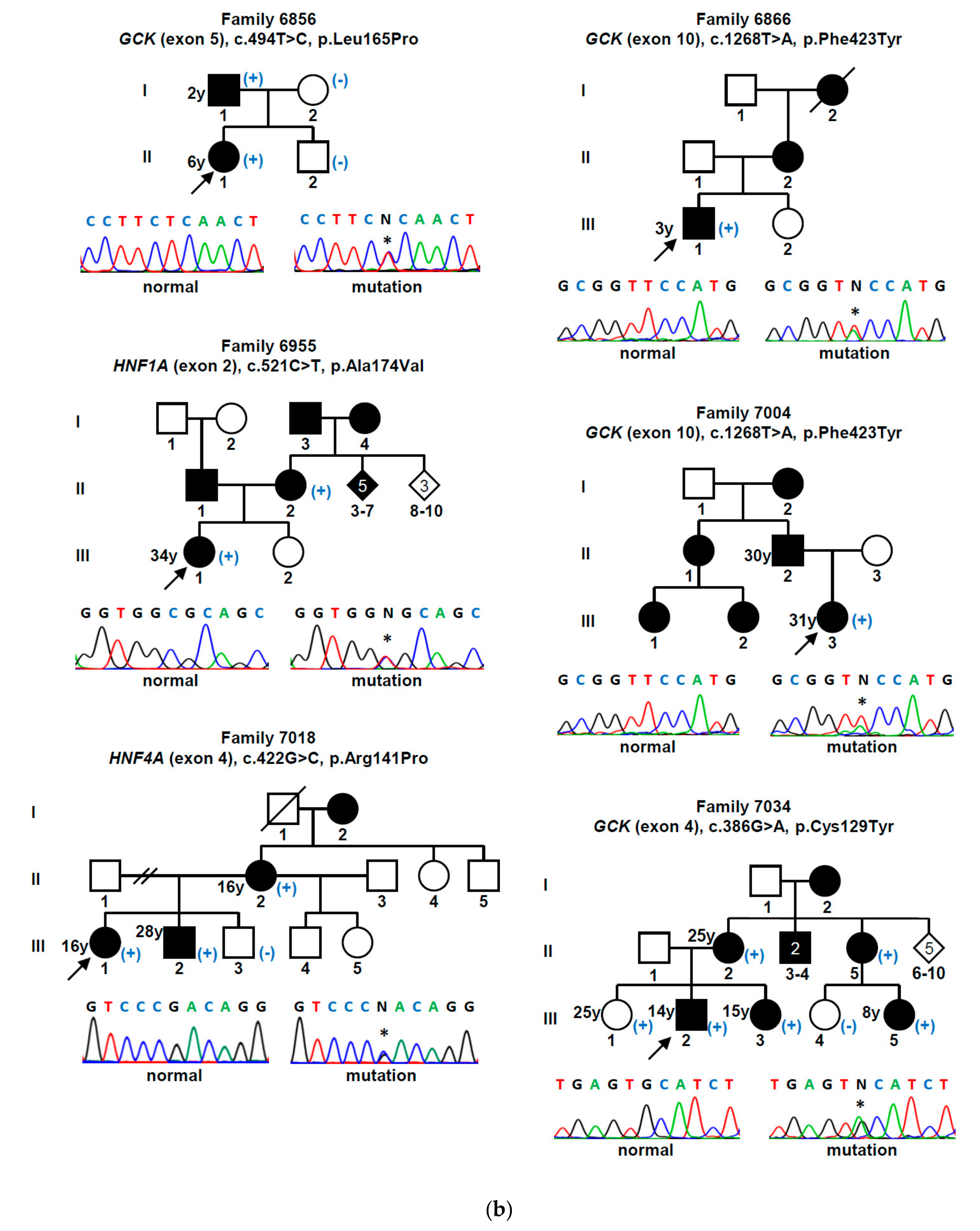
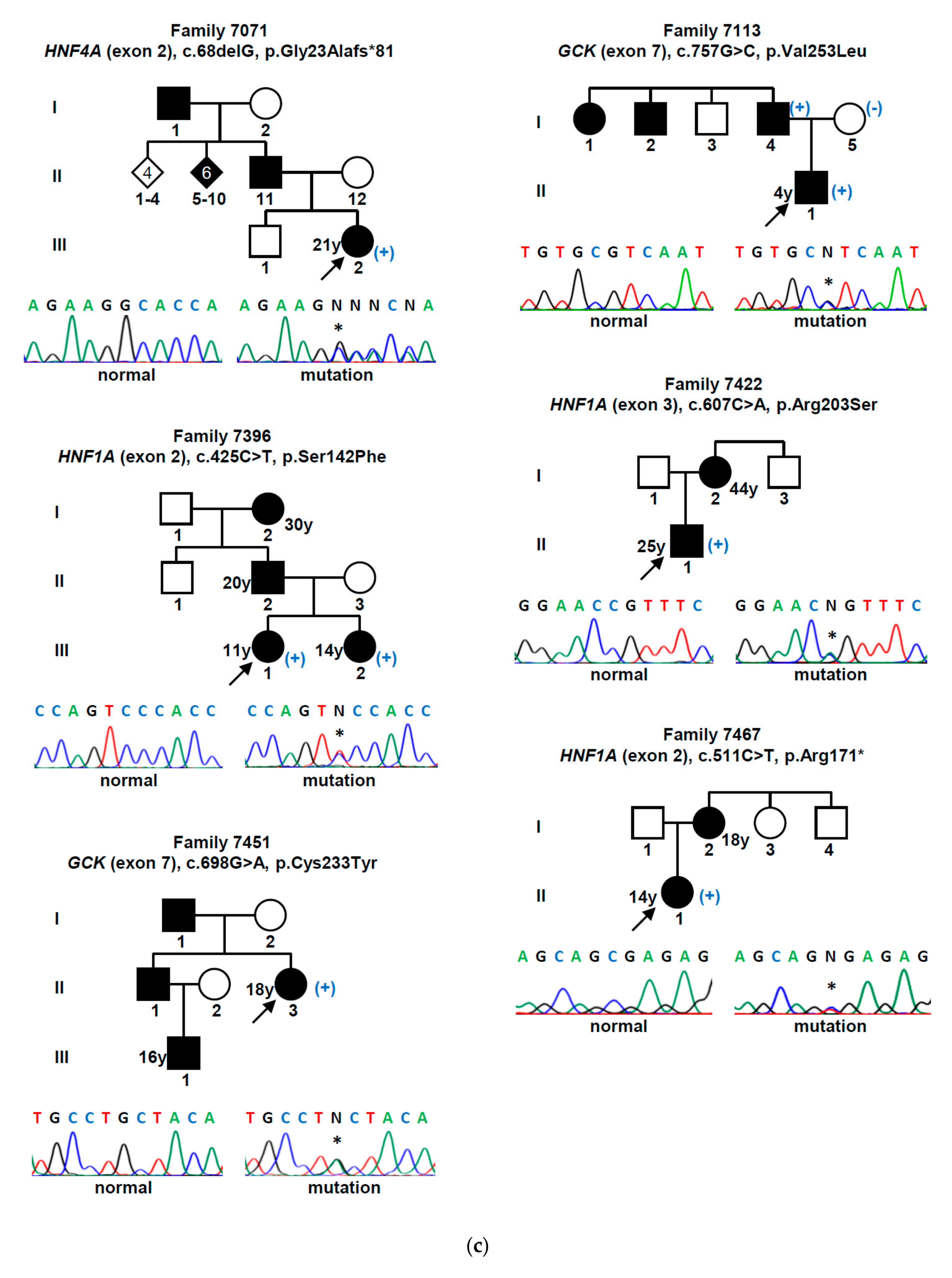
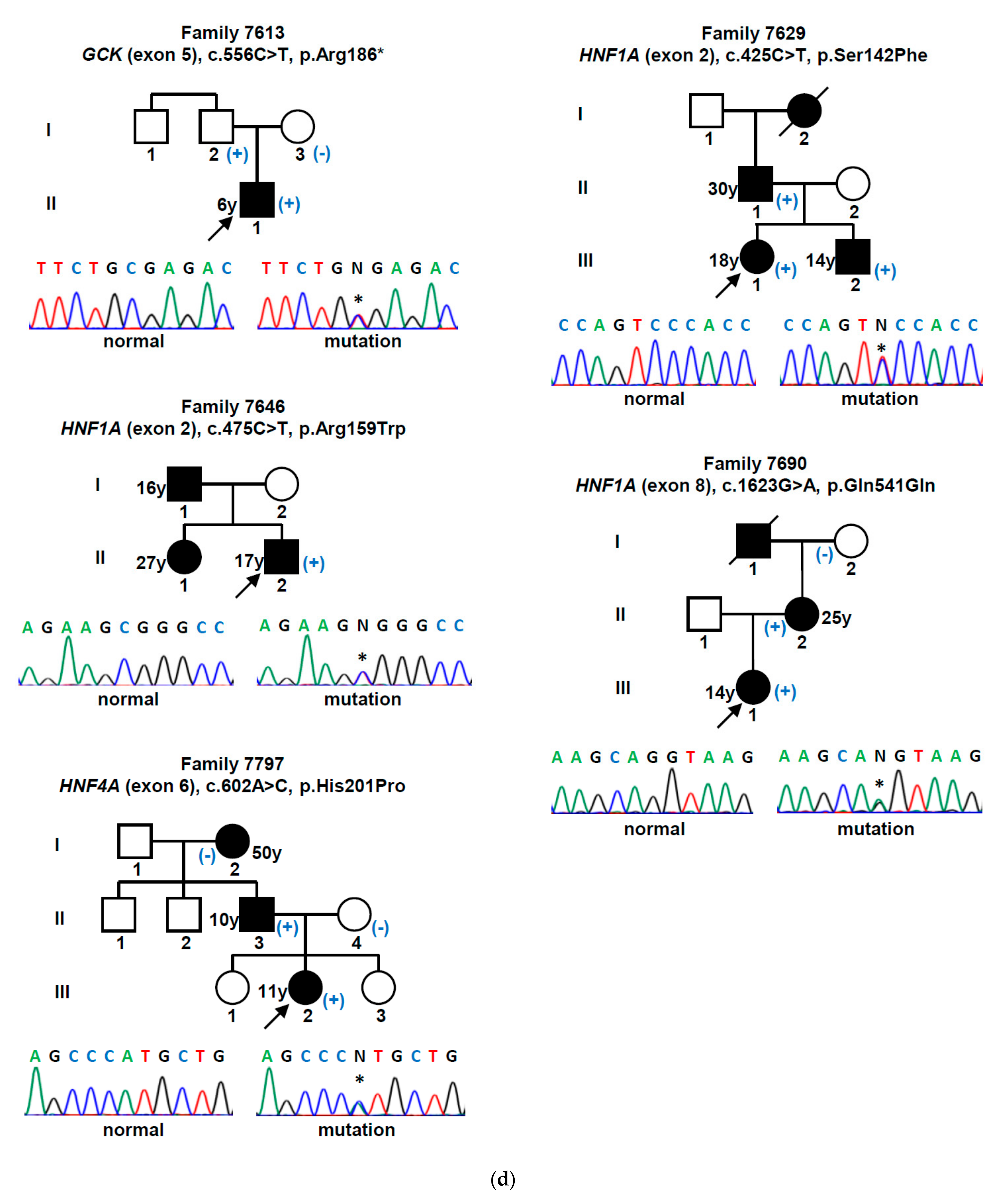
| Gene (a) | Nucleotide Change | Amino Acid Change | Mutation Type | Population Allele Frequency (gnomAD/Portuguese Controls) | Computational Programs That Support a Pathogenic Effect (b) | Classification (ACMG Criteria) (c) | Previously Reported (Reference) |
|---|---|---|---|---|---|---|---|
| GCK | c.130G>A | p.Gly44Ser | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PM5, PP2, PP3) | Yes [15] |
| c.386G>A | p.Cys129Tyr | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PP2, PP3) | Yes [16] | |
| c.494T>C | p.Leu165Pro | missense | 0/0 (d) | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PP2, PP3, PP5) | No | |
| c.544G>A | p.Val182Met | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PP2, PP3, PP5) | Yes [17] | |
| c.556C>T | p.Arg186* | nonsense | 0 | MT | Pathogenic (PVS1, PM1, PM2, PP3, PP5) | Yes [18] | |
| c.563C>G | p.Ala188Gly | missense | 0/0 (h) | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PM5, PP2, PP3) | No | |
| c.698G>A | p.Cys233Tyr | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PP2, PP3) | Yes [19] | |
| c.757G>C | p.Val253Leu | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PM5, PM6, PP2, PP3) | Yes [20] | |
| c.1099G>A | p.Val367Met | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PP2, PP3, PP5) | Yes [21] | |
| c.1268T>A | p.Phe423Tyr | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PP2, PP3, PP5) | Yes [22] | |
