Role of Non-Coding RNAs in the Development of Targeted Therapy and Immunotherapy Approaches for Chronic Lymphocytic Leukemia

Abstract
:1. Chronic Lymphocytic Leukemia
2. Non-coding RNAs in CLL
3. NcRNAs, Tumor Microenvironment, and Extracellular Vesicles in CLL
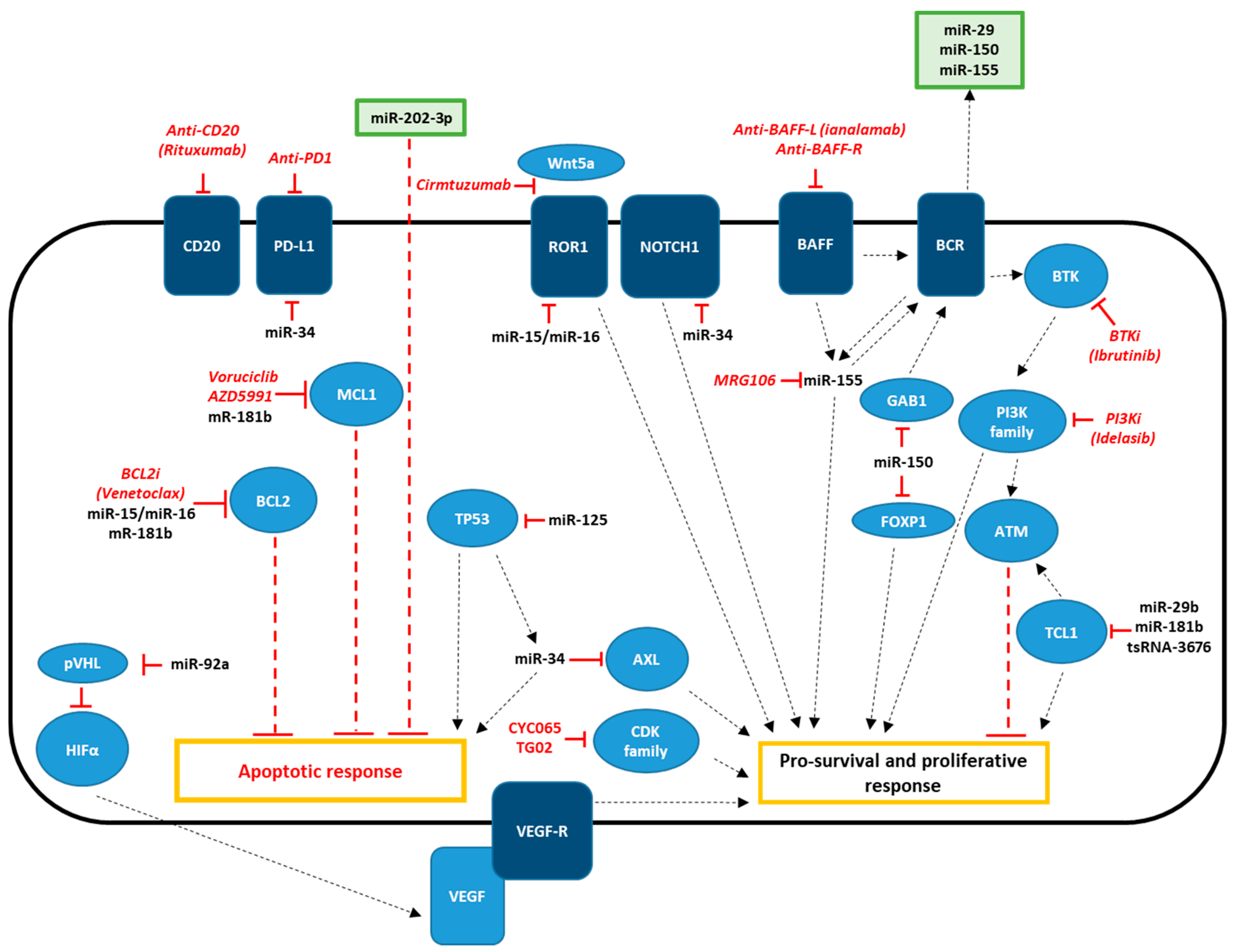
4. Treatment Options for CLL
4.1. Targeted Therapy for CLL
4.1.1. Bruton’s Tyrosine Kinase Inhibitors (BTKis)
4.1.2. Phosphoinositide 3-Kinase Inhibitors (PI3Ki)
4.1.3. Bcl2 Blockers
4.1.4. Other Targets: CDK Inhibitors, Mcl1 Inhibitors, Axl Inhibitors
4.2. Immunotherapy for CLL
4.2.1. Monoclonal Antibodies
4.2.2. CAR-T Cells
4.2.3. Immune Checkpoint Inhibitors: Anti-PD1/PD-L1, Anti-CTLA-4, and Anti-CD47-SIRPα Monoclonal Antibodies
4.2.4. Immunomodulatory Drugs (IMiDs)
5. Role of Non-Coding RNAs in Targeted and Immunotherapeutic Strategies for CLL
6. Conclusions
Author Contributions
Conflicts of Interest
References
- DeSantis, C.E.; Lin, C.C.; Mariotto, A.B.; Siegel, R.L.; Stein, K.D.; Kramer, J.L.; Alteri, R.; Robbins, A.S.; Jemal, A. Cancer treatment and survivorship statistics, 2014. CA Cancer J. Clin. 2014, 64, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Pekarsky, Y.; Zanesi, N.; Croce, C.M. Molecular basis of CLL. Semin. Cancer Biol. 2010, 20, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Kalil, N.; Cheson, B.D. Chronic lymphocytic leukemia. Oncologist 1999, 4, 352–369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rai, K.R.; Jain, P. Advances in the Clinical Staging of Chronic Lymphocytic Leukemia. Clin. Chem. 2011, 57, 1771–1772. [Google Scholar] [CrossRef] [Green Version]
- Binet, J.L.; Auquier, A.; Dighiero, G.; Chastang, C.; Piguet, H.; Goasguen, J.; Vaugier, G.; Potron, G.; Colona, P.; Oberling, F.; et al. A new prognostic classification of chronic lymphocytic leukemia derived from a multivariate survival analysis. Cancer 1981, 48, 198–206. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.J.; Montserrat, E.; Rai, K.R.; et al. Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: A report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute–Working Group 1996 guidelines. Blood 2008, 111, 5446–5456. [Google Scholar] [CrossRef] [Green Version]
- Jaseb, K.; Purrahman, D.; Shahrabi, S.; Ghanavat, M.; Rezaeean, H.; Saki, N. Prognostic significance of aberrant CD5 expression in B-cell leukemia. Oncol. Rev. 2019, 13, 400. [Google Scholar] [CrossRef]
- Rassenti, L.Z.; Jain, S.; Keating, M.J.; Wierda, W.G.; Grever, M.R.; Byrd, J.C.; Kay, N.E.; Brown, J.R.; Gribben, J.G.; Neuberg, D.S.; et al. Relative value of ZAP-70, CD38, and immunoglobulin mutation status in predicting aggressive disease in chronic lymphocytic leukemia. Blood 2008, 112, 1923–1930. [Google Scholar] [CrossRef] [Green Version]
- Edelmann, J.; Holzmann, K.; Miller, F.; Winkler, D.; Bühler, A.; Zenz, T.; Bullinger, L.; Kühn, M.W.M.; Gerhardinger, A.; Bloehdorn, J.; et al. High-resolution genomic profiling of chronic lymphocytic leukemia reveals new recurrent genomic alterations. Blood 2012, 120, 4783–4794. [Google Scholar] [CrossRef] [Green Version]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef] [Green Version]
- Hallek, M. Chronic lymphocytic leukemia: 2015 Update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2015, 90, 446–460. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Croce, C.M. MicroRNA signatures in human cancers. Nat. Rev. Cancer 2006, 6, 857–866. [Google Scholar] [CrossRef] [PubMed]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzifi, F.; Economopoulou, C.; Gourgiotis, D.; Ardavanis, A.; Papageorgiou, S.; Scorilas, A. The Role of BCL2 Family of Apoptosis Regulator Proteins in Acute and Chronic Leukemias. Adv. Hematol. 2012, 2012, 524308. [Google Scholar] [CrossRef] [Green Version]
- Pekarsky, Y.; Croce, C.M. Role of miR-15/16 in CLL. Cell Death Differ. 2015, 22, 6–11. [Google Scholar] [CrossRef] [Green Version]
- Borcherding, N.; Kusner, D.; Liu, G.-H.; Zhang, W. ROR1, an embryonic protein with an emerging role in cancer biology. Protein Cell 2014, 5, 496–502. [Google Scholar] [CrossRef] [Green Version]
- Savitsky, K.; Sfez, S.; Tagle, D.A.; Ziv, Y.; Sartiel, A.; Collins, F.S.; Shiloh, Y.; Rotman, G. The complete sequence of the coding region of the ATM gene reveals similarity to cell cycle regulators in different species. Hum. Mol. Genet. 1995, 4, 2025–2032. [Google Scholar] [CrossRef]
- Zenz, T.; Eichhorst, B.; Busch, R.; Denzel, T.; Häbe, S.; Winkler, D.; Bühler, A.; Edelmann, J.; Bergmann, M.; Hopfinger, G.; et al. TP53 mutation and survival in chronic lymphocytic leukemia. J. Clin. Oncol. 2010, 28, 4473–4479. [Google Scholar] [CrossRef]
- Rosati, E.; Baldoni, S.; De Falco, F.; Del Papa, B.; Dorillo, E.; Rompietti, C.; Albi, E.; Falzetti, F.; Di Ianni, M.; Sportoletti, P. NOTCH1 Aberrations in Chronic Lymphocytic Leukemia. Front. Oncol. 2018, 8, 229. [Google Scholar] [CrossRef] [Green Version]
- Schaffner, C.; Stilgenbauer, S.; Rappold, G.A.; Döhner, H.; Lichter, P. Somatic ATM mutations indicate a pathogenic role of ATM in B-cell chronic lymphocytic leukemia. Blood 1999, 94, 748–753. [Google Scholar] [CrossRef]
- Calin, G.A.; Cimmino, A.; Fabbri, M.; Ferracin, M.; Wojcik, S.E.; Shimizu, M.; Taccioli, C.; Zanesi, N.; Garzon, R.; Aqeilan, R.I.; et al. MiR-15a and miR-16-1 cluster functions in human leukemia. Proc. Natl. Acad. Sci. USA 2008, 105, 5166–5171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed] [Green Version]
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; et al.; ENCODE Project Consortium Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar] [PubMed] [Green Version]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 2011, 12, 861–874. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bastide, A.; David, A. Interaction of rRNA with mRNA and tRNA in Translating Mammalian Ribosome: Functional Implications in Health and Disease. Biomolecules 2018, 8, 100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balatti, V.; Pekarsky, Y.; Croce, C.M. Role of the tRNA-Derived Small RNAs in Cancer: New Potential Biomarkers and Target for Therapy. Adv. Cancer Res. 2017, 135, 173–187. [Google Scholar]
- Slack, F.J.; Chinnaiyan, A.M. The Role of Non-coding RNAs in Oncology. Cell 2019, 179, 1033–1055. [Google Scholar] [CrossRef]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef]
