The Inflammatory Milieu of Adamantinomatous Craniopharyngioma and Its Implications for Treatment


Abstract
:1. Introduction
1.1. The Central Role of WNT Pathway Overactivation in the Tumorigenesis of ACP
1.2. The role of the Inflammatory Response in Generating the Cystic Compartment in ACP
1.3. The Solid Component of ACP Also Demonstrates Elevated Levels of Several Inflammatory Markers
1.4. Immune Checkpoint Inhibitors and Their Potential Use in ACP
1.5. CTLA-4 Inhibition and Its Potential Use in the Treatment of ACP
1.6. The Role of Senescence and the Senescence Associated Secretory Phenotype (SASP) in the Pathogenesis of ACP
2. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Martinez-Barbera, J.P.; Buslei, R. Adamantinomatous craniopharyngioma: Pathology, molecular genetics and mouse models. J. Pediatr. Endocrinol. Metab. 2015, 28, 7–17. [Google Scholar] [CrossRef]
- Kasai, H.; Hirano, A.; Llena, J.F.; Kawamoto, K. A histopathological study of craniopharyngioma with special reference to its stroma and surrounding tissue. Brain Tumor Pathol. 1997, 14, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Daubenbüchel, A.; Müller, H. Neuroendocrine Disorders in Pediatric Craniopharyngioma Patients. J. Clin. Med. 2015, 4, 389–413. [Google Scholar] [CrossRef] [PubMed]
- Müller, H.L.; Gebhardt, U.; Teske, C.; Faldum, A.; Zwiener, I.; Warmuth-Metz, M.; Pietsch, T.; Pohl, F.; Sörensen, N.; Calaminus, G. Post-operative hypothalamic lesions and obesity in childhood craniopharyngioma: Results of the multinational prospective trial KRANIOPHARYNGEOM 2000 after 3-year follow-up. Eur. J. Endocrinol. 2011, 165, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Heinks, K.; Boekhoff, S.; Hoffmann, A.; Warmuth-Metz, M.; Eveslage, M.; Peng, J.; Calaminus, G.; Müller, H.L. Quality of life and growth after childhood craniopharyngioma: Results of the multinational trial KRANIOPHARYNGEOM 2007. Endocrine 2018, 59, 364–372. [Google Scholar] [CrossRef]
- Müller, H.L.; Merchant, T.E.; Puget, S.; Martinez-Barbera, J.P. New outlook on the diagnosis, treatment and follow-up of childhood-onset craniopharyngioma. Nat. Rev. Endocrinol. 2017, 13, 299–312. [Google Scholar] [CrossRef]
- Brastianos, P.K.; Shankar, G.M.; Gill, C.M.; Taylor-Weiner, A.; Nayyar, N.; Panka, D.J.; Sullivan, R.J.; Frederick, D.T.; Abedalthagafi, M.; Jones, P.S.; et al. Dramatic Response of BRAF V600E Mutant Papillary Craniopharyngioma to Targeted Therapy. J. Natl. Cancer Inst. 2015, 108. [Google Scholar] [CrossRef]
- Aylwin, S.J.B.; Bodi, I.; Beaney, R. Pronounced response of papillary craniopharyngioma to treatment with vemurafenib, a BRAF inhibitor. Pituitary 2016, 19, 544–546. [Google Scholar] [CrossRef] [Green Version]