| HNF1A | c.425C>T | p.Ser142Phe | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM2, PP1, PP2, PP3, PP5) | Yes [23] |
| c.475C>T | p.Arg159Trp | missense | 0.0000039 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PM5, PP2, PP3, PP5) | Yes [24] | |
| c.511C>T | p.Arg171* | nonsense | 0 | MT | Pathogenic (PVS1, PM2, PP3, PP5) | Yes [23] | |
| c.521C>T | p.Ala174Val | missense | 0.0001951 | MT | VUS (PP2, PP3, BS2) | Yes [25] | |
| c.607C>A | p.Arg203Ser | missense | 0 | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PM5, PP2, PP3) | Yes [26] | |
| c.1623G>A | p.Gln541Gln | synonymous | 0/0 (e) | MT, HSF | VUS (PM2, PP1, PP3) | No | |
| c.1729C>G | p.His577Asp | missense | 0.0001429/0.001 (i) | SIFT, MT | VUS (PP1, PP2, PP3, BS2) | No | |
| HNF4A | c.68delG | p.Gly23Alafs*81 | frameshift | 0 | MT | Pathogenic (PVS1, PM2, PP3) | No |
| c.422G>C | p.Arg141Pro | missense | 0/0 (f) | SIFT, MT | L. Pathogenic (PM1, PM2, PP1, PP2, PP3) | No | |
| c.602A>C | p.His201Pro | missense | 0/0 (g) | SIFT, PPh-2, MT | L. Pathogenic (PM1, PM2, PP2, PP3) | No |
| Patient ID | Sex/Age (yrs) | Age at Diagnosis (yrs) | Family History (a) | Presenting Signs and Symptoms | BMI (kg/m2) | A1c (%) | C-Peptide (ng/mL) (b) | Abs | Treatment | Complications | Last A1c (%) | Mutation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2476 | M/28 | 9 | Yes | Asymptomatic | 21.8 (*) | 7.2 | 0.10 | Yes | Insulin | Retinopathy, neuropathy, nephropathy (kidney transplant) | 5.2 | HNF1A c.1729C>G (p.His577Asp) |
| 5035 | F/48 | 15 | No | Asymptomatic | 19.4 (*) | n/a | n/a | n/a | No | No | 6.5 | GCK c.563C>G (p.Ala188Gly) |
| 5227 | M/9 | 8 | No | Asymptomatic | 17.5 | 6.4 | 0.84 | No | No | No | 6.7 | GCK c.130G>A (p.Gly44Ser) |
| 6243 | F/22 | 10 | Yes | Asymptomatic | 22.3 (*) | n/a | 7.20 | No | OHA | No | 6.6 | GCK c.544G>A (p.Val182Met) |
| 6291 | F/12 | 11 | No | Weight loss | 17.9 | 6.0 | 4.80 | No | No | No | 6.0 | GCK c.757G>C (p.Val253Leu) |
| 6518 | F/15 | 14 | Yes | Polyuria/polydipsia | 25.5 | 6.5 | 4.50 | No | OHA | No | 6.4 | GCK c.1099G>A (p.Val367Met) |
| 6856 | F/16 | 6 | No | Asymptomatic | 15.5 | 6.6 | 0.82 | No | OHA | No | 6.3 | GCK c.494T>C (p.Leu165Pro) |
| 6866 | M/33 | 3 | Yes | Asymptomatic | 25.2 (*) | 6.6 | 2.52 | n/a | No | No | 6.2 | GCK c.1268T>A (p.Phe423Tyr) |
| 6955 | F/34 | 34 | Yes | Asymptomatic | 26.2 | 6.4 | 3.40 | No | OHA | No | 5.9 | HNF1A c.521C>T (p.Ala174Val) |
| 7004 | F/32 | 31 | Yes | Gestational diabetes | 20.7 (*) | 6.1 | 0.80 | No | No | No | 6.1 | GCK c.1268T>A (p.Phe423Tyr) |
| 7018 | F/31 | 16 | Yes | Polyuria/polydipsia | 28.9 | 9.0 | 1.00 | No | Insulin | Retinopathy | 6.7 | HNF4A c.422G>C (p.Arg141Pro) |
| 7034 | M/20 | 14 | Yes | Asymptomatic | 23.4 (*) | 6.5 | 2.39 | n/a | No | No | 6.1 | GCK c.386G>A (p.Cys129Tyr) |
| 7071 | F/39 | 21 | Yes | Gestational diabetes | 19.1 (*) | n/a | 0.70 | No | No | No | 6.4 | HNF4A c.68delG (p.Gly23Alafs *81) |
| 7113 | M/7 | 4 | No | Asymptomatic | 17.4 (*) | 6.4 | 1.17 | No | No | No | 6.7 | GCK c.757G>C (p.Val253Leu) |
| 7396 | F/13 | 11 | Yes | Weight loss | 16.2 | 8.7 | 1.80 | No | Insulin | No | 5.9 | HNF1A c.425C>T (p.Ser142Phe) |
| 7422 | M/27 | 25 | No | Asymptomatic | 22.7 | n/a | 1.70 | No | OHA + Insulin | No | 5.1 | HNF1A c.607C>A (p.Arg203Ser) |
| 7451 | F/39 | 18 | Yes | Asymptomatic | 25.8 | 6.4 | 2.30 | No | OHA | No | 7.3 | GCK c.698G>A (p.Cys233Tyr) |
| 7467 | F/14 | 14 | No | Asymptomatic | 22.2 | 8.0 | 1.37 | No | OHA | No | 8.0 | HNF1A c.511C>T (p.Arg171 *) |
| 7613 | M/7 | 6 | No | Asymptomatic | 15.6 | 6.9 | 0.90 | No | No | No | 6.7 | GCK c.556C>T (p.Arg186 *) |
| 7629 | F/32 | 18 | Yes | Polyuria/polydipsia | 22.9 (*) | n/a | 1.50 | No | Insulin | No | 5.6 | HNF1A c.425C>T (p.Ser142Phe) |
| 7646 | M/35 | 17 | Yes | Polyuria/polydipsia | 19.3 (*) | n/a | 0.10 | No | Insulin | No | 5.5 | HNF1A c.475C>T (p.Arg159Trp) |
| 7690 | F/18 | 14 | No | Asymptomatic | 30.7 | 6.8 | 3.60 | No | OHA | No | 7.7 | HNF1A c.1623G>A (p.Gln541Gln) |
| 7797 | F/12 | 11 | Yes | Polyuria/polydipsia | 24.1 | 12.0 | 2.22 | No | OHA + Insulin | No | 8.1 | HNF4A c.602A>C (p.His201Pro) |
| Mutation-Positive (n = 23) | Mutation-Negative (n = 23) | p-Value (c) | |
|---|---|---|---|
| Age at diagnosis (mean ± SD) (y) | 14.3 ± 7.7 | 23.0 ± 13.2 | 0.011* |
| Family history (a) (n, %) | 14 (60.9) | 15 (65.2) | 1.000 |
| Clinical presentation and course (b) (n, %) | 22 (95.7) | 22 (95.7) | 1.000 |
| Presence of 2 inclusion criteria (n, %) | 12 (52.2) | 16 (69.6) | 0.365 |
| Presence of 3 inclusion criteria (n, %) | 11 (47.8) | 7 (30.4) | 0.365 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvelos, M.I.; Gonçalves, C.I.; Coutinho, E.; Almeida, J.T.; Bastos, M.; Sampaio, M.L.; Melo, M.; Martins, S.; Dinis, I.; Mirante, A.; et al. Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations. J. Clin. Med. 2020, 9, 288. https://doi.org/10.3390/jcm9010288
Alvelos MI, Gonçalves CI, Coutinho E, Almeida JT, Bastos M, Sampaio ML, Melo M, Martins S, Dinis I, Mirante A, et al. Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations. Journal of Clinical Medicine. 2020; 9(1):288. https://doi.org/10.3390/jcm9010288
Chicago/Turabian StyleAlvelos, Maria I., Catarina I. Gonçalves, Eduarda Coutinho, Joana T. Almeida, Margarida Bastos, Maria L. Sampaio, Miguel Melo, Sofia Martins, Isabel Dinis, Alice Mirante, and et al. 2020. "Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations" Journal of Clinical Medicine 9, no. 1: 288. https://doi.org/10.3390/jcm9010288
APA StyleAlvelos, M. I., Gonçalves, C. I., Coutinho, E., Almeida, J. T., Bastos, M., Sampaio, M. L., Melo, M., Martins, S., Dinis, I., Mirante, A., Gomes, L., Saraiva, J., Pereira, B. D., Gama-de-Sousa, S., Moreno, C., Guelho, D., Martins, D., Baptista, C., Barros, L., ... Lemos, M. C. (2020). Maturity-Onset Diabetes of the Young (MODY) in Portugal: Novel GCK, HNFA1 and HNFA4 Mutations. Journal of Clinical Medicine, 9(1), 288. https://doi.org/10.3390/jcm9010288