- Calin, G.A.; Ferracin, M.; Cimmino, A.; Di Leva, G.; Shimizu, M.; Wojcik, S.E.; Iorio, M.V.; Visone, R.; Sever, N.I.; Fabbri, M.; et al. A MicroRNA Signature Associated with Prognosis and Progression in Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2005, 353, 1793–1801. [Google Scholar] [CrossRef]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.V.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rassenti, L.Z.; Balatti, V.; Ghia, E.M.; Palamarchuk, A.; Tomasello, L.; Fadda, P.; Pekarsky, Y.; Widhopf, G.F.; Kipps, T.J.; Croce, C.M. MicroRNA dysregulation to identify therapeutic target combinations for chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2017, 114, 10731–10736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aqeilan, R.I.; Calin, G.A.; Croce, C.M. miR-15a and miR-16-1 in cancer: Discovery, function and future perspectives. Cell Death Differ. 2010, 17, 215–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.; Xu, Z.; Ou, D.; Liu, J.; Zhang, J. The miR-15a/16 gene cluster in human cancer: A systematic review. J. Cell Physiol. 2019, 234, 5496–5506. [Google Scholar] [CrossRef] [PubMed]
- Pekarsky, Y.; Santanam, U.; Cimmino, A.; Palamarchuk, A.; Efanov, A.; Maximov, V.; Volinia, S.; Alder, H.-G.; Liu, C.; Rassenti, L.; et al. Tcl1 Expression in Chronic Lymphocytic Leukemia Is Regulated by miR-29 and miR-181. Cancer Res. 2006, 66, 11590–11593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekarsky, Y.; Croce, C.M. Is miR-29 an oncogene or tumor suppressor in CLL? Oncotarget 2010, 1, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Mott, J.L.; Kobayashi, S.; Bronk, S.F.; Gores, G.J. mir-29 regulates Mcl-1 protein expression and apoptosis. Oncogene 2007, 26, 6133–6140. [Google Scholar] [CrossRef] [Green Version]
- Auer, R.L.; Riaz, S.; Cotter, F.E. The 13q and 11q B-cell chronic lymphocytic leukaemia-associated regions derive from a common ancestral region in the zebrafish. Br. J. Haematol. 2007, 137, 443–453. [Google Scholar] [CrossRef]
- Lehmann, S.; Ogawa, S.; Raynaud, S.D.; Sanada, M.; Nannya, Y.; Ticchioni, M.; Bastard, C.; Kawamata, N.; Phillip Koeffler, H. Molecular allelokaryotyping of early-stage, untreated chronic lymphocytic leukemia. Cancer 2008, 112, 1296–1305. [Google Scholar] [CrossRef]
- Deneberg, S.; Kanduri, M.; Ali, D.; Bengtzen, S.; Karimi, M.; Qu, Y.; Kimby, E.; Mansouri, L.; Rosenquist, R.; Lennartsson, A.; et al. microRNA-34b/con chromosome 11q23 is aberrantly methylated in chronic lymphocytic leukemia. Epigenetics 2014, 9, 910–917. [Google Scholar] [CrossRef] [Green Version]
- Li, X.J.; Ren, Z.J.; Tang, J.H. MicroRNA-34a: A potential therapeutic target in human cancer. Cell Death Dis. 2014, 5, e1327. [Google Scholar] [CrossRef] [PubMed]
- Ferracin, M.; Zagatti, B.; Rizzotto, L.; Cavazzini, F.; Veronese, A.; Ciccone, M.; Saccenti, E.; Lupini, L.; Grilli, A.; De Angeli, C.; et al. MicroRNAs involvement in fludarabine refractory chronic lymphocytic leukemia. Mol. Cancer 2010, 9, 123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fabbri, M. Association of a MicroRNA/TP53 Feedback Circuitry With Pathogenesis and Outcome of B-Cell Chronic Lymphocytic Leukemia. JAMA 2011, 305, 59. [Google Scholar] [CrossRef] [PubMed]
- Hermeking, H. The miR-34 family in cancer and apoptosis. Cell Death Differ. 2010, 17, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Boysen, J.; Sinha, S.; Price-Troska, T.; Warner, S.L.; Bearss, D.J.; Viswanatha, D.; Shanafelt, T.D.; Kay, N.E.; Ghosh, A.K. The tumor suppressor axis p53/miR-34a regulates Axl expression in B-cell chronic lymphocytic leukemia: Implications for therapy in p53-defective CLL patients. Leukemia 2014, 28, 451–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.; Liu, X.; Koul, S.; Lee, C.Y.; Zhang, Z.; Halmos, B. AXL kinase as a novel target for cancer therapy. Oncotarget 2014, 5, 9546–9563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balatti, V.; Tomasello, L.; Rassenti, L.Z.; Veneziano, D.; Nigita, G.; Wang, H.-Y.; Thorson, J.A.; Kipps, T.J.; Pekarsky, Y.; Croce, C.M. miR-125a and miR-34a expression predicts Richter syndrome in chronic lymphocytic leukemia patients. Blood 2018, 132, 2179–2182. [Google Scholar] [CrossRef] [Green Version]
- Calin, G.A.; Sevignani, C.; Dumitru, C.D.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar] [CrossRef] [Green Version]
- Visone, R.; Rassenti, L.Z.; Veronese, A.; Taccioli, C.; Costinean, S.; Aguda, B.D.; Volinia, S.; Ferracin, M.; Palatini, J.; Balatti, V.; et al. Karyotype-specific microRNA signature in chronic lymphocytic leukemia. Blood 2009, 114, 3872–3879. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, A.K.; Shanafelt, T.D.; Cimmino, A.; Taccioli, C.; Volinia, S.; Liu, C.-G.; Calin, G.A.; Croce, C.M.; Chan, D.A.; Giaccia, A.J.; et al. Aberrant regulation of pVHL levels by microRNA promotes the HIF/VEGF axis in CLL B cells. Blood 2009, 113, 5568–5574. [Google Scholar] [CrossRef] [Green Version]
- Kay, N.E.; Bone, N.D.; Tschumper, R.C.; Howell, K.H.; Geyer, S.M.; Dewald, G.W.; Hanson, C.A.; Jelinek, D.F. B-CLL cells are capable of synthesis and secretion of both pro- and anti-angiogenic molecules. Leukemia 2002, 16, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paesler, J.; Gehrke, I.; Gandhirajan, R.K.; Filipovich, A.; Hertweck, M.; Erdfelder, F.; Uhrmacher, S.; Poll-Wolbeck, S.J.; Hallek, M.-A.; Kreuzer, K. The Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitors Vatalanib and Pazopanib Potently Induce Apoptosis in Chronic Lymphocytic Leukemia Cells In vitro and In vivo. Clin. Cancer Res. 2010, 16, 3390–3398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Visone, R.; Veronese, A.; Rassenti, L.Z.; Balatti, V.; Pearl, D.K.; Acunzo, M.; Volinia, S.; Taccioli, C.; Kipps, T.J.; Croce, C.M. miR-181b is a biomarker of disease progression in chronic lymphocytic leukemia. Blood 2011, 118, 3072–3079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Y.-B.; Lu, Y.; Yue, S.; Giffard, R.G. miR-181 targets multiple Bcl-2 family members and influences apoptosis and mitochondrial function in astrocytes. Mitochondrion 2012, 12, 213–219. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Yu, J.; Zhao, C.; Ren, H.; Yuan, Z.; Zhang, B.; Zhuang, J.; Wang, J.; Feng, B. MiR-181b-5p modulates chemosensitivity of glioma cells to temozolomide by targeting Bcl-2. Biomed Pharm. 2019, 109, 2192–2202. [Google Scholar] [CrossRef]
- Zhu, D.-X.; Zhu, W.; Fang, C.; Fan, L.; Zou, Z.-J.; Wang, Y.-H.; Liu, P.; Hong, M.; Miao, K.-R.; Liu, P.; et al. miR-181a/b significantly enhances drug sensitivity in chronic lymphocytic leukemia cells via targeting multiple anti-apoptosis genes. Carcinogenesis 2012, 33, 1294–1301. [Google Scholar] [CrossRef] [Green Version]