- Brastianos, P.K.; Taylor-Weiner, A.; Manley, P.E.; Jones, R.T.; Dias-Santagata, D.; Thorner, A.R.; Lawrence, M.S.; Rodriguez, F.J.; Bernardo, L.A.; Schubert, L.; et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat. Genet. 2014, 46, 161–165. [Google Scholar] [CrossRef]
- Apps, J.R.; Carreno, G.; Gonzalez-Meljem, J.M.; Haston, S.; Guiho, R.; Cooper, J.E.; Manshaei, S.; Jani, N.; Hölsken, A.; Pettorini, B.; et al. Tumour compartment transcriptomics demonstrates the activation of inflammatory and odontogenic programmes in human adamantinomatous craniopharyngioma and identifies the MAPK/ERK pathway as a novel therapeutic target. Acta Neuropathol. 2018, 135, 757–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gump, J.M.; Donson, A.M.; Birks, D.K.; Amani, V.M.; Rao, K.K.; Griesinger, A.M.; Kleinschmidt-DeMasters, B.K.; Johnston, J.M.; Anderson, R.C.E.; Rosenfeld, A.; et al. Identification of targets for rational pharmacological therapy in childhood craniopharyngioma. Acta Neuropathol. Commun. 2015, 3, 30. [Google Scholar] [CrossRef] [Green Version]
- Hölsken, A.; Sill, M.; Merkle, J.; Schweizer, L.; Buchfelder, M.; Flitsch, J.; Fahlbusch, R.; Metzler, M.; Kool, M.; Pfister, S.M.; et al. Adamantinomatous and papillary craniopharyngiomas are characterized by distinct epigenomic as well as mutational and transcriptomic profiles. Acta Neuropathol. Commun. 2016, 4, 20. [Google Scholar] [CrossRef] [Green Version]
- Goschzik, T.; Gessi, M.; Dreschmann, V.; Gebhardt, U.; Wang, L.; Yamaguchi, S.; Wheeler, D.A.; Lauriola, L.; Lau, C.C.; Müller, H.L.; et al. Genomic Alterations of Adamantinomatous and Papillary Craniopharyngioma. J. Neuropathol. Exp. Neurol. 2017, 76, 126–134. [Google Scholar] [CrossRef]
- Andoniadou, C.L.; Gaston-Massuet, C.; Reddy, R.; Schneider, R.P.; Blasco, M.A.; Le Tissier, P.; Jacques, T.S.; Pevny, L.H.; Dattani, M.T.; Martinez-Barbera, J.P. Identification of novel pathways involved in the pathogenesis of human adamantinomatous craniopharyngioma. Acta Neuropathol. 2012, 124, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Massimi, L.; Martelli, C.; Caldarelli, M.; Castagnola, M.; Desiderio, C. Proteomics in pediatric cystic craniopharyngioma. Brain Pathol. 2017, 27, 370–376. [Google Scholar] [CrossRef]
- Robinson, L.C.; Santagata, S.; Hankinson, T.C. Potential evolution of neurosurgical treatment paradigms for craniopharyngioma based on genomic and transcriptomic characteristics. Neurosurg. Focus 2016, 41, E3. [Google Scholar] [CrossRef] [Green Version]
- Buslei, R.; Nolde, M.; Hofmann, B.; Meissner, S.; Eyupoglu, I.Y.; Siebzehnrübl, F.; Hahnen, E.; Kreutzer, J.; Fahlbusch, R. Common mutations of β-catenin in adamantinomatous craniopharyngiomas but not in other tumours originating from the sellar region. Acta Neuropathol. 2005, 109, 589–597. [Google Scholar] [CrossRef]
- Martinez-Barbera, J.P. Molecular and cellular pathogenesis of adamantinomatous craniopharyngioma. Neuropathol. Appl. Neurobiol. 2015, 41, 721–732. [Google Scholar] [CrossRef] [Green Version]
- Apps, J.R.; Martinez-Barbera, J.P. Genetically engineered mouse models of craniopharyngioma: An opportunity for therapy development and understanding of tumor biology. Brain Pathol. 2017, 27, 364–369. [Google Scholar] [CrossRef] [Green Version]
- Hölsken, A.; Buchfelder, M.; Fahlbusch, R.; Blümcke, I.; Buslei, R. Tumour cell migration in adamantinomatous craniopharyngiomas is promoted by activated Wnt-signalling. Acta Neuropathol. 2010, 119, 631–639. [Google Scholar] [CrossRef]
- Martinez-Barbera, J.P.; Andoniadou, C.L. Concise Review: Paracrine Role of Stem Cells in Pituitary Tumors: A Focus on Adamantinomatous Craniopharyngioma. Stem Cells 2016, 34, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stache, C.; Hölsken, A.; Schlaffer, S.M.; Hess, A.; Metzler, M.; Frey, B.; Fahlbusch, R.; Flitsch, J.; Buchfelder, M.; Buslei, R. Insights into the infiltrative behavior of adamantinomatous craniopharyngioma in a new xenotransplant mouse model. Brain Pathol. 2015, 25, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Apps, J.R.; Hutchinson, J.C.; Arthurs, O.J.; Virasami, A.; Joshi, A.; Zeller-Plumhoff, B.; Moulding, D.; Jacques, T.S.; Sebire, N.J.; Martinez-Barbera, J.P. Imaging Invasion: Micro-CT imaging of adamantinomatous craniopharyngioma highlights cell type specific spatial relationships of tissue invasion. Acta Neuropathol. Commun. 2016, 4, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darnel, A.D.; Behmoaram, E.; Vollmer, R.T.; Corcos, J.; Bijian, K.; Sircar, K.; Su, J.; Jiao, J.; Alaoui-Jamali, M.A.; Bismar, T.A. Fascin regulates prostate cancer cell invasion and is associated with metastasis and biochemical failure in prostate cancer. Clin. Cancer Res. 2009, 15, 1376–1383. [Google Scholar] [CrossRef] [Green Version]
- Kureishy, N.; Sapountzi, V.; Prag, S.; Anilkumar, N.; Adams, J.C. Fascins, and their roles in cell structure and function. BioEssays 2002, 24, 350–361. [Google Scholar] [CrossRef]
- Chen, S.F.; Lin, C.Y.; Chang, Y.C.; Li, J.W.; Fu, E.; Chang, F.N.; Lin, Y.L.; Nieh, S. Effects of small interfering RNAs targeting Fascin on gene expression in oral cancer cells. J. Oral Pathol. Med. 2009, 38, 722–730. [Google Scholar] [CrossRef]
- Krishnamurthy, N.; Kurzrock, R. Targeting the Wnt/beta-catenin pathway in cancer: Update on effectors and inhibitors. Cancer Treat. Rev. 2018, 62, 50–60. [Google Scholar] [CrossRef]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef]
- Staal, F.J.T.; Sen, J.M. The canonical Wnt signaling pathway plays an important role in lymphopoiesis and hematopoiesis. Eur. J. Immunol. 2008, 38, 1788–1794. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Y.; Katavolos, P.; Nguyen, T.; Lau, T.; Boggs, J.; Sambrone, A.; Kan, D.; Merchant, M.; Harstad, E.; Diaz, D.; et al. Tankyrase Inhibition Causes Reversible Intestinal Toxicity in Mice with a Therapeutic Index < 1. Toxicol. Pathol. 2016, 44, 267–278. [Google Scholar]
- Donson, A.M.; Apps, J.; Griesinger, A.M.; Amani, V.; Witt, D.A.; Anderson, R.C.E.; Niazi, T.N.; Grant, G.; Souweidane, M.; Johnston, J.M.; et al. Molecular analyses reveal inflammatory mediators in the solid component and cyst fluid of human adamantinomatous craniopharyngioma. J. Neuropathol. Exp. Neurol. 2017, 76, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Martelli, C.; Serra, R.; Inserra, I.; Rossetti, D.V.; Iavarone, F.; Vincenzoni, F.; Castagnola, M.; Urbani, A.; Tamburrini, G.; Caldarelli, M.; et al. Investigating the Protein Signature of Adamantinomatous Craniopharyngioma Pediatric Brain Tumor Tissue: Towards the Comprehension of Its Aggressive Behavior. Dis. Markers 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Coy, S.; Rashid, R.; Lin, J.R.; Du, Z.; Donson, A.M.; Hankinson, T.C.; Foreman, N.K.; Manley, P.E.; Kieran, M.W.; Reardon, D.A.; et al. Multiplexed immunofluorescence reveals potential PD-1/PD-L1 pathway vulnerabilities in craniopharyngioma. Neuro. Oncol. 2018, 20, 1101–1112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martha Lilia, T.S.; Citlaltepelt, S.L.; Ma Elena, H.C.; Carlos, S.G.; Manuel, C.L. Running Head: Craniopharyngioma and Immune Response. J. Neurol. Neurosci. 2015, 06, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kilday, J.P.; Caldarelli, M.; Massimi, L.; Chen, R.H.H.; Lee, Y.Y.; Liang, M.L.; Parkes, J.; Naiker, T.; Van Veelen, M.L.; Michiels, E.; et al. Intracystic interferon-alpha in pediatric craniopharyngioma patients: An international multicenter assessment on behalf of SIOPE and ISPN. Neuro. Oncol. 2017, 19, 1398–1407. [Google Scholar] [CrossRef] [PubMed]
- Pettorini, B.L.; Inzitari, R.; Massimi, L.; Tamburrini, G.; Caldarelli, M.; Fanali, C.; Cabras, T.; Messana, I.; Castagnola, M.; Di Rocco, C. The role of inflammation in the genesis of the cystic component of craniopharyngiomas. Child’s Nerv. Syst. 2010, 26, 1779–1784. [Google Scholar] [CrossRef]
- Mori, M.; Takeshima, H.; Kuratsu, J.I. Expression of interleukin-6 in human craniopharyngiomas: A possible inducer of tumor-associated inflammation. Int. J. Nol. Med. 2004, 14, 505–509. [Google Scholar] [CrossRef]
- Zitvogel, L.; Galluzzi, L.; Kepp, O.; Smyth, M.J.; Kroemer, G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015, 15, 405–414. [Google Scholar] [CrossRef]
- Cavalheiro, S.; Dastoli, P.A.; Silva, N.S.; Toledo, S.; Lederman, H.; da Silva, M.C. Use of interferon alpha in intratumoral chemotherapy for cystic craniopharyngioma. Child’s Nerv. Syst. 2005, 21, 719–724. [Google Scholar] [CrossRef]
- Bartels, U.; Laperriere, N.; Bouffet, E.; Drake, J. Intracystic therapies for cystic craniopharyngioma in childhood. Front. Endocrinol. (Lausanne) 2012, 3, 39. [Google Scholar] [CrossRef] [Green Version]
- Garon, E.B.; Rizvi, N.A.; Hui, R.; Leighl, N.; Balmanoukian, A.S.; Eder, J.P.; Patnaik, A.; Aggarwal, C.; Gubens, M.; Horn, L.; et al. Pembrolizumab for the Treatment of Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Robert, C.; Long, G.V.; Brady, B.; Dutriaux, C.; Maio, M.; Mortier, L.; Hassel, J.C.; Rutkowski, P.; McNeil, C.; Kalinka-Warzocha, E.; et al. Nivolumab in previously untreated melanoma without BRAF mutation. N. Engl. J. Med. 2015, 372, 320–330. [Google Scholar] [CrossRef] [Green Version]
- Seidel, J.A.; Otsuka, A.; Kabashima, K. Anti-PD-1 and Anti-CTLA-4 Therapies in Cancer: Mechanisms of Action, Efficacy, and Limitations. Front. Oncol. 2018, 8, 86. [Google Scholar] [CrossRef]
- Taube, J.M.; Klein, A.; Brahmer, J.R.; Xu, H.; Pan, X.; Kim, J.H.; Chen, L.; Pardoll, D.M.; Topalian, S.L.; Anders, R.A. Association of PD-1, PD-1 ligands, and other features of the tumor immune microenvironment with response to anti-PD-1 therapy. Clin. Cancer Res. 2014, 20, 5064–5074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Topalian, S.L.; Hodi, F.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2554. [Google Scholar] [CrossRef] [PubMed]
- Witt, D.A.; Donson, A.M.; Amani, V.; Moreira, D.C.; Sanford, B.; Hoffman, L.M.; Handler, M.H.; Levy, J.M.M.; Jones, K.L.; Nellan, A.; et al. Specific expression of PD-L1 in RELA-fusion supratentorial ependymoma: Implications for PD-1-targeted therapy. Pediatr. Blood Cancer 2018, 65, e26960. [Google Scholar] [CrossRef]
- Hodi, F.S.; O’Day, S.J.; McDermott, D.F.; Weber, R.W.; Sosman, J.A.; Haanen, J.B.; Gonzalez, R.; Robert, C.; Schadendorf, D.; Hassel, J.C.; et al. Improved survival with ipilimumab in patients with metastatic melanoma. N. Engl. J. Med. 2010, 363, 711–723. [Google Scholar] [CrossRef]
- Ji, R.R.; Chasalow, S.D.; Wang, L.; Hamid, O.; Schmidt, H.; Cogswell, J.; Alaparthy, S.; Berman, D.; Jure-Kunkel, M.; Siemers, N.O.; et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol. Immunother. 2012, 61, 1019–1031. [Google Scholar] [CrossRef]
- Hellmann, M.D.; Ciuleanu, T.E.; Pluzanski, A.; Lee, J.S.; Otterson, G.A.; Audigier-Valette, C.; Minenza, E.; Linardou, H.; Burgers, S.; Salman, P.; et al. Nivolumab plus ipilimumab in lung cancer with a high tumor mutational burden. N. Engl. J. Med. 2018, 378, 2093–2104. [Google Scholar] [CrossRef]
- Motzer, R.J.; Tannir, N.M.; McDermott, D.F.; Arén Frontera, O.; Melichar, B.; Choueiri, T.K.; Plimack, E.R.; Barthélémy, P.; Porta, C.; George, S.; et al. Nivolumab plus Ipilimumab versus Sunitinib in advanced renal-cell carcinoma. N. Engl. J. Med. 2018, 378, 1277–1290. [Google Scholar] [CrossRef]
- Hodi, F.S.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Nivolumab plus ipilimumab or nivolumab alone versus ipilimumab alone in advanced melanoma (CheckMate 067): 4-year outcomes of a multicentre, randomised, phase 3 trial. Lancet Oncol. 2018, 19, 1480–1492. [Google Scholar] [CrossRef]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Rutkowski, P.; Lao, C.D.; Cowey, C.L.; Schadendorf, D.; Wagstaff, J.; Dummer, R.; et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N. Engl. J. Med. 2019, 381, 1535–1546. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppé, J.-P.; Desprez, P.-Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef] [Green Version]
- Rodier, F.; Coppé, J.P.; Patil, C.K.; Hoeijmakers, W.A.M.; Muñoz, D.P.; Raza, S.R.; Freund, A.; Campeau, E.; Davalos, A.R.; Campisi, J. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat. Cell Biol. 2009, 11, 973–979. [Google Scholar] [CrossRef] [PubMed]
- Mario Gonzalez-Meljem, J.; Haston, S.; Carreno, G.; Apps, J.R.; Pozzi, S.; Stache, C.; Kaushal, G.; Virasami, A.; Panousopoulos, L.; Neda Mousavy-Gharavy, S.; et al. Stem cell senescence drives age-attenuated induction of pituitary tumours in mouse models of paediatric craniopharyngioma. Nat. Commun. 2017, 8, 1819. [Google Scholar] [CrossRef]
- Hickson, L.T.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganau, L.; Prisco, L.; Ligarotti, G.K.I.; Ambu, R.; Ganau, M. Understanding the Pathological Basis of Neurological Diseases Through Diagnostic Platforms Based on Innovations in Biomedical Engineering: New Concepts and Theranostics Perspectives. Medicines (Basel) 2018, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Ganau, M.; Paris, M.; Syrmos, N.; Ganau, L.; Ligarotti, G.K.I.; Moghaddamjou, A.; Prisco, L.; Ambu, R.; Chibbaro, S. How nanotechnology and biomedical engineering are supporting the identification of predictive biomarkers in neuro-oncology. Medicines (Basel) 2018, 5, 23. [Google Scholar] [CrossRef] [Green Version]




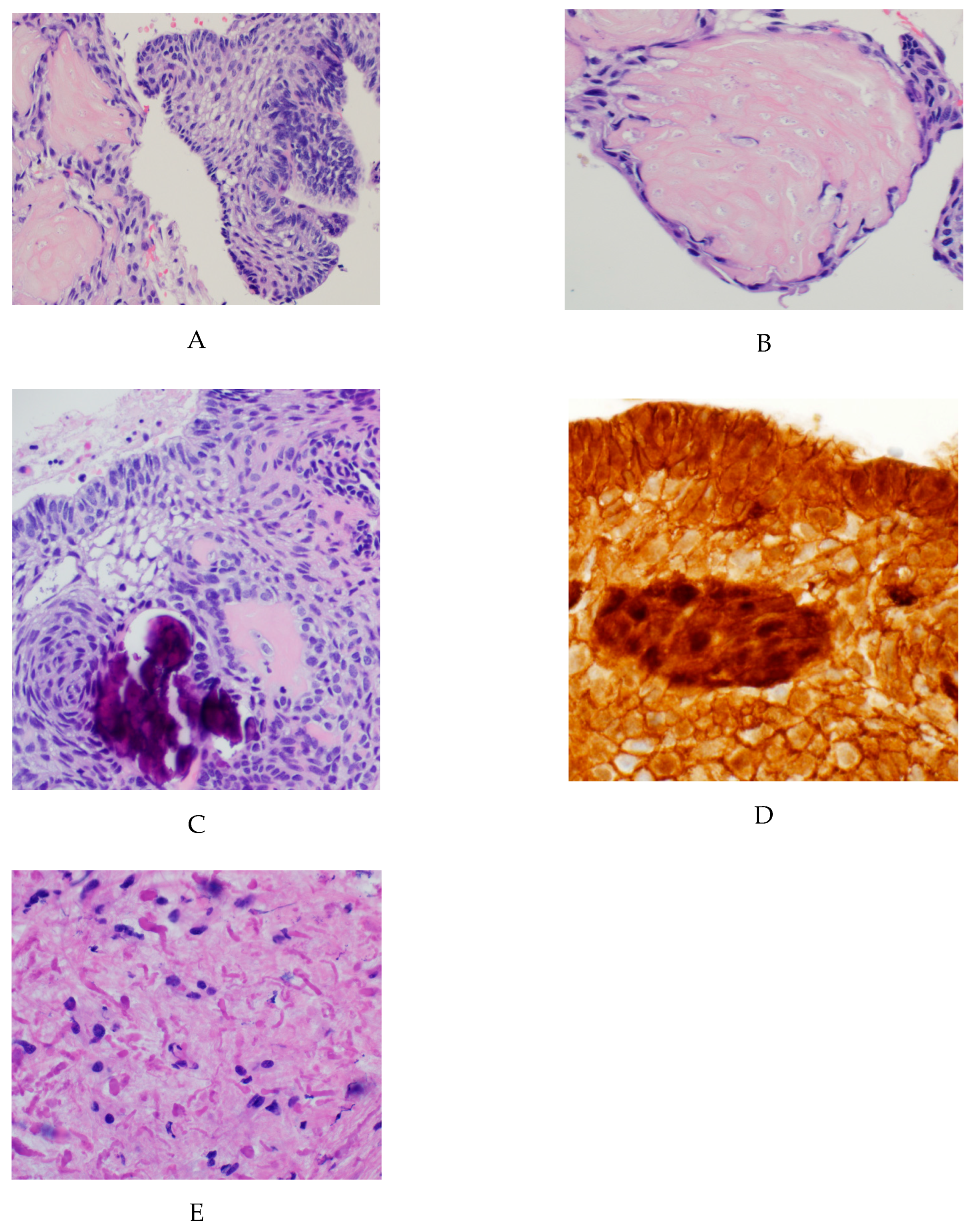{kind=link}
{kind=link}
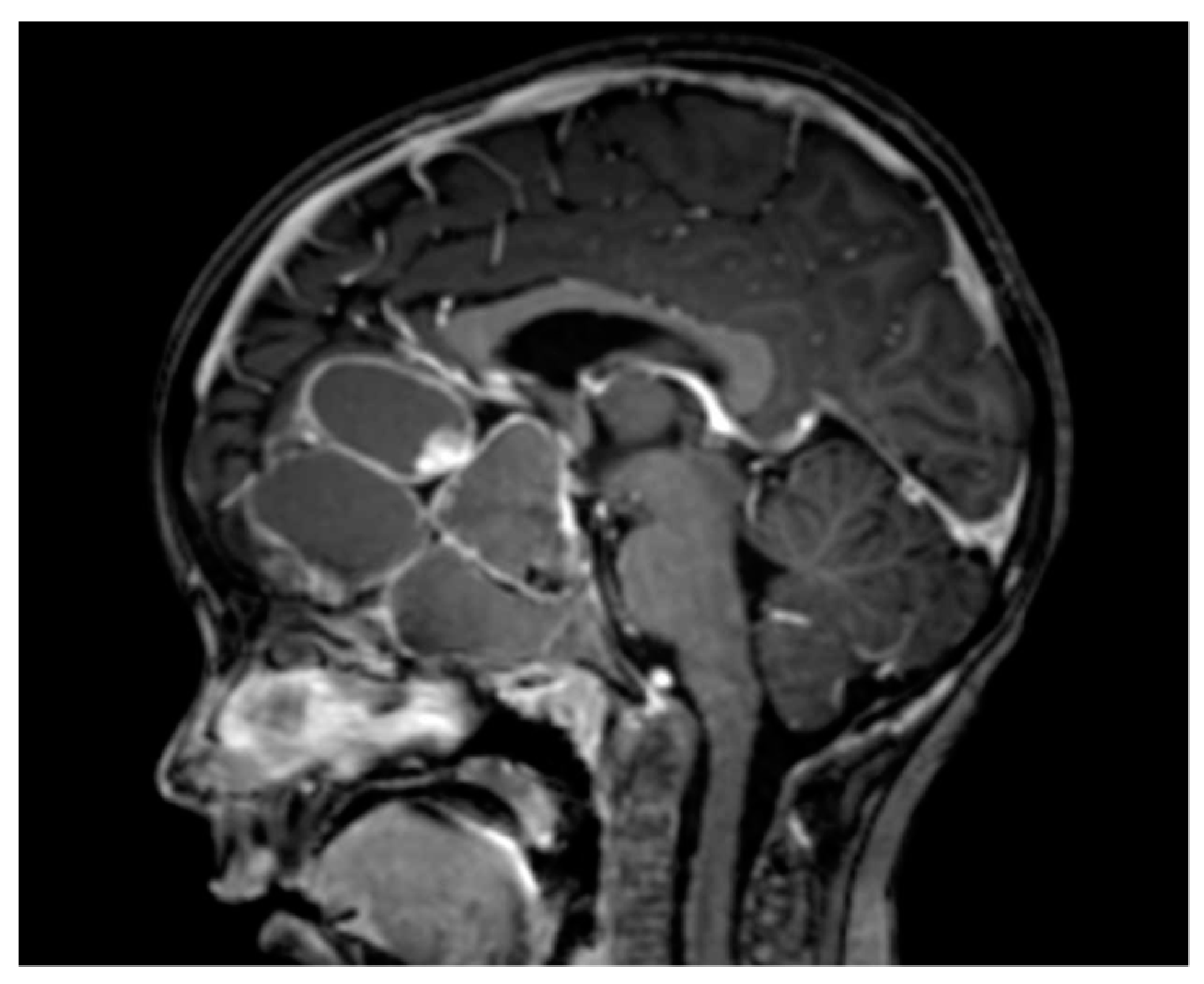{kind=link}
| Study | Summary of Study | Findings |
|---|---|---|
| Kilday et al. 2017 [35] | Multinational study assessing the efficacy of intra-cystic IFN-alpha in treating ACP | Demonstrated a progression free survival advantage for intracystic IFN-alpha |
| Pettorini et al. 2010 [36] | Identified the presence of alpha-defensins 1–3 in ACP cyst fluid | Demonstrated the importance of inflammation the genesis of ACP cysts |
| Gump et al. 2015 [11] | Used mRNA microarray analysis to identify the overexpression of multiple inflammatory markers in ACP relative to other tumors | Identifies IL6R and IL2RB to be overexpressed in ACP relative to normal brain and other tumors |
| Donson et al. 2017 [31] | Identified elevated levels of severeal inflammatory markers in both ACP cyst fluid and solid tumor | Overexpressed inflammatory markers identified included IL-6, IL-8, CXCL1, and IL-10 |
| Apps et al. 2018 [10] | Used various methods including RNA sequencing to identify activation of the inflammasome in ACP cyst fluid and solid tumor | Imflamatory genes that were overexpressed included IL-1B, IL-18, IL-6, IL-8, IL-10 |
| Coy et al. 2018 [33] | demonstrated the expression of PD-L1 in epithelial cells lining the cysts and intrinsic PD-1 expression in the beta-catenin over expressing whorl-like epithelial cell clusters in ACP | The first paper to demonstrate that immune checkpoint inhibitors may play a role in ACP treatments |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whelan, R.; Prince, E.; Gilani, A.; Hankinson, T. The Inflammatory Milieu of Adamantinomatous Craniopharyngioma and Its Implications for Treatment. J. Clin. Med. 2020, 9, 519. https://doi.org/10.3390/jcm9020519
Whelan R, Prince E, Gilani A, Hankinson T. The Inflammatory Milieu of Adamantinomatous Craniopharyngioma and Its Implications for Treatment. Journal of Clinical Medicine. 2020; 9(2):519. https://doi.org/10.3390/jcm9020519
Chicago/Turabian StyleWhelan, Ros, Eric Prince, Ahmed Gilani, and Todd Hankinson. 2020. "The Inflammatory Milieu of Adamantinomatous Craniopharyngioma and Its Implications for Treatment" Journal of Clinical Medicine 9, no. 2: 519. https://doi.org/10.3390/jcm9020519
APA StyleWhelan, R., Prince, E., Gilani, A., & Hankinson, T. (2020). The Inflammatory Milieu of Adamantinomatous Craniopharyngioma and Its Implications for Treatment. Journal of Clinical Medicine, 9(2), 519. https://doi.org/10.3390/jcm9020519