Sorting Rare ALS Genetic Variants by Targeted Re-Sequencing Panel in Italian Patients: OPTN, VCP, and SQSTM1 Variants Account for 3% of Rare Genetic Forms


 , ,
, ,
Abstract
:1. Introduction
2. Methods
2.1. Patients
2.2. Genetic Screening
2.3. Targeted Next-Generation Sequencing and Bioinformatic Analysis
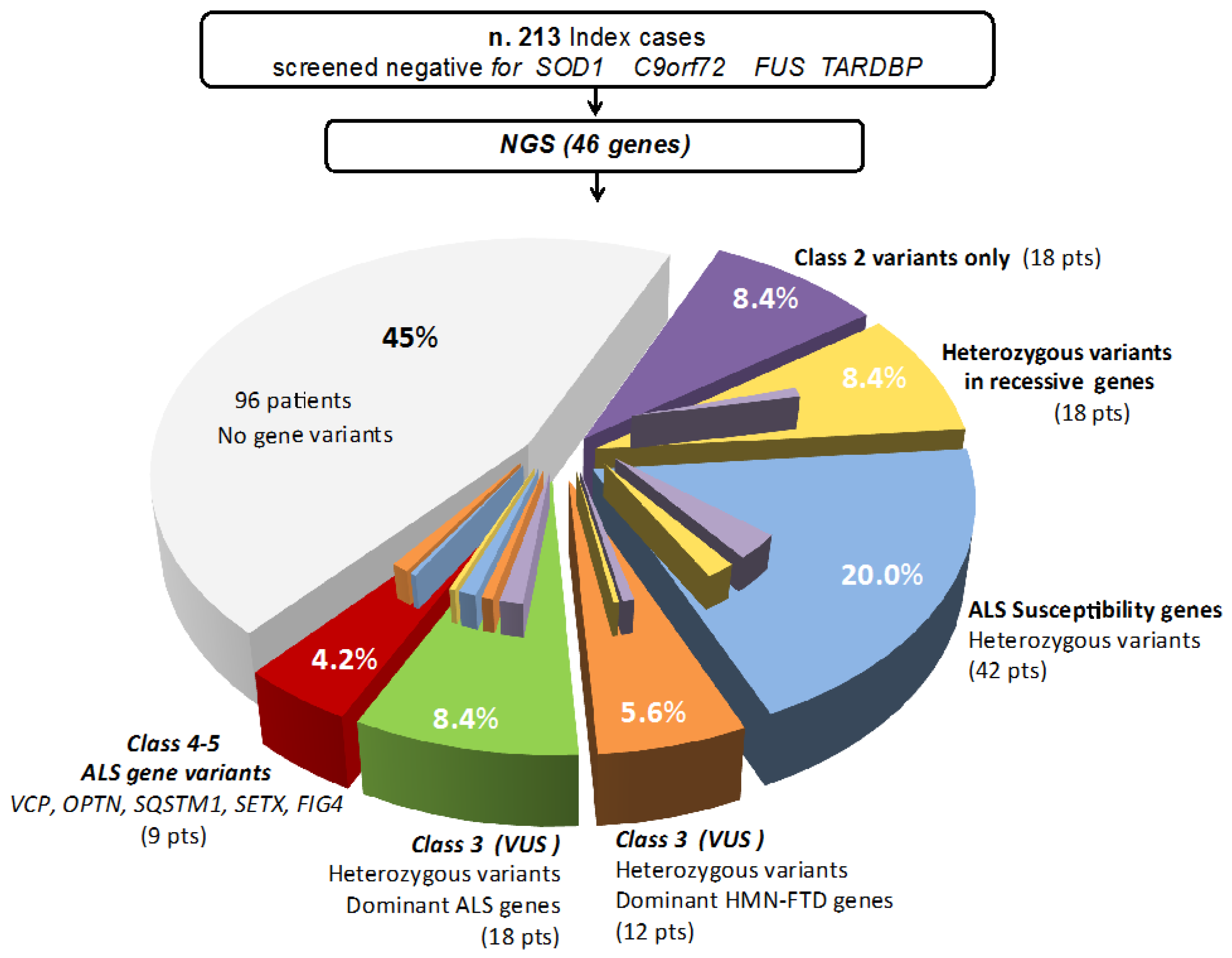
3. Results
Clinical Phenotypes of Patients with Class-3 to Class-5 Gene Variants
OPTN Gene
VCP Gene
SQSTM1 Gene
FIG4 Gene
SETX Gene
GARS1 Gene
4. Discussion and Conclusion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hardiman, O.; Al-Chalabi, A.; Chio, A.; Corr, E.M.; Logroscino, G.; Robberecht, W.; Shaw, P.J.; Simmons, Z.; Van Den Berg, L.H. Amyotrophic lateral sclerosis. Nat. Rev. Dis. Prim. 2017, 3, 17071. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O.; Kiernan, M.C.; Chiò, A.; Rix-Brooks, B.; Van Den Berg, L.H. Amyotrophic lateral sclerosis: Moving towards a new classification system. Lancet Neurol. 2016, 15, 1182–1194. [Google Scholar] [CrossRef]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic lateral sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Renton, A.E.; Chiò, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Cady, J.; Allred, P.; Bali, T.; Pestronk, A.; Goate, A.; Miller, T.M.; Mitra, R.D.; Ravits, J.; Harms, M.B.; Baloh, R.H. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann. Neurol. 2015, 77, 100–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.; Hentati, A.; Donaldson, D.; Goto, J.; O’Regan, J.P.; Deng, H.X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Su, X.W.; Broach, J.R.; Connor, J.R.; Gerhard, G.S.; Simmons, Z. Genetic heterogeneity of amyotrophic lateral sclerosis: Implications for clinical practice and research. Muscle Nerve 2014, 49, 786–803. [Google Scholar] [CrossRef]
- Al-Chalabi, A.; Hardiman, O. The epidemiology of ALS: A conspiracy of genes, environment and time. Nat. Rev. Neurol. 2013, 9, 617–628. [Google Scholar] [CrossRef]
- Cruts, M.; Gijselinck, I.; Van Langenhove, T.; van der Zee, J.; Van Broeckhoven, C. Current insights into the C9orf72 repeat expansion diseases of the FTLD/ALS spectrum. Trends Neurosci. 2013, 36, 450–459. [Google Scholar] [CrossRef]
- Ratti, A.; Corrado, L.; Castellotti, B.; Del Bo, R.; Fogh, I.; Cereda, C.; Tiloca, C.; D’Ascenzo, C.; Bagarotti, A.; Pensato, V.; et al. C9ORF72 repeat expansion in a large Italian ALS cohort: Evidence of a founder effect. Neurobiol. Aging 2012, 33, 2528.e7–2528.e14. [Google Scholar] [CrossRef]
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E.; et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corrado, L.; Ratti, A.; Gellera, C.; Buratti, E.; Castellotti, B.; Carlomagno, Y.; Ticozzi, N.; Mazzini, L.; Testa, L.; Taroni, F.; et al. High frequency of TARDBP gene mutations in italian patients with amyotrophic lateral sclerosis. Hum. Mutat. 2009, 30, 688–694. [Google Scholar] [CrossRef] [PubMed]
- Corrado, L.; Del Bo, R.; Castellotti, B.; Ratti, A.; Cereda, C.; Penco, S.; Sorarù, G.; Carlomagno, Y.; Ghezzi, S.; Pensato, V.; et al. Mutations of FUS gene in sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2010, 47, 190–194. [Google Scholar] [CrossRef]
- Ticozzi, N.; Silani, V.; LeClerc, A.L.; Keagle, P.; Gellera, C.; Ratti, A.; Taroni, F.; Kwiatkowski, T.J.; McKenna-Yasek, D.M.; Sapp, P.C.; et al. Analysis of FUS gene mutation in familial amyotrophic lateral sclerosis within an Italian cohort. Neurology 2009, 73, 1180–1185. [Google Scholar] [CrossRef] [Green Version]
- Marangi, G.; Traynor, B.J. Genetic causes of amyotrophic lateral sclerosis: New genetic analysis methodologies entailing new opportunities and challenges. Brain Res. 2015, 1607, 75–93. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Calvo, A.; Chio, A.; Colville, S.; Ellis, C.M.; Hardiman, O.; Heverin, M.; Howard, R.S.; Huisman, M.H.B.; Keren, N.; et al. Analysis of amyotrophic lateral sclerosis as a multistep process: A population-based modelling study. Lancet Neurol. 2014, 13, 1108–1113. [Google Scholar] [CrossRef]
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M.; et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642. [Google Scholar] [CrossRef] [Green Version]
- Campanari, M.-L.; Bourefis, A.-R.; Kabashi, E. Diagnostic challenge and neuro-muscular junction contribution to ALS pathogenesis. Front. Neurol. 2019, 10, 68. [Google Scholar] [CrossRef] [Green Version]
- Al-Chalabi, A.; Van Den Berg, L.H.; Veldink, J. Gene discovery in amyotrophic lateral sclerosis: Implications for clinical management. Nat. Rev. Neurol. 2017, 13, 96–104. [Google Scholar] [CrossRef] [Green Version]
- Antoniadi, T.; Buxton, C.; Dennis, G.; Forrester, N.; Smith, D.; Lunt, P.; Burton-Jones, S. Application of targeted multi-gene panel testing for the diagnosis of inherited peripheral neuropathy provides a high diagnostic yield with unexpected phenotype-genotype variability. BMC Med. Genet. 2015, 16, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Bo, R.; Tiloca, C.; Pensato, V.; Corrado, L.; Ratti, A.; Ticozzi, N.; Corti, S.; Castellotti, B.; Mazzini, L.; Sorarù, G.; et al. Novel optineurin mutations in patients with familial and sporadic amyotrophic lateral sclerosis. J. Neurol. Neurosurg. Psychiatry 2011, 82, 1239–1243. [Google Scholar] [CrossRef] [PubMed]
- Black, H.A.; Leighton, D.J.; Cleary, E.M.; Rose, E.; Stephenson, L.; Colville, S.; Ross, D.; Warner, J.; Porteous, M.; Gorrie, G.H.; et al. Genetic epidemiology of motor neuron disease-associated variants in the Scottish population. Neurobiol. Aging 2017, 51, e11–e178. [Google Scholar] [CrossRef] [PubMed]
- Belzil, V.V.; Daoud, H.; Desjarlais, A.; Bouchard, J.P.; Dupré, N.; Camu, W.; Dion, P.A.; Rouleau, G.A. Analysis of OPTN as a causative gene for amyotrophic lateral sclerosis. Neurobiol. Aging 2011, 32, 555.e13–555.e14. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Bieniek, K.F.; Finch, N.C.; van de Vorst, M.; Baker, M.; Perkersen, R.; Brown, P.; Ravenscroft, T.; van Blitterswijk, M.; Nicholson, A.M.; et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015, 130, 77–92. [Google Scholar] [CrossRef]
- Guyant-Maréchal, L.; Laquerrière, A.; Duyckaerts, C.; Dumanchin, C.; Bou, J.; Dugny, F.; Le Ber, I.; Frébourg, T.; Hannequin, D.; Campion, D. Valosin-containing protein gene mutations: Clinical and neuropathologic features. Neurology 2006, 67, 644–651. [Google Scholar] [CrossRef]
- Shi, Z.; Hayashi, Y.K.; Mitsuhashi, S.; Goto, K.; Kaneda, D.; Choi, Y.C.; Toyoda, C.; Hieda, S.; Kamiyama, T.; Sato, H.; et al. Characterization of the Asian myopathy patients with VCP mutations. Eur. J. Neurol. 2012, 19, 501–509. [Google Scholar] [CrossRef]
- Stojkovic, T.; Hammouda, E.H.; Richard, P.; de Munain, A.L.; Ruiz-Martinez, J.; Gonzalez, P.C.; Laforêt, P.; Pénisson-Besnier, I.; Ferrer, X.; Lacour, A.; et al. Clinical outcome in 19 French and Spanish patients with valosin-containing protein myopathy associated with Paget’s disease of bone and frontotemporal dementia. Neuromuscul. Disord. 2009, 19, 316–323. [Google Scholar] [CrossRef]
- Watts, G.D.J.; Wymer, J.; Kovach, M.J.; Mehta, S.G.; Mumm, S.; Darvish, D.; Pestronk, A.; Whyte, M.P.; Kimonis, V.E. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet. 2004, 36, 377–381. [Google Scholar] [CrossRef]
- Bertolin, C.; Querin, G.; Bozzoni, V.; Martinelli, I.; De Bortoli, M.; Rampazzo, A.; Gellera, C.; Pegoraro, E.; Sorarù, G. New FIG4 gene mutations causing aggressive ALS. Eur. J. Neurol. 2018, 25, e41–e42. [Google Scholar] [CrossRef]
- Chow, C.Y.; Zhang, Y.; Dowling, J.J.; Jin, N.; Adamska, M.; Shiga, K.; Szigeti, K.; Shy, M.E.; Li, J.; Zhang, X.; et al. Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature 2007, 448, 68–72. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, G.; Lenk, G.M.; Reddel, S.W.; Grant, A.E.; Towne, C.F.; Ferguson, C.J.; Simpson, E.; Scheuerle, A.; Yasick, M.; Hoffman, S.; et al. Distinctive genetic and clinical features of CMT4J: A severe neuropathy caused by mutations in the PI(3,5)P 2 phosphatase FIG4. Brain 2011, 134, 1959–1971. [Google Scholar] [CrossRef] [Green Version]
- Zou, Z.Y.; Zhou, Z.R.; Che, C.H.; Liu, C.Y.; He, R.L.; Huang, H.P. Genetic epidemiology of amyotrophic lateral sclerosis: A systematic review and meta-analysis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and Guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Kenna, K.P.; McLaughlin, R.L.; Byrne, S.; Elamin, M.; Heverin, M.; Kenny, E.M.; Cormican, P.; Morris, D.W.; Donaghy, C.G.; Bradley, D.G.; et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J. Med. Genet. 2013, 50, 776–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, K.; Brenner, D.; Weydt, P.; Meyer, T.; Grehl, T.; Petri, S.; Grosskreutz, J.; Schuster, J.; Volk, A.E.; Borck, G.; et al. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J. Neurol. Neurosurg. Psychiatry 2018, 89, 817–827. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.; Shoai, M.; Fratta, P.; Sidle, K.; Orrell, R.; Sweeney, M.G.; Shatunov, A.; Sproviero, W.; Jones, A.; Al-Chalabi, A.; et al. Investigation of next-generation sequencing technologies as a diagnostic tool for amyotrophic lateral sclerosis. Neurobiol. Aging 2015, 36, 1600.e5–1600.e8. [Google Scholar] [CrossRef]
- Nishiyama, A.; Niihori, T.; Warita, H.; Izumi, R.; Akiyama, T.; Kato, M.; Suzuki, N.; Aoki, Y.; Aoki, M. Comprehensive targeted next-generation sequencing in Japanese familial amyotrophic lateral sclerosis. Neurobiol. Aging 2017, 53, 194.e1–194.e8. [Google Scholar] [CrossRef]
- Maruyama, H.; Morino, H.; Ito, H.; Izumi, Y.; Kato, H.; Watanabe, Y.; Kinoshita, Y.; Kamada, M.; Nodera, H.; Suzuki, H.; et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010, 465, 223–226. [Google Scholar] [CrossRef]
- Johnson, J.O.; Mandrioli, J.; Benatar, M.; Abramzon, Y.; Van Deerlin, V.M.; Trojanowski, J.Q.; Gibbs, J.R.; Brunetti, M.; Gronka, S.; Wuu, J.; et al. Exome Sequencing Reveals VCP Mutations as a Cause of Familial ALS. Neuron 2010, 68, 857–864. [Google Scholar] [CrossRef] [Green Version]
- Al-Obeidi, E.; Al-Tahan, S.; Surampalli, A.; Goyal, N.; Wang, A.K.; Hermann, A.; Omizo, M.; Smith, C.; Mozaffar, T.; Kimonis, V. Genotype-phenotype study in patients with valosin-containing protein mutations associated with multisystem proteinopathy. Clin. Genet. 2018, 93, 119–125. [Google Scholar] [CrossRef]
- Laurin, N.; Brown, J.P.; Morissette, J.; Raymond, V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in paget disease of bone. Am. J. Hum. Genet. 2002, 70, 1582–1588. [Google Scholar] [CrossRef] [Green Version]
- Fecto, F.; Yan, J.; Vemula, S.P.; Liu, E.; Yang, Y.; Chen, W.; Zheng, J.G.; Shi, Y.; Siddique, N.; Arrat, H.; et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch. Neurol. 2011, 68, 1440–1446. [Google Scholar] [CrossRef]
- Rubino, E.; Rainero, I.; Chio, A.; Rogaeva, E.; Galimberti, D.; Fenoglio, P.; Grinberg, Y.; Isaia, G.; Calvo, A.; Gentile, S.; et al. SQSTM1 mutations in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurology 2012, 79, 1556–1562. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Nakamura, Y.; Saigoh, K.; Sakamoto, H.; Ueno, S.; Isono, C.; Miyamoto, K.; Akamatsu, M.; Mitsui, Y.; Kusunoki, S. Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology 2013, 80, 458–463. [Google Scholar] [CrossRef]
- Le Ber, I.; Camuzat, A.; Guerreiro, R.; Bouya-Ahmed, K.; Bras, J.; Nicolas, G.; Gabelle, A.; Didic, M.; De Septenville, A.; Millecamps, S.; et al. SQSTM1 Mutations in french patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol. 2013, 70, 1403–1410. [Google Scholar]
- Bucelli, R.C.; Arhzaouy, K.; Pestronk, A.; Pittman, S.K.; Rojas, L.; Sue, C.M.; Evilä, A.; Hackman, P.; Udd, B.; Harms, M.B.; et al. SQSTM1 splice site mutation in distal myopathy with rimmed vacuoles. Neurology 2015, 85, 665–674. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef] [Green Version]
- Hirano, M.; Quinzii, C.M.; Mitsumoto, H.; Hays, A.P.; Roberts, J.K.; Richard, P.; Rowland, L.P. Senataxin mutations and amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2011, 12, 223–227. [Google Scholar] [CrossRef]
- Chow, C.Y.; Landers, J.E.; Bergren, S.K.; Sapp, P.C.; Grant, A.E.; Jones, J.M.; Everett, L.; Lenk, G.M.; McKenna-Yasek, D.M.; Weisman, L.S.; et al. Deleterious Variants of FIG4, a Phosphoinositide Phosphatase, in Patients with ALS. Am. J. Hum. Genet. 2009, 84, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.-Q.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA Synthetase Mutations in Charcot-Marie-Tooth Disease Type 2D and Distal Spinal Muscular Atrophy Type V. Am. J. Hum. Genet. 2003, 72, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Corcia, P.; Brulard, C.; Beltran, S.; Marouillat, S.; Bakkouche, S.E.; Andres, C.R.; Blasco, H.; Vourc’h, P. Typical bulbar ALS can be linked to GARS mutation. Amyotroph. Lateral Scler. Front. Degener. 2019, 20, 275–277. [Google Scholar] [CrossRef]
- Sproviero, W.; Shatunov, A.; Stahl, D.; Shoai, M.; van Rheenen, W.; Jones, A.R.; Al-Sarraj, S.; Andersen, P.M.; Bonini, N.M.; Conforti, F.L.; et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol. Aging 2017, 51, 178.e1–178.e9. [Google Scholar] [CrossRef]


{kind=link}
| ALS | Gene | Protein | Disease[OMIM] | Inherit. |
|---|---|---|---|---|
| ALS1 | SOD1 | Superoxide Dismutase [Cu-Zn] | Amyotrophic lateral sclerosis 1 [MIM:105400] | AD |
| Amyotrophic lateral sclerosis 1 [MIM:105400] D90A; D96N | AR | |||
| ALS2 | ALS2 | Alsin | Amyotrophic lateral sclerosis 2 [MIM:205100] | AR |
| Infantile-onset ascending spastic paralysis (IAHSP) [MIM:607225] | AR | |||
| Juvenile primary lateral sclerosis (JPLS) [MIM:606353] | AR | |||
| ALS4 | SETX | Probable helicase senataxin | Amyotrophic lateral sclerosis 4, juvenile ALS [MIM:602433] | AD |
| Spinocerebellar ataxia, 1 (SCAR1) [MIM:606002] | AR | |||
| ALS5 | SPG11 | Spatacsin | Amyotrophic lateral sclerosis 5, juvenile [MIM:602099] | AR |
| Charcot–Marie–Tooth disease, axonal type 2X(CMT2X) [MIM:616668] | AR | |||
| Spastic paraplegia 11, (SPG11) [MIM:604360] | AR | |||
| ALS6 | FUS | RNA-binding protein FUS | Amyotrophic lateral sclerosis 6, +/- FTD [MIM:608030] | AD |
| Tremor, hereditary essential 4 (ETM4) [MIM:614782] | AD | |||
| ALS8 | VAPB | Vesicle-associated membrane protein-B/C | Amyotrophic lateral sclerosis 8 [MIM:608627] | AD |
| Spinal muscular atrophy, late-onset, (SMAFK) [MIM:182980] | AD | |||
| ALS9 | ANG | Angiogenin | Amyotrophic lateral sclerosis 9; [MIM:611895] | AD |
| ALS10 | TARDBP | TAR DNA-binding protein 43 | Amyotrophic lateral sclerosis 10, +/- FTD [MIM:612069] | AD |
| ALS11 | FIG4 | Polyphospho inositide phosphatase | Amyotrophic lateral sclerosis 11 [MIM:612577] | AD |
| Charcot–Marie–Tooth disease 4J (CMT4J) [MIM:611228] | AR | |||
| Polymicrogyria, bilateral temporooccipital (BTOP) [MIM:612691] | AR | |||
| ALS12 | OPTN | Optineurin | Amyotrophic lateral sclerosis 12 [MIM:613435] | AR/AD |
| Glaucoma 1, open angle, E (GLC1E) [MIM:137760] | AD | |||
| ALS14 | VCP | Transitional endoplasmic reticulum ATPase | Amyotrophic lateral sclerosis 14, +/- FTD [MIM:613954] | AD |
| Charcot–Marie–Tooth disease, type 2Y (CMT2Y) [MIM:616687] | AD | |||
| Inclusion body myopathy with Paget disease +/- FTD [MIM:167320] | AD | |||
| ALS15 | UBQLN2 | Ubiquilin-2 | Amyotrophic lateral sclerosis 15, +/- FTD [MIM:300857] | XLD |
| ALS16 | SIGMAR1 | Sigma non-opioid intracellular receptor 1 | Amyotrophic lateral sclerosis 16, juvenile [MIM:614373] | AR |
| Distal spinal muscular atrophy, 2 (DSMA2) [MIM:605726] | AR | |||
| ALS17 | CHMP2B | Charged multivesicular body protein 2b | Amyotrophic lateral sclerosis 17 [MIM:614696] | AD |
| FTD chromosome 3-linked (FTD3) [MIM:600795] | AD | |||
| ALS18 | PFN1 | Profilin-1 | Amyotrophic lateral sclerosis 18 [MIM:614808] | AD |
| ALS19 | ERBB4 | Receptor tyrosine-protein kinase erbB-4 | Amyotrophic lateral sclerosis 19 [MIM:615515] | AD |
| ALS20 | HNRNPA1 | Heterogeneous nuclear ribonucleoprotein A1 | Amyotrophic lateral sclerosis 20 [MIM:615426] | AD |
| Inclusion body myopathy with Paget disease +/- FTD3 [MIM:615424] | AD | |||
| ALS21 | MATR3 | Matrin-3 | Amyotrophic lateral sclerosis 21 [MIM:606070] | AD |
| ALS22 | TUBA4A | Tubulin alpha-4A chain | Amyotrophic lateral sclerosis 22, +/- FTD [MIM:616208] | AD |
| FTDALS2 | CHCHD10 | Coiled-coil-helix-coiled-coil-helix domain-containing protein 10, mitochondrial | FTD +/- amyotrophic lateral sclerosis 2 [MIM:615911] | AD |
| Isolated Mitochondrial Myopathy (IMMD) [MIM:616209] | AD | |||
| Spinal muscular atrophy, Jokela type (SMAJ) [MIM:615048] | AD | |||
| FTDALS3 | SQSTM1 | Sequestosome-1 | FTD +/- amyotrophic lateral sclerosis 3 [MIM:616437] | AD |
| Ataxia, dystonia, gaze palsy[MIM:617145] | AR | |||
| Myopathy distal, with rimmed vacuoles [MIM:617158] | AD | |||
| Paget disease of bone 3 (PDB3) [MIM:167250] | AD | |||
| ALS suscep | ATXN2 | Ataxin-2 | {Amyotrophic lateral sclerosis, susceptibility to, 13} [MIM:183090] | AD |
| Spinocerebellar ataxia 2 (SCA2) [MIM:183090] | AD | |||
| ALS suscep | APEX1 | apurinic/apyrimidinic endodeoxyribonuclease1 | ||
| ALS suscep | CRYM | Ketimine reductase mu-crystallin | Deafness, autosomal dominant 40 (DFNA40) [MIM:616357] | AD |
| ALS suscep | CYP27A1 | Sterol 26-hydroxylase, | Cerebrotendinous xanthomatosis (CTX) [MIM:213700] | AR |
| ALS suscep | DAO | D-amino-acid oxidase | ||
| ALS suscep | DCTN1 | Dynactin subunit 1 | {Amyotrophic lateral sclerosis, susceptibility to} [MIM:105400] | |
| Neuronopathy, distal hereditary motor, 7B (HMN7B) [MIM:607641] | AD | |||
| Perry syndrome (PERRYS) [MIM:168605] | AD | |||
| ALS suscep | DPP6 | Dipeptidyl-aminope-peptidase-like protein 6 | Mental retardation, autosomal dominant 33 (MRD33) [MIM:616311] | AD |
| ALS suscep | ELP3 | Elongator Acetyltransferase Complex Subunit 3 | ||
| ALS suscep | EPHA4 | Ephrin type-A receptor 4 | ||
| ALS suscep | HNRNPA2B1 | Heterogeneous nuclear ribonucleoproteinsA2/B1 | Inclusion body myopathy and Paget disease +/- FTD [MIM:615422] | |
| ALS suscep | HNRNPA3 | Heterogeneous nuclear ribonucleo-protein A3 | ||
| ALS suscep | NEFH | Neurofilament heavy polypeptide | {Amyotrophic lateral sclerosis, susceptibility to} [MIM:105400] | |
| Charcot–Marie–Tooth, axonal, type 2CC (CMT2CC) [MIM:616924] | AD | |||
| ALS suscep | NEK1 | Serine/threonine-protein kinase Nek1 | Short-rib thoracic dysplasia 6 +/- polydactyly(SRTD6) [MIM:263520] | AR |
| ALS suscep | TAF15 | TATA-binding protein-associated factor 2N | Chondrosarcoma, extraskeletal myxoid [MIM:612237] | |
| ALS suscep | TREM2 | Triggering receptor expressed myeloid cells2 | Polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL) [MIM:221770] | AR |
| FTD | GRN | Granulins | FTD with ubiquitin-positive inclusions ([MIM:607485] | AD |
| Aphasia, primary progressive [MIM:607485] | AD | |||
| Ceroid lipofuscinosis, neuronal, 11 (CLN11) [MIM:614706] | AR | |||
| FTD | MAPT | Microtubule-associated protein tau | Frontotemporal dementia (FTD) [MIM:600274] | AD |
| Pick disease of the brain (PIDB) [MIM:172700] | AD | |||
| Progressive supranuclear palsy 1 (PSNP1) [MIM:601104] | AD | |||
| Parkinson-dementia syndrome (PARDE) [MIM:260540] | AR | |||
| SMA | ASAH1 | Acid ceramidase | Farber lipogranulomatosis (FRBRL) [MIM:228000] | AR |
| Spinal muscular atrophy, progressive myoclonic epilepsy[MIM:159950] | AR | |||
| HMN/HSP | BSCL2 | Seipin | Lipodystrophy congenital generalized type 2 (CGL2) [MIM:269700] | AR |
| Encephalopathy progressive, +/- lipodystrophy (PELD) [MIM:615924] | AR | |||
| Neuronopathy, distal hereditary motor, 5A (HMN5A) [MIM:600794] | AD | |||
| Spastic paraplegia 17, (SPG17) [MIM:270685] | AD | |||
| HMN | GARS1 | Glycyl-tRNA synthetase 1 | Charcot-Marie-Tooth disease 2D (CMT2D) [MIM:601472] | AD |
| Neuronopathy, distal hereditary motor, 5A (HMN5A) [MIM:600794] | AD | |||
| HMN | IGHMBP2 | Immunoglobulin mu-binding protein 2 | Charcot–Marie–Tooth disease 2S (CMT2S) [MIM:616155] | AR |
| Neuronopathy, distal hereditary motor, 6 (HMN6) [MIM:604320] | AR | |||
| HMN/HSP | PNPLA6 | Neuropathy target esterase | Boucher–Neuhauser syndrome (BNHS) [MIM:215470] | AR |
| Laurence-Moon [MIM:245800];Oliver-McFarlane [MIM:275400] | AR | |||
| Spastic paraplegia 39 (SPG39) [MIM:612020] | AR | |||
| Neuronopathy, distal hereditary motor, 5B (HMN5B) [MIM:614751] | AR | |||
| HSP | REEP1 | Receptor expression-enhancing protein1 | Spastic paraplegia 31 (SPG31) [MIM:610250] | AD |
| HSP | SPAST | Spastin | Spastic paraplegia 4 (SPG4) [MIM:182601] | AD |
| HMN | TRPV4 | Transient receptor potential cation channel subfamily V4 | Digital arthropathy-brachydactyly, familial (FDAB) [MIM:606835] | AD |
| Neuronopathy, distal hereditary motor, 8 (HMN8) [MIM:600175] | AD | |||
| Scapuloperoneal spinal muscular atrophy (SPSMA) [MIM:181405] | AD | |||
| Gene | N. of Variants | Class 5 | Class 4 | Class 3 | Class 2 | Recurrent Variants | |
|---|---|---|---|---|---|---|---|
| ALS (AD) | ANG | 4 | 1 | 3 * | * 1 recurrent in 3 pts | ||
| CHCHD10 | 0 | ||||||
| CHMP2B | 1 | 1 | |||||
| ERBB4 | 4 | 2 | 2 * | * 1 recurrent in 2 pts | |||
| FIG4 | 4 | 1 | 1 | 2 | |||
| HNRNPA1 | 0 | ||||||
| MATR3 | 1 | 1 | |||||
| OPTN | 3 | 2 | 1 | ||||
| PFN1 | 0 | ||||||
| SETX | 21 | 1 | 6 | 14 * | * 1 recurrent in 7 pts * 1 recurrent in 5 pts | ||
| SQSTM1 | 6 | 2 | 1 | 3 * | * 1 recurrent in 3 pts | ||
| TUBA4A | 0 | ||||||
| VAPB | 3 | 3 * | * 1 recurrent in 3 pts | ||||
| VCP | 4 | 2 | 2 | ||||
| ALS (XLD) | UBQLN2 | 1 | 1 | ||||
| ALS (AR) | ALS2 | 4 | 1 | 3 | |||
| SIGMAR1 | 1 | 1 | |||||
| SPG11 | 7 | 3 | 4 | ||||
| HMN/HSP(AD) | BSCL2 | 0 | |||||
| GARS1 | 9 | 1 | 5 § | 3 * | § 1 recurrent in 3 pts * 1 recurrent in 3 pts | ||
| REEP1 | 0 | ||||||
| TRPV4 | 9 | 3 | 6 * | * 1 recurrent in 3 pts | |||
| SPAST | 1 | 1 | |||||
| SMA/HMN/HSP(AR) | ASAH1 | 3 | 1 | 2 | |||
| IGHMBP2 | 10 | 4 | 1 | 4 | 1 | ||
| PNPLA6 | 6 | 1 | 5 | ||||
| FTD (AD) | GRN | 8 | 4 | 4* | * 1 recurrent in 4 pts | ||
| MAPT | 2 | 2 | |||||
| Total | 112 | 11 | 8 | 49 | 44 | 10 recurrent variants (9 Class-2, 1 Class-3) |
| Gene | Number of Variants | ||
|---|---|---|---|
| ALS susceptibility genes | APEX1 | ||
| ATXN2 | 14 * | * 1 recurrent in 2 pts * 1 recurrent in 7 pts | |
| CRYM | 2 * | * 1 recurrent in 2 pts | |
| CYP27A1 | 4 | ||
| DAO | 1 | ||
| DCTN1 | 10 * | * 1 recurrent in 3 pts | |
| DPP6 | 1 | ||
| ELP3 | 2 | ||
| EPHA4 | 2 | ||
| HNRNPA2B1 | |||
| HNRNPA3 | 5 * | * 1 recurrent in 3 pts | |
| NEFH | 4 * | * 1 recurrent in 4 pts | |
| NEK1 | 1 | ||
| TAF15 | |||
| TREM2 | 2 | ||
| Total | 48 | 6 recurrent variants |
| Gene | Class | Variant DNA | Variant Protein | MAF % * | dbSNP | Additional Variants | Clinical Notes | Patient | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| OPTN | 5 | c.941A>T | p.Gln314Leu | 0%-0.02%-0.01% | rs142812715 | (AR) ALS2: c.331G>A (p.Val111Ile) het [rs61745503, MAF *: 0.04%-0.02%-0.03%] (Class3) | Bulbar-Spinal ALS laboratory supported | P087 | [22,23] |
| OPTN | 5 | c.1442C>T | p.Ala481Val | 0%-0.01%-0% | rs377219791 | Spinal ALS-laboratory supported | P015 | [24,25] | |
| VCP | 5 | c.277C>T | p.Arg93Cys | 0%-0%-0% | Spinal ALS, no FTD, no Paget | P072 | [26,27] | ||
| VCP | 5 | c.463C>T | p.Arg155Cys | 0%-0%-0% | rs121909330 | (AR) IGHMBP2: c.2911_2912delAG (p.Arg971GlufsTer4) het [rs572973851; rs724159994, MAF *: 0%-0.82%-0.02%] (Class5) | Spinal ALS, no FTD, no Paget | P008 | [28,29] |
| SQSTM1 | 4 | c.695C>T | p.Pro232Leu | 0%-0%-0% | rs757778292 | (Suscep.) DAO: c.627G>A (p.Trp209Ter) het [rs766258671, MAF*: 0%-0%-0% ] (Suscep.) DCTN1: c.652G>A(p.Glu218Lys) het [MAF *: 0%-0%-0%] | Bulbar ALS, onset 43 years | P002 | |
| SQSTM1 | 4 | c.301 + 4delA | p.? HSF: Alteration of the WT donor site, most probably affecting splicing. | 0%-0%-0% | (AR) IGHMBP2: c.2176G>A (p.Val726Met) het [rs143986510, MAF *: 0.02%-0.08%-0.04%] (Class3) | ALS | P103 | ||
| FIG4 | 5 4 | c.122T>C c.1667C>T | p.Ile41Thr p.Thr556Ile | 0.08%-0.11%-0.1% 0%-0%-0% | rs121908287 | Juvenile ALS with predominant upper motor neuron involvement | P100 | [30] | |
| SETX | 4 | c.2750T>C | p.Met917Thr | 0%-0.01%-0.01% | rs376022544 | ALS | P022 | [5] | |
| GARS1 | 4 | c.1955G>A | p.Gly652Glu | 0%-0%-0% | rs747080824 | ALS Pseudo-polyneuritic type | P073 |
| Gene | Class | Variant DNA | Variant Protein | MAF % * | dbSNP | Additional Variants | Clinical Notes | Patient |
|---|---|---|---|---|---|---|---|---|
| OPTN | 3 | c.910C>T | p.Leu304Phe | 0%-0%-0% | (Suscep.) ATXN2: c.3000A > G (p =) het [rs140262591, MAF *: 0.2%-0.31%-0.3%] | Spinal ALS | P075 | |
| VCP | 3 | c.179A>G | p.Lys60Arg | 0%-0%-0% | (Suscep.) ATXN2: c.3322C > T (p.Pro1108Ser) het [rs140242317, MAF *: 0.04%-0.11%-0.07% ] | Slowly progressive ALS with cognitive impairment | P112 | |
| VCP | 3 | c.1696-3C>T | p.? | 0%-0.01%-0.01% | rs372638909 | Rapidly progressive ALS with cognitive impairment | P025 | |
| FIG4 | 3 | c.646G>A | p.Gly216Arg | 0%-0%-0% | rs759566206 | (Class 2) SQSTM1: c.712A > G (p.Lys238Glu) het [rs11548633, MAF *: 0.24%-0.26%-0.24%] | Progressive muscular weakness and sensory neuropathy | P105 |
| FIG4 | 3 | c.1243C>G | p.Pro415Ala | 0%-0%-0% | (Class 3) GARS1: c.302G > A (p.Arg101His) het [rs200887429, MAF *: 0.02%-0.02%-0.04%]; (Class 2) SETX: c.4660T > G (p.Cys1554Gly) het [rs112089123, MAF *: 0.58%-0.31%-0.58%] | Spastic quadriplegia | P106 | |
| SQSTM1 | 3 | c.833C>T | p.Thr278Ile | 0%-0%-0% | rs200445838 | Myopathy | P043 | |
| UBQLN2 | 3 | c.809G>A hem | p.Arg270His | 0%-0%-0% | rs767597171 | Spinal ALS (Flail arm) | P017 | |
| SETX | 3 | c.-114-2A>G | p.? | 0.08%-0%-0% | rs560095915 | Bulbar ALS, onset 43 years | P079 | |
| SETX | 3 | c.934A>G | p.Ile312Val | 0%-0%-0% | Spinal ALS, onset 67 years | P045 | ||
| SETX | 3 | c.2344G>T | p.Val782Leu | 0%-0%-0% | (AR) ALS2: c.37G > A (p.Gly13Arg) het [rs367871772, MAF *: 0%-0.01%-0.01%] (Class 4); | Lower motor neuron involvement, onset 21 years | P071 | |
| SETX | 3 | c.3494C>G | p.Ser1165Cys | 0%-0%-0% | Spinal ALS, onset 72 years | P029 | ||
| SETX | 3 | c.4220A>G | p.Asn1407Ser | 0%-0%-0% | rs747050949 | Bulbar ALS, onset 79 years | P069 | |
| SETX | 3 | c.4957A>G | p.Lys1653Glu | 0%-0%-0% | Spinal ALS, onset 66 years | P088 | ||
| ANG | 3 | c.61C>T | p.Pro21Ser | 0%-0.02%-0.02% | rs149672657 | (HMN) TRPV4: c.1006C > T (p.Arg336Cys) het [rs781229110, MAF *: 0%-0%-0%] (Class 3); (Class 2) SQSTM1: c.712A > G (p.Lys238Glu) het [rs11548633, MAF *: 0.24%-0.26%-0.24%] (Class 2) SETX: c.3229G > A (p.Asp1077Asn) het [rs145097270, MAF *: 0.08%-0.07%-0.11%] | Spinal ALS | P094 |
| CHMP2B | 3 | c.36T>G | p.Asp12Glu | 0%-0%-0% | (Class 2) GRN: c.264+7G > A (p = ?)het [rs60100877, MAF *: 0.5%-0.6%-0.53%] | Spinal ALS, onset 71 years | P074 | |
| ERBB4 | 3 | c.421 + 5G>A | p.? HSF: Alteration of the WT donor site, most probably affecting splicing | 0%-0%-0% | rs778195807 | - | P024 | |
| NRBB4 | 3 | c.1441A>G | p.Ile481Val | 0%-0.01%-0% | rs368860175 | Bulbar ALS, onset 70. One affected sibling | P092 | |
| MATR3 | 3 | c.1132G>A | p.Ala378Thr | 0%-0.03%-0.01% | rs201075828 | Spinal ALS, onset 37 years | P099 | |
| GARS1 | 3 | c.302G>A | p.Arg101His | 0.02%-0.02%-0.04% | rs200887429 | - | P005 | |
| GARS1 | 3 | c.302G>A | p.Arg101His | 0.02%-0.02%-0.04% | rs200887429 | Spinal ALS | P030 | |
| GARS1 | 3 | c.571A>T | p.Thr191Ser | 0%-0%-0% | rs760133861 | (Class 2) GARS1: c.803C > T (p.Thr268Ile) het [rs2230310, MAF *: 0.12%-0.48% 0.32% | Spinal ALS | P062 |
| GARS1 | 3 | c.1159G>A | p.Ala387Thr | 0%-0%-0% | rs776528885 | Bulbar ALS and FTD | P065 | |
| GRN | 3 | c.-8 + 7G>C | p? | 0%-0%-0% | Bulbar ALS | P054 | ||
| GRN | 3 | c.329G>A | p.Arg110Gln | 0.02%-0.01%-0.01% | rs375439809 | Neuroacanthocytosis | P110 | |
| GRN | 3 | c.1528C>T | p.Arg510Cys | 0%-0%-0% | rs747873577 | Spinal ALS | P034 | |
| GRN | 3 | c.1691G>A | p.Arg564His | 0%-0%-0% | Spinal ALS | P035 | ||
| MAPT | 3 | c.64G>A | p.Asp22Asn | 0%-0%-0% | rs745662662 | (AR) IGHMBP2: c.2362C > T (p.Arg788Ter) het [rs199839840, MAF *: 0%-0.01%-0% (Class5) | Spinal ALS | P059 |
| MAPT | 3 | c.319G>A | p.Gly107Ser | 0%-0%-0% | rs769901930 | Spinal ALS and mild cognitive impairment | P057 | |
| TRPV4 | 3 | c.113A>G | p.Asn38Ser | 0.02%-0%-0% | rs527355587 | (Class 2) TRPV4: c.2518G > A (p.Glu840Lys) het [rs55728855, MAF *: 0.24%-0.74%-0.63% | ALS monomelic type | P033 |
| TRPV4 | 3 | c.1496C>T | p.Pro499Leu | 0.02%-0%-0% | rs115358347 | - | P093 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pensato, V.; Magri, S.; Dalla Bella, E.; Tannorella, P.; Bersano, E.; Sorarù, G.; Gatti, M.; Ticozzi, N.; Taroni, F.; Lauria, G.; et al. Sorting Rare ALS Genetic Variants by Targeted Re-Sequencing Panel in Italian Patients: OPTN, VCP, and SQSTM1 Variants Account for 3% of Rare Genetic Forms. J. Clin. Med. 2020, 9, 412. https://doi.org/10.3390/jcm9020412
Pensato V, Magri S, Dalla Bella E, Tannorella P, Bersano E, Sorarù G, Gatti M, Ticozzi N, Taroni F, Lauria G, et al. Sorting Rare ALS Genetic Variants by Targeted Re-Sequencing Panel in Italian Patients: OPTN, VCP, and SQSTM1 Variants Account for 3% of Rare Genetic Forms. Journal of Clinical Medicine. 2020; 9(2):412. https://doi.org/10.3390/jcm9020412
Chicago/Turabian StylePensato, Viviana, Stefania Magri, Eleonora Dalla Bella, Pierpaola Tannorella, Enrica Bersano, Gianni Sorarù, Marta Gatti, Nicola Ticozzi, Franco Taroni, Giuseppe Lauria, and et al. 2020. "Sorting Rare ALS Genetic Variants by Targeted Re-Sequencing Panel in Italian Patients: OPTN, VCP, and SQSTM1 Variants Account for 3% of Rare Genetic Forms" Journal of Clinical Medicine 9, no. 2: 412. https://doi.org/10.3390/jcm9020412
APA StylePensato, V., Magri, S., Dalla Bella, E., Tannorella, P., Bersano, E., Sorarù, G., Gatti, M., Ticozzi, N., Taroni, F., Lauria, G., Mariotti, C., & Gellera, C. (2020). Sorting Rare ALS Genetic Variants by Targeted Re-Sequencing Panel in Italian Patients: OPTN, VCP, and SQSTM1 Variants Account for 3% of Rare Genetic Forms. Journal of Clinical Medicine, 9(2), 412. https://doi.org/10.3390/jcm9020412