- Tili, E.; Michaille, J.-J.; Luo, Z.; Volinia, S.; Rassenti, L.Z.; Kipps, T.J.; Croce, C.M. The down-regulation of miR-125b in chronic lymphocytic leukemias leads to metabolic adaptation of cells to a transformed state. Blood 2012, 120, 2631–2638. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.-M.; Lin, K.-Y.; Chen, Y.-Q. Diverse functions of miR-125 family in different cell contexts. J. Hematol. Oncol. 2013, 6, 6. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Gao, J.-S.; Tang, X.; Tucker, L.D.; Quesenberry, P.; Rigoutsos, I.; Ramratnam, B. MicroRNA 125a and its regulation of the p53 tumor suppressor gene. FEBS Lett. 2009, 583, 3725–3730. [Google Scholar] [CrossRef] [Green Version]
- Ferrajoli, A.; Shanafelt, T.D.; Ivan, C.; Shimizu, M.; Rabe, K.G.; Nouraee, N.; Ikuo, M.; Ghosh, A.K.; Lerner, S.; Rassenti, L.Z.; et al. Prognostic value of miR-155 in individuals with monoclonal B-cell lymphocytosis and patients with B chronic lymphocytic leukemia. Blood 2013, 122, 1891–1899. [Google Scholar] [CrossRef] [Green Version]
- Cui, B.; Chen, L.; Zhang, S.; Mraz, M.; Fecteau, J.-F.; Yu, J.; Ghia, E.M.; Zhang, L.; Bao, L.; Rassenti, L.Z.; et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014, 124, 546–554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pagotto, S.; Veronese, A.; Soranno, A.; Lanuti, P.; Di Marco, M.; Russo, M.V.; Ramassone, A.; Marchisio, M.; Simeone, P.; Guanciali Franchi, P.E.; et al. Hsa-miR-155-5p drives aneuploidy at early stages of cellular transformation. Oncotarget 2018, 9, 13036–13047. [Google Scholar] [CrossRef] [Green Version]
- Mraz, M.; Chen, L.; Rassenti, L.Z.; Ghia, E.M.; Li, H.; Jepsen, K.; Smith, E.N.; Messer, K.; Frazer, K.A.; Kipps, T.J. miR-150 influences B-cell receptor signaling in chronic lymphocytic leukemia by regulating expression of GAB1 and FOXP1. Blood 2014, 124, 84–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balatti, V.; Rizzotto, L.; Miller, C.; Palamarchuk, A.; Fadda, P.; Pandolfo, R.; Rassenti, L.Z.; Hertlein, E.; Ruppert, A.S.; Lozanski, A.; et al. TCL1 targeting miR-3676 is codeleted with tumor protein p53 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2015, 112, 2169–2174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekarsky, Y.; Balatti, V.; Palamarchuk, A.; Rizzotto, L.; Veneziano, D.; Nigita, G.; Rassenti, L.Z.; Pass, H.I.; Kipps, T.J.; Liu, C.-G.; et al. Dysregulation of a family of short noncoding RNAs, tsRNAs, in human cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5071–5076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balatti, V.; Nigita, G.; Veneziano, D.; Drusco, A.; Stein, G.S.; Messier, T.L.; Farina, N.H.; Lian, J.B.; Tomasello, L.; Liu, C.-G.; et al. tsRNA signatures in cancer. Proc. Natl. Acad. Sci. USA 2017, 114, 8071–8076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veneziano, D.; Tomasello, L.; Balatti, V.; Palamarchuk, A.; Rassenti, L.Z.; Kipps, T.J.; Pekarsky, Y.; Croce, C.M. Dysregulation of different classes of tRNA fragments in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2019, 116, 24252–24258. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [Green Version]
- Fabbri, M.; Paone, A.; Calore, F.; Galli, R.; Gaudio, E.; Santhanam, R.; Lovat, F.; Fadda, P.; Mao, C.; Nuovo, G.J.; et al. MicroRNAs bind to Toll-like receptors to induce prometastatic inflammatory response. Proc. Natl. Acad. Sci. USA 2012, 109, E2110–E2116. [Google Scholar] [CrossRef] [Green Version]
- Burger, J.A.; Tsukada, N.; Burger, M.; Zvaifler, N.J.; Dell’Aquila, M.; Kipps, T.J. Blood-derived nurse-like cells protect chronic lymphocytic leukemia B cells from spontaneous apoptosis through stromal cell–derived factor-1. Blood 2000, 96, 2655–2663. [Google Scholar] [CrossRef]
- Jia, L.; Clear, A.; Liu, F.-T.; Matthews, J.; Uddin, N.; McCarthy, A.; Hoxha, E.; Durance, C.; Iqbal, S.; Gribben, J.G. Extracellular HMGB1 promotes differentiation of nurse-like cells in chronic lymphocytic leukemia. Blood 2014, 123, 1709–1719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umezu, T.; Ohyashiki, K.; Kuroda, M.; Ohyashiki, J.H. Leukemia cell to endothelial cell communication via exosomal miRNAs. Oncogene 2013, 32, 2747–2755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.-Y.; Ozer, H.G.; Lehman, A.M.; Maddocks, K.; Yu, L.; Johnson, A.J.; Byrd, J.C. Characterization of CLL exosomes reveals a distinct microRNA signature and enhanced secretion by activation of BCR signaling. Blood 2015, 125, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Stamatopoulos, B.; Van Damme, M.; Crompot, E.; Dessars, B.; El Housni, H.; Mineur, P.; Meuleman, N.; Bron, D.; Lagneaux, L. Opposite Prognostic Significance of Cellular and Serum Circulating MicroRNA-150 in Patients with Chronic Lymphocytic Leukemia. Mol. Med. 2015, 21, 123–133. [Google Scholar] [CrossRef]
- Kumar, V.; Patel, S.; Tcyganov, E.; Gabrilovich, D.I. The Nature of Myeloid-Derived Suppressor Cells in the Tumor Microenvironment. Trends Immunol. 2016, 37, 208–220. [Google Scholar] [CrossRef]
- Bruns, H.; Böttcher, M.; Qorraj, M.; Fabri, M.; Jitschin, S.; Dindorf, J.; Busch, L.; Jitschin, R.; Mackensen, A.; Mougiakakos, D. CLL-cell-mediated MDSC induction by exosomal miR-155 transfer is disrupted by vitamin D. Leukemia 2017, 31, 985–988. [Google Scholar] [CrossRef]
- Farahani, M.; Rubbi, C.; Liu, L.; Slupsky, J.R.; Kalakonda, N. CLL Exosomes Modulate the Transcriptome and Behaviour of Recipient Stromal Cells and Are Selectively Enriched in miR-202-3p. PLoS ONE 2015, 10, e0141429. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, A.E.; Liu, R.; Fu, A.; Zheng, T.; Slack, F.; Zhu, Y. Targetome Profiling, Pathway Analysis and Genetic Association Study Implicate miR-202 in Lymphomagenesis. Cancer Epidemiol. Biomark. Prev. 2013, 22, 327–336. [Google Scholar] [CrossRef] [Green Version]
- Paggetti, J.; Haderk, F.; Seiffert, M.; Janji, B.; Distler, U.; Ammerlaan, W.; Kim, Y.J.; Adam, J.; Lichter, P.; Solary, E.; et al. Exosomes released by chronic lymphocytic leukemia cells induce the transition of stromal cells into cancer-associated fibroblasts. Blood 2015, 126, 1106–1117. [Google Scholar] [CrossRef] [Green Version]
- Surman, M.; Drożdż, A.; Stępień, E.; Przybyło, M. Extracellular Vesicles as Drug Delivery Systems - Methods of Production and Potential Therapeutic Applications. Curr. Pharm. Des. 2019, 25, 132–154. [Google Scholar] [CrossRef]
- Lamichhane, T.N.; Jay, S.M. Production of Extracellular Vesicles Loaded with Therapeutic Cargo. Methods Mol. Biol. 2018, 1831, 37–47. [Google Scholar] [PubMed]
- Flinn, I.W.; Neuberg, D.S.; Grever, M.R.; Dewald, G.W.; Bennett, J.M.; Paietta, E.M.; Hussein, M.A.; Appelbaum, F.R.; Larson, R.A.; Moore, D.F.; et al. Phase III Trial of Fludarabine Plus Cyclophosphamide Compared With Fludarabine for Patients With Previously Untreated Chronic Lymphocytic Leukemia: US Intergroup Trial E2997. J. Clin. Oncol. 2007, 25, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Woyach, J.A.; Johnson, A.J. Targeted therapies in CLL: Mechanisms of resistance and strategies for management. Blood 2015, 126, 471–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nowell, P. The clonal evolution of tumor cell populations. Science 1976, 194, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Greaves, M.; Maley, C.C. Clonal evolution in cancer. Nature 2012, 481, 306–313. [Google Scholar] [CrossRef] [PubMed]
- Leeksma, A.C.; Taylor, J.; Wu, B.; Gardner, J.R.; He, J.; Nahas, M.; Gonen, M.; Alemayehu, W.G.; te Raa, D.; Walther, T.; et al. Clonal diversity predicts adverse outcome in chronic lymphocytic leukemia. Leukemia 2019, 33, 390–402. [Google Scholar] [CrossRef]
- Singh, S.P.; Dammeijer, F.; Hendriks, R.W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 2018, 17. [Google Scholar] [CrossRef]
- Davis, R.E.; Eric Davis, R.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080. [Google Scholar] [CrossRef] [Green Version]
- Itchaki, G.; Brown, J.R. Experience with ibrutinib for first-line use in patients with chronic lymphocytic leukemia. Ther. Adv. Hematol. 2018, 9, 3–19. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Targeting BTK with Ibrutinib in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2013, 369, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Herman, S.E.M.; Gordon, A.L.; Hertlein, E.; Ramanunni, A.; Zhang, X.; Jaglowski, S.; Flynn, J.; Jones, J.; Blum, K.A.; Buggy, J.J.; et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood 2011, 117, 6287–6296. [Google Scholar] [CrossRef] [PubMed]
- Owen, C.; Berinstein, N.L.; Christofides, A.; Sehn, L.H. Review of Bruton tyrosine kinase inhibitors for the treatment of relapsed or refractory mantle cell lymphoma. Curr. Oncol. 2019, 26, e233–e240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, J.C.; Harrington, B.; O’Brien, S.; Jones, J.A.; Schuh, A.; Devereux, S.; Chaves, J.; Wierda, W.G.; Awan, F.T.; Brown, J.R.; et al. Acalabrutinib (ACP-196) in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Awan, F.T.; Schuh, A.; Brown, J.R.; Furman, R.R.; Pagel, J.M.; Hillmen, P.; Stephens, D.M.; Woyach, J.; Bibikova, E.; Charuworn, P.; et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019, 3, 1553–1562. [Google Scholar] [CrossRef] [Green Version]
- Maddocks, K.J.; Ruppert, A.S.; Lozanski, G.; Heerema, N.A.; Zhao, W.; Abruzzo, L.; Lozanski, A.; Davis, M.; Gordon, A.; Smith, L.L.; et al. Etiology of Ibrutinib Therapy Discontinuation and Outcomes in Patients With Chronic Lymphocytic Leukemia. JAMA Oncol. 2015, 1, 80–87. [Google Scholar] [CrossRef]
- Jain, P.; Keating, M.; Wierda, W.; Estrov, Z.; Ferrajoli, A.; Jain, N.; George, B.; James, D.; Kantarjian, H.; Burger, J.; et al. Outcomes of patients with chronic lymphocytic leukemia after discontinuing ibrutinib. Blood 2015, 125, 2062–2067. [Google Scholar] [CrossRef] [Green Version]
- Walliser, C.; Hermkes, E.; Schade, A.; Wiese, S.; Deinzer, J.; Zapatka, M.; Désiré, L.; Mertens, D.; Stilgenbauer, S.; Gierschik, P. The Phospholipase Cγ2Mutants R665W and L845F Identified in Ibrutinib-resistant Chronic Lymphocytic Leukemia Patients Are Hypersensitive to the Rho GTPase Rac2 Protein. J. Biol. Chem. 2016, 291, 22136–22148. [Google Scholar] [CrossRef] [Green Version]
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond Ibrutinib. Curr. Hematol. Malig. Rep. 2019, 14, 197–205. [Google Scholar] [CrossRef]
- Bunney, T.D.; Katan, M. Phosphoinositide signalling in cancer: Beyond PI3K and PTEN. Nat. Rev. Cancer 2010, 10, 342–352. [Google Scholar] [CrossRef]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.E.; Kahl, B.S. PI3-Kinase Inhibitors in Chronic Lymphocytic Leukemia. Curr. Hematol. Malig. Rep. 2014, 9, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Vanhaesebroeck, B.; Welham, M.J.; Kotani, K.; Stein, R.; Warne, P.H.; Zvelebil, M.J.; Higashi, K.; Volinia, S.; Downward, J.; Waterfield, M.D. p110, a novel phosphoinositide 3-kinase in leukocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 4330–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.Y.; Wu, X.; Eissa, N.; Hou, S.; Ghia, J.-E.; Murooka, T.T.; Banerji, V.; Johnston, J.B.; Lin, F.; Gibson, S.B.; et al. Distinct roles for phosphoinositide 3-kinases γ and δ in malignant B cell migration. Leukemia 2018, 32, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Hoellenriegel, J.; Meadows, S.A.; Sivina, M.; Wierda, W.G.; Kantarjian, H.; Keating, M.J.; Giese, N.; O’Brien, S.; Yu, A.; Miller, L.L.; et al. The phosphoinositide 3′-kinase delta inhibitor, CAL-101, inhibits B-cell receptor signaling and chemokine networks in chronic lymphocytic leukemia. Blood 2011, 118, 3603–3612. [Google Scholar] [CrossRef] [Green Version]
- Zelenetz, A.D.; Barrientos, J.C.; Brown, J.R.; Coiffier, B.; Delgado, J.; Egyed, M.; Ghia, P.; Illés, Á.; Jurczak, W.; Marlton, P.; et al. Idelalisib or placebo in combination with bendamustine and rituximab in patients with relapsed or refractory chronic lymphocytic leukaemia: Interim results from a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2017, 18, 297–311. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, D.A.; Sagrillo, F.S.; Fraga, C.A.M. Duvelisib: A 2018 Novel FDA-Approved Small Molecule Inhibiting Phosphoinositide 3-Kinases. Pharmaceuticals 2019, 12, 69. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.; Danilov, A.V.; Pagel, J.M. Duvelisib for CLL/SLL and follicular non-Hodgkin lymphoma. Blood 2019, 134, 1573–1577. [Google Scholar] [CrossRef]
- Mato, A.R.; Nabhan, C.; Barr, P.M.; Ujjani, C.S.; Hill, B.T.; Lamanna, N.; Skarbnik, A.P.; Howlett, C.; Pu, J.J.; Sehgal, A.R.; et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: A real world experience. Blood 2016, 128, 2199–2205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reed, J.C.; Miyashita, T.; Krajewski, S.; Takayama, S.; Aime-Sempe, C.; Kitada, S.; Sato, T.; Wang, H.-G.; Harigai, M.; Hanada, M.; et al. Bcl-2 family proteins and the regulation of programmed cell death in leukemia and lymphoma. Cancer Treat. Res. 1996, 84, 31–72. [Google Scholar]
- Chao, D.T.; Korsmeyer, S.J. BCL-2 FAMILY: Regulators of Cell Death. Annu. Rev. Immunol. 1998, 16, 395–419. [Google Scholar] [CrossRef] [PubMed]
- Balatti, V.; Acunzo, M.; Pekarky, Y.; Croce, C.M. Novel Mechanisms of Regulation of miRNAs in CLL. Trends Cancer 2016, 2, 134–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rampazzo, E.; Bojnik, E.; Trentin, L.; Bonaldi, L.; Del Bianco, P.; Frezzato, F.; Visentin, A.; Facco, M.; Semenzato, G.; De Rossi, A. Role of miR-15a/miR-16-1 and the TP53 axis in regulating telomerase expression in chronic lymphocytic leukemia. Haematologica 2017, 102, e253–e256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pekarsky, Y.; Balatti, V.; Croce, C.M. BCL2 and miR-15/16: From gene discovery to treatment. Cell Death Differ. 2018, 25, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Oltersdorf, T.; Elmore, S.W.; Shoemaker, A.R.; Armstrong, R.C.; Augeri, D.J.; Belli, B.A.; Bruncko, M.; Deckwerth, T.L.; Dinges, J.; Hajduk, P.J.; et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [Google Scholar] [CrossRef]
- Tse, C.; Shoemaker, A.R.; Adickes, J.; Anderson, M.G.; Chen, J.; Jin, S.; Johnson, E.F.; Marsh, K.C.; Mitten, M.J.; Nimmer, P.; et al. ABT-263: A Potent and Orally Bioavailable Bcl-2 Family Inhibitor. Cancer Res. 2008, 68, 3421–3428. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Seymour, J.F.; Brown, J.R.; Wierda, W.G.; Kipps, T.J.; Khaw, S.L.; Carney, D.A.; He, S.Z.; Huang, D.C.S.; Xiong, H.; et al. Substantial Susceptibility of Chronic Lymphocytic Leukemia to BCL2 Inhibition: Results of a Phase I Study of Navitoclax in Patients With Relapsed or Refractory Disease. J. Clin. Oncol. 2012, 30, 488–496. [Google Scholar] [CrossRef] [Green Version]
- Souers, A.J.; Leverson, J.D.; Boghaert, E.R.; Ackler, S.L.; Catron, N.D.; Chen, J.; Dayton, B.D.; Ding, H.; Enschede, S.H.; Fairbrother, W.J.; et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [Google Scholar] [CrossRef]
- Cheson, B.D.; Enschede, S.H.; Cerri, E.; Desai, M.; Potluri, J.; Lamanna, N.; Tam, C. Tumor Lysis Syndrome in Chronic Lymphocytic Leukemia with Novel Targeted Agents. Oncologist 2017, 22, 1283–1291. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.W.; Davids, M.S.; Pagel, J.M.; Kahl, B.S.; Puvvada, S.D.; Gerecitano, J.F.; Kipps, T.J.; Anderson, M.A.; Brown, J.R.; Gressick, L.; et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med. 2016, 374, 311–322. [Google Scholar] [CrossRef]
- Deeks, E.D. Venetoclax: First Global Approval. Drugs 2016, 76, 979–987. [Google Scholar] [CrossRef]
- Malumbres, M. Cyclin-dependent kinases. Genome Biol. 2014, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Goh, K.C.; Novotny-Diermayr, V.; Hart, S.; Ong, L.C.; Loh, Y.K.; Cheong, A.; Tan, Y.C.; Hu, C.; Jayaraman, R.; William, A.D.; et al. TG02, a novel oral multi-kinase inhibitor of CDKs, JAK2 and FLT3 with potent anti-leukemic properties. Leukemia 2012, 26, 236–243. [Google Scholar] [CrossRef] [Green Version]
- Craig, R.W.; Jabs, E.W.; Zhou, P.; Kozopas, K.M.; Hawkins, A.L.; Rochelle, J.M.; Seldin, M.F.; Griffins, C.A. Human and Mouse Chromosomal Mapping of the Myeloid Cell Leukemia-1 Gene: MCL1 Maps to Human Chromosome 1q21, a Region That Is Frequently Altered in Preneoplastic and Neoplastic Disease. Genomics 1994, 23, 457–463. [Google Scholar] [CrossRef]
- Choudhary, G.S.; Al-Harbi, S.; Mazumder, S.; Hill, B.T.; Smith, M.R.; Bodo, J.; Hsi, E.D.; Almasan, A. MCL-1 and BCL-xL-dependent resistance to the BCL-2 inhibitor ABT-199 can be overcome by preventing PI3K/AKT/mTOR activation in lymphoid malignancies. Cell Death Dis. 2015, 6, e1593. [Google Scholar] [CrossRef]
- Dey, J.; Deckwerth, T.L.; Kerwin, W.S.; Casalini, J.R.; Merrell, A.J.; Grenley, M.O.; Burns, C.; Ditzler, S.H.; Dixon, C.P.; Beirne, E.; et al. Voruciclib, a clinical stage oral CDK9 inhibitor, represses MCL-1 and sensitizes high-risk Diffuse Large B-cell Lymphoma to BCL2 inhibition. Sci. Rep. 2017, 7, 18007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tron, A.E.; Belmonte, M.A.; Adam, A.; Aquila, B.M.; Boise, L.H.; Chiarparin, E.; Cidado, J.; Embrey, K.J.; Gangl, E.; Gibbons, F.D.; et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat. Commun. 2018, 9, 5341. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Boysen, J.C.; Chaffee, K.G.; Kabat, B.F.; Slager, S.L.; Parikh, S.A.; Secreto, C.R.; Call, T.; Shanafelt, T.D.; Leis, J.F.; et al. Chronic lymphocytic leukemia cells from ibrutinib treated patients are sensitive to Axl receptor tyrosine kinase inhibitor therapy. Oncotarget 2018, 9, 37173–37184. [Google Scholar] [CrossRef] [PubMed]
- Jaglowski, S.M.; Alinari, L.; Lapalombella, R.; Muthusamy, N.; Byrd, J.C. The clinical application of monoclonal antibodies in chronic lymphocytic leukemia. Blood 2010, 116, 3705–3714. [Google Scholar] [CrossRef] [Green Version]
- Maher, J.; Davies, E.T. Targeting cytotoxic T lymphocytes for cancer immunotherapy. Br. J. Cancer 2004, 91, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Coiffier, B.; Haioun, C.; Ketterer, N.; Engert, A.; Tilly, H.; Ma, D.; Johnson, P.; Lister, A.; Feuring-Buske, M.; Radford, J.A.; et al. Rituximab (anti-CD20 monoclonal antibody) for the treatment of patients with relapsing or refractory aggressive lymphoma: A multicenter phase II study. Blood 1998, 92, 1927–1932. [Google Scholar] [PubMed]
- Hallek, M.; Fischer, K.; Fingerle-Rowson, G.; Fink, A.M.; Busch, R.; Mayer, J.; Hensel, M.; Hopfinger, G.; Hess, G.; von Grünhagen, U.; et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: A randomised, open-label, phase 3 trial. Lancet 2010, 376, 1164–1174. [Google Scholar] [CrossRef]
- Fischer, K.; Al-Sawaf, O.; Bahlo, J.; Fink, A.-M.; Tandon, M.; Dixon, M.; Robrecht, S.; Warburton, S.; Humphrey, K.; Samoylova, O.; et al. Venetoclax and Obinutuzumab in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2019, 380, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Castillo, J.; Perez, K. The role of ofatumumab in the treatment of chronic lymphocytic leukemia resistant to previous therapies. J. Blood Med. 2010, 1, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Teeling, J.L.; Mackus, W.J.M.; Luus, J.J.; van den Brakel, J.H.N.; Beers, S.A.; French, R.R.; van Meerten, T.; Ebeling, S.; Vink, T.; Slootstra, J.W.; et al. The Biological Activity of Human CD20 Monoclonal Antibodies Is Linked to Unique Epitopes on CD20. J. Immunol. 2006, 177, 362–371. [Google Scholar] [CrossRef] [Green Version]
- Bologna, L.; Gotti, E.; Manganini, M.; Rambaldi, A.; Intermesoli, T.; Introna, M.; Golay, J. Mechanism of Action of Type II, Glycoengineered, Anti-CD20 Monoclonal Antibody GA101 in B-Chronic Lymphocytic Leukemia Whole Blood Assays in Comparison with Rituximab and Alemtuzumab. J. Immunol. 2011, 186, 3762–3769. [Google Scholar] [CrossRef] [Green Version]
- Coiffier, B.; Lepretre, S.; Pedersen, L.M.; Gadeberg, O.; Fredriksen, H.; van Oers, M.H.J.; Wooldridge, J.; Kloczko, J.; Holowiecki, J.; Hellmann, A.; et al. Safety and efficacy of ofatumumab, a fully human monoclonal anti-CD20 antibody, in patients with relapsed or refractory B-cell chronic lymphocytic leukemia: A phase 1-2 study. Blood 2008, 111, 1094–1100. [Google Scholar] [CrossRef] [Green Version]
- Hillmen, P.; Robak, T.; Janssens, A.; Govind Babu, K.; Kloczko, J.; Grosicki, S.; Doubek, M.; Panagiotidis, P.; Kimby, E.; Schuh, A.; et al. Chlorambucil plus ofatumumab versus chlorambucil alone in previously untreated patients with chronic lymphocytic leukaemia (COMPLEMENT 1): A randomised, multicentre, open-label phase 3 trial. Lancet 2015, 385, 1873–1883. [Google Scholar] [CrossRef]
- Laurenti, L.; Innocenti, I.; Autore, F.; Sica, S.; Efremov, D. New developments in the management of chronic lymphocytic leukemia: Role of ofatumumab. Onco Targets Ther. 2016, 9, 421–429. [Google Scholar] [CrossRef] [Green Version]
- Sawas, A.; Farber, C.M.; Schreeder, M.T.; Khalil, M.Y.; Mahadevan, D.; Deng, C.; Amengual, J.E.; Nikolinakos, P.G.; Kolesar, J.M.; Kuhn, J.G.; et al. A phase 1/2 trial of ublituximab, a novel anti-CD20 monoclonal antibody, in patients with B-cell non-Hodgkin lymphoma or chronic lymphocytic leukaemia previously exposed to rituximab. Br. J. Haematol. 2017, 177, 243–253. [Google Scholar] [CrossRef]
- Tedder, T.F.; Inaoki, M.; Sato, S. The CD19–CD21 Complex Regulates Signal Transduction Thresholds Governing Humoral Immunity and Autoimmunity. Immunity 1997, 6, 107–118. [Google Scholar] [CrossRef] [Green Version]
- Woyach, J.A.; Awan, F.; Flinn, I.W.; Berdeja, J.G.; Wiley, E.; Mansoor, S.; Huang, Y.; Lozanski, G.; Foster, P.A.; Byrd, J.C. A phase 1 trial of the Fc-engineered CD19 antibody XmAb5574 (MOR00208) demonstrates safety and preliminary efficacy in relapsed CLL. Blood 2014, 124, 3553–3560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaplon, H.; Reichert, J.M. Antibodies to watch in 2019. mAbs 2019, 11, 219–238. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Pepper, C.; Brennan, P.; Nagorsen, D.; Man, S.; Fegan, C. Blinatumomab induces autologous T-cell killing of chronic lymphocytic leukemia cells. Haematologica 2013, 98, 1930–1938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardarelli, P.M.; Rao-Naik, C.; Chen, S.; Huang, H.; Pham, A.; Moldovan-Loomis, M.-C.; Pan, C.; Preston, B.; Passmore, D.; Liu, J.; et al. A nonfucosylated human antibody to CD19 with potent B-cell depletive activity for therapy of B-cell malignancies. Cancer Immunol. Immunother. 2010, 59, 257–265. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.; Wang, Y.; Gallagher, S.; Mittereder, N.; Kuta, E.; Damschroder, M.; Woods, R.; Rowe, D.C.; Cheng, L.; Cook, K.; et al. B-Cell Depletion In Vitro and In Vivo with an Afucosylated Anti-CD19 Antibody. J. Pharmacol. Exp. Ther. 2010, 335, 213–222. [Google Scholar] [CrossRef] [Green Version]
- Awan, F.T.; Lapalombella, R.; Trotta, R.; Butchar, J.P.; Yu, B.; Benson, D.M.; Roda, J.M.; Cheney, C.; Mo, X.; Lehman, A.; et al. CD19 targeting of chronic lymphocytic leukemia with a novel Fc-domain–engineered monoclonal antibody. Blood 2010, 115, 1204–1213. [Google Scholar] [CrossRef] [Green Version]
- Robinson, H.R.; Qi, J.; Cook, E.M.; Nichols, C.; Dadashian, E.L.; Underbayev, C.; Herman, S.E.M.; Saba, N.S.; Keyvanfar, K.; Sun, C.; et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood 2018, 132, 521–532. [Google Scholar] [CrossRef]
- Golay, J.; Manganini, M.; Rambaldi, A.; Introna, M. Effect of alemtuzumab on neoplastic B cells. Haematologica 2004, 89, 1476–1483. [Google Scholar]
- Zhao, X.; Lapalombella, R.; Joshi, T.; Cheney, C.; Gowda, A.; Hayden-Ledbetter, M.S.; Baum, P.R.; Lin, T.S.; Jarjoura, D.; Lehman, A.; et al. Targeting CD37-positive lymphoid malignancies with a novel engineered small modular immunopharmaceutical. Blood 2007, 110, 2569–2577. [Google Scholar] [CrossRef] [Green Version]
- Robak, T.; Hellmann, A.; Kloczko, J.; Loscertales, J.; Lech-Maranda, E.; Pagel, J.M.; Mato, A.; Byrd, J.C.; Awan, F.T.; Hebart, H.; et al. Randomized phase 2 study of otlertuzumab and bendamustineversusbendamustine in patients with relapsed chronic lymphocytic leukaemia. Br. J. Haematol. 2017, 176, 618–628. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and Function of the ADP Ribosyl Cyclase/CD38 Gene Family in Physiology and Pathology. Physiol. Rev. 2008, 88, 841–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Funaro, A.; De Monte, L.B.; Dianzani, U.; Forni, M.; Malavasi, F. Human CD38 is associated to distinct molecules which mediate transmembrane signaling in different lineages. Eur. J. Immunol. 1993, 23, 2407–2411. [Google Scholar] [CrossRef] [PubMed]
- Dürig, J.; Naschar, M.; Schmücker, U.; Renzing-Köhler, K.; Hölter, T.; Hüttmann, A.; Dührsen, U. CD38 expression is an important prognostic marker in chronic lymphocytic leukaemia. Leukemia 2002, 16, 30–35. [Google Scholar] [CrossRef] [Green Version]
- Manna, A.; Aulakh, S.; Jani, P.; Ahmed, S.; Akhtar, S.; Coignet, M.; Heckman, M.; Meghji, Z.; Bhatia, K.; Sharma, A.; et al. Targeting CD38 Enhances the Antileukemic Activity of Ibrutinib in Chronic Lymphocytic Leukemia. Clin. Cancer Res. 2019, 25, 3974–3985. [Google Scholar] [CrossRef] [Green Version]
- Hulkkonen, J.; Vilpo, L.; Hurme, M.; Vilpo, J. Surface antigen expression in chronic lymphocytic leukemia: Clustering analysis, interrelationships and effects of chromosomal abnormalities. Leukemia 2002, 16, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C.; Kipps, T.J.; Flinn, I.W.; Cooper, M.; Odenike, O.; Bendiske, J.; Rediske, J.; Bilic, S.; Dey, J.; Baeck, J.; et al. Phase I study of the anti-CD40 humanized monoclonal antibody lucatumumab (HCD122) in relapsed chronic lymphocytic leukemia. Leuk Lymphoma 2012, 53, 2136–2142. [Google Scholar] [CrossRef] [Green Version]
- Schneider, P.; MacKay, F.; Steiner, V.; Hofmann, K.; Bodmer, J.-L.; Holler, N.; Ambrose, C.; Lawton, P.; Bixler, S.; Acha-Orbea, H.; et al. BAFF, a Novel Ligand of the Tumor Necrosis Factor Family, Stimulates B Cell Growth. J. Exp. Med. 1999, 189, 1747–1756. [Google Scholar] [CrossRef]
- Rodig, S.J.; Shahsafaei, A.; Li, B.; Mackay, C.R.; Dorfman, D.M. BAFF-R, the major B cell–activating factor receptor, is expressed on most mature B cells and B-cell lymphoproliferative disorders. Hum. Pathol. 2005, 36, 1113–1119. [Google Scholar] [CrossRef]
- Burger, J.A.; Ghia, P.; Rosenwald, A.; Caligaris-Cappio, F. The microenvironment in mature B-cell malignancies: A target for new treatment strategies. Blood 2009, 114, 3367–3375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McWilliams, E.M.; Lucas, C.R.; Chen, T.; Harrington, B.K.; Wasmuth, R.; Campbell, A.; Rogers, K.A.; Cheney, C.M.; Mo, X.; Andritsos, L.A.; et al. Anti–BAFF-R antibody VAY-736 demonstrates promising preclinical activity in CLL and enhances effectiveness of ibrutinib. Blood Adv. 2019, 3, 447–460. [Google Scholar] [CrossRef] [PubMed]
- Broome, H.E.; Elizabeth Broome, H.; Rassenti, L.Z.; Wang, H.-Y.; Meyer, L.M.; Kipps, T.J. ROR1 is expressed on hematogones (non-neoplastic human B-lymphocyte precursors) and a minority of precursor-B acute lymphoblastic leukemia. Leukemia Res. 2011, 35, 1390–1394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, B.; Ghia, E.M.; Chen, L.; Rassenti, L.Z.; DeBoever, C.; Widhopf, G.F.; Yu, J.; Neuberg, D.S.; Wierda, W.G.; Rai, K.R.; et al. High-level ROR1 associates with accelerated disease progression in chronic lymphocytic leukemia. Blood 2016, 128, 2931–2940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DaneshManesh, A.H.; Mikaelsson, E.; Jeddi-Tehrani, M.; Bayat, A.A.; Ghods, R.; Ostadkarampour, M.; Akhondi, M.; Lagercrantz, S.; Larsson, C.; Österborg, A.; et al. Ror1, a cell surface receptor tyrosine kinase is expressed in chronic lymphocytic leukemia and may serve as a putative target for therapy. Int. J. Cancer 2008, 123, 1190–1195. [Google Scholar] [CrossRef] [PubMed]
- Balatti, V.; Croce, C.M. MicroRNA dysregulation and multi-targeted therapy for cancer treatment. Adv. Biol. Regul. 2019, 2019, 100669. [Google Scholar] [CrossRef] [PubMed]
- Choi, M.Y.; Widhopf, G.F.; Ghia, E.M.; Kidwell, R.L.; Hasan, M.K.; Yu, J.; Rassenti, L.Z.; Chen, L.; Chen, Y.; Pittman, E.; et al. Phase I Trial: Cirmtuzumab Inhibits ROR1 Signaling and Stemness Signatures in Patients with Chronic Lymphocytic Leukemia. Cell Stem Cell 2018, 22, 951–959. [Google Scholar] [CrossRef] [Green Version]
- June, C.H.; O’Connor, R.S.; Kawalekar, O.U.; Ghassemi, S.; Milone, M.C. CAR T cell immunotherapy for human cancer. Science 2018, 359, 1361–1365. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Levine, B.L.; Kalos, M.; Bagg, A.; June, C.H. Chimeric Antigen Receptor–Modified T Cells in Chronic Lymphoid Leukemia. N. Engl. J. Med. 2011, 365, 725–733. [Google Scholar] [CrossRef] [Green Version]
- Porter, D.L.; Hwang, W.-T.; Frey, N.V.; Lacey, S.F.; Shaw, P.A.; Loren, A.W.; Bagg, A.; Marcucci, K.T.; Shen, A.; Gonzalez, V.; et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015. [Google Scholar] [CrossRef] [Green Version]
- van Bruggen, J.A.C.; Martens, A.W.J.; Fraietta, J.A.; Hofland, T.; Tonino, S.H.; Eldering, E.; Levin, M.-D.; Siska, P.J.; Endstra, S.; Rathmell, J.C.; et al. Chronic lymphocytic leukemia cells impair mitochondrial fitness in CD8 T cells and impede CAR T-cell efficacy. Blood 2019, 134, 44–58. [Google Scholar] [CrossRef]
- Maude, S.L.; Frey, N.; Shaw, P.A.; Aplenc, R.; Barrett, D.M.; Bunin, N.J.; Chew, A.; Gonzalez, V.E.; Zheng, Z.; Lacey, S.F.; et al. Chimeric Antigen Receptor T Cells for Sustained Remissions in Leukemia. N. Engl. J. Med. 2014, 371, 1507–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloess, S.; Kretschmer, A.; Stahl, L.; Fricke, S.; Koehl, U. CAR-Expressing Natural Killer Cells for Cancer Retargeting. Transfus. Med. Hemother. 2019, 46, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Hu, L.; Zhang, X.; Jiang, S.; Li, J.; Zhang, Z.; Wang, X. The Diverse Function of PD-1/PD-L Pathway Beyond Cancer. Front. Immunol. 2019, 10, 2298. [Google Scholar] [CrossRef] [PubMed]
- Greaves, P.; Gribben, J.G. The role of B7 family molecules in hematologic malignancy. Blood 2013, 121, 734–744. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.; Heo, Y.-S. Molecular Interactions of Antibody Drugs Targeting PD-1, PD-L1, and CTLA-4 in Immuno-Oncology. Molecules 2019, 24, 1190. [Google Scholar] [CrossRef] [Green Version]
- Ding, W.; LaPlant, B.R.; Call, T.G.; Parikh, S.A.; Leis, J.F.; He, R.; Shanafelt, T.D.; Sinha, S.; Le-Rademacher, J.; Feldman, A.L.; et al. Pembrolizumab in patients with CLL and Richter transformation or with relapsed CLL. Blood 2017, 129, 3419–3427. [Google Scholar] [CrossRef]
- Sagiv-Barfi, I.; Kohrt, H.E.K.; Czerwinski, D.K.; Ng, P.P.; Chang, B.Y.; Levy, R. Therapeutic antitumor immunity by checkpoint blockade is enhanced by ibrutinib, an inhibitor of both BTK and ITK. Proc. Natl. Acad. Sci. USA 2015, 112, E966–E972. [Google Scholar] [CrossRef] [Green Version]
- Younes, A.; Brody, J.; Carpio, C.; Lopez-Guillermo, A.; Ben-Yehuda, D.; Ferhanoglu, B.; Nagler, A.; Ozcan, M.; Avivi, I.; Bosch, F.; et al. Safety and activity of ibrutinib in combination with nivolumab in patients with relapsed non-Hodgkin lymphoma or chronic lymphocytic leukaemia: A phase 1/2a study. Lancet Haematol. 2019, 6, e67–e78. [Google Scholar] [CrossRef]
- Mittal, A.K.; Chaturvedi, N.K.; Rohlfsen, R.A.; Gupta, P.; Joshi, A.D.; Hegde, G.V.; Gregory Bociek, R.; Joshi, S.S. Role of CTLA4 in the Proliferation and Survival of Chronic Lymphocytic Leukemia. PLoS ONE 2013, 8, e70352. [Google Scholar] [CrossRef] [Green Version]
- Schubert, D.; Bode, C.; Kenefeck, R.; Hou, T.Z.; Wing, J.B.; Kennedy, A.; Bulashevska, A.; Petersen, B.-S.; Schäffer, A.A.; Grüning, B.A.; et al. Autosomal dominant immune dysregulation syndrome in humans with CTLA4 mutations. Nat. Med. 2014, 20, 1410–1416. [Google Scholar] [CrossRef]
- Syn, N.L.; Teng, M.W.L.; Mok, T.S.K.; Soo, R.A. De-novo and acquired resistance to immune checkpoint targeting. Lancet Oncol. 2017, 18, e731–e741. [Google Scholar] [CrossRef]
- Ciszak, L.; Frydecka, I.; Wolowiec, D.; Szteblich, A.; Kosmaczewska, A. Patients with chronic lymphocytic leukaemia (CLL) differ in the pattern of CTLA-4 expression on CLL cells: The possible implications for immunotherapy with CTLA-4 blocking antibody. Tumour. Biol. 2016, 37, 4143–4157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bashey, A.; Medina, B.; Corringham, S.; Pasek, M.; Carrier, E.; Vrooman, L.; Lowy, I.; Solomon, S.R.; Morris, L.E.; Kent Holland, H.; et al. CTLA4 blockade with ipilimumab to treat relapse of malignancy after allogeneic hematopoietic cell transplantation. Blood 2009, 113, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Russ, A.; Hua, A.B.; Montfort, W.R.; Rahman, B.; Riaz, I.B.; Khalid, M.U.; Carew, J.S.; Nawrocki, S.T.; Persky, D.; Anwer, F. Blocking “don’t eat me” signal of CD47-SIRPα in hematological malignancies, an in-depth review. Blood Rev. 2018, 32, 480–489. [Google Scholar] [CrossRef]
- Jaiswal, S.; Jamieson, C.H.M.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 Is Upregulated on Circulating Hematopoietic Stem Cells and Leukemia Cells to Avoid Phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [Green Version]
- Mateo, V.; Lagneaux, L.; Bron, D.; Biron, G.; Armant, M.; Delespesse, G.; Sarfati, M. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat. Med. 1999, 5, 1277–1284. [Google Scholar] [CrossRef]
- Sikic, B.I.; Narayanan, S.; Dimitrios Colevas, A.; Padda, S.K.; Fisher, G.A.; Supan, D.; Wakelee, H.A.; Aoki, R.; Pegram, M.D.; Villalobos, V.M.; et al. A first-in-human, first-in-class phase I trial of the anti-CD47 antibody Hu5F9-G4 in patients with advanced cancers. J. Clin. Oncol. 2019, 37, 946–953. [Google Scholar] [CrossRef]
- Andrechak, J.C.; Dooling, L.J.; Discher, D.E. The macrophage checkpoint CD47 : SIRPα for recognition of “self” cells: From clinical trials of blocking antibodies to mechanobiological fundamentals. Philos. Trans. R. Soc. Lond B Biol. Sci. 2019, 374, 20180217. [Google Scholar] [CrossRef]
- Kater, A.P.; Tonino, S.H.; Egle, A.; Ramsay, A.G. How does lenalidomide target the chronic lymphocytic leukemia microenvironment? Blood 2014, 124, 2184–2189. [Google Scholar] [CrossRef]
- Ramsay, A.G.; Clear, A.J.; Fatah, R.; Gribben, J.G. Multiple inhibitory ligands induce impaired T-cell immunologic synapse function in chronic lymphocytic leukemia that can be blocked with lenalidomide: Establishing a reversible immune evasion mechanism in human cancer. Blood 2012, 120, 1412–1421. [Google Scholar] [CrossRef]
- Giuliani, M.; Janji, B.; Berchem, G. Activation of NK cells and disruption of PD-L1/PD-1 axis: Two different ways for lenalidomide to block myeloma progression. Oncotarget 2017, 8, 24031–24044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byrd, J.C.; Ruppert, A.S.; Heerema, N.A.; Halvorson, A.E.; Hoke, E.; Smith, M.R.; Godwin, J.E.; Couban, S.; Fehniger, T.A.; Thirman, M.J.; et al. Lenalidomide consolidation benefits patients with CLL receiving chemoimmunotherapy: Results for CALGB 10404 (Alliance). Blood Adv. 2018, 2, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Egle, A.; Steurer, M.; Melchardt, T.; Weiss, L.; Gassner, F.J.; Zaborsky, N.; Geisberger, R.; Catakovic, K.; Hartmann, T.N.; Pleyer, L.; et al. Fludarabine and rituximab with escalating doses of lenalidomide followed by lenalidomide/rituximab maintenance in previously untreated chronic lymphocytic leukaemia (CLL): The REVLIRIT CLL-5 AGMT phase I/II study. Ann. Hematol. 2018, 97, 1825–1839. [Google Scholar] [CrossRef] [PubMed]
- Rasco, D.W.; Papadopoulos, K.P.; Pourdehnad, M.; Gandhi, A.K.; Hagner, P.R.; Li, Y.; Wei, X.; Chopra, R.; Hege, K.; DiMartino, J.; et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin. Cancer Res. 2019, 25, 90–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kremmidiotis, G.; Leske, A.F.; Lavranos, T.C.; Beaumont, D.; Gasic, J.; Hall, A.; O’Callaghan, M.; Matthews, C.A.; Flynn, B. BNC105: A Novel Tubulin Polymerization Inhibitor That Selectively Disrupts Tumor Vasculature and Displays Single-Agent Antitumor Efficacy. Mol. Cancer Ther. 2010, 9, 1562–1573. [Google Scholar] [CrossRef] [Green Version]
- Eichhorst, B.; Fink, A.-M.; Bahlo, J.; Busch, R.; Kovacs, G.; Maurer, C.; Lange, E.; Köppler, H.; Kiehl, M.; Sökler, M.; et al. First-line chemoimmunotherapy with bendamustine and rituximab versus fludarabine, cyclophosphamide, and rituximab in patients with advanced chronic lymphocytic leukaemia (CLL10): An international, open-label, randomised, phase 3, non-inferiority trial. Lancet Oncol. 2016, 17, 928–942. [Google Scholar] [CrossRef]
- Goede, V.; Fischer, K.; Busch, R.; Engelke, A.; Eichhorst, B.; Wendtner, C.M.; Chagorova, T.; de la Serna, J.; Dilhuydy, M.-S.; Illmer, T.; et al. Obinutuzumab plus Chlorambucil in Patients with CLL and Coexisting Conditions. N. Engl. J. Med. 2014, 370, 1101–1110. [Google Scholar] [CrossRef] [Green Version]
- Byrd, J.C. Randomized phase 2 study of fludarabine with concurrent versus sequential treatment with rituximab in symptomatic, untreated patients with B-cell chronic lymphocytic leukemia: Results from Cancer and Leukemia Group B 9712 (CALGB 9712). Blood 2003, 101, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Samoilova, O.; Novak, J.; Ben-Yehuda, D.; et al. Ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab in first-line treatment of chronic lymphocytic leukaemia (iLLUMINATE): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019, 20, 43–56. [Google Scholar] [CrossRef]
- Lamanna, N.; Kalaycio, M.; Maslak, P.; Jurcic, J.G.; Heaney, M.; Brentjens, R.; Zelenetz, A.D.; Horgan, D.; Gencarelli, A.; Panageas, K.S.; et al. Pentostatin, Cyclophosphamide, and Rituximab Is an Active, Well-Tolerated Regimen for Patients With Previously Treated Chronic Lymphocytic Leukemia. J. Clin. Oncol. 2006, 24, 1575–1581. [Google Scholar] [CrossRef] [Green Version]
- Hillmen, P.; Gribben, J.G.; Follows, G.A.; Milligan, D.; Sayala, H.A.; Moreton, P.; Oscier, D.G.; Dearden, C.E.; Kennedy, D.B.; Pettitt, A.R.; et al. Rituximab Plus Chlorambucil As First-Line Treatment for Chronic Lymphocytic Leukemia: Final Analysis of an Open-Label Phase II Study. J. Clin. Oncol. 2014, 32, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Hill, S.L.; Davies, A. Subcutaneous rituximab with recombinant human hyaluronidase in the treatment of non-Hodgkin lymphoma and chronic lymphocytic leukemia. Future Oncol. 2018, 14, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Dürig, J.; Dührsen, U.; Klein-Hitpass, L.; Worm, J.; Rode Hansen, J.B.; Ørum, H.; Wissenbach, M. The novel antisense Bcl-2 inhibitor SPC2996 causes rapid leukemic cell clearance and immune activation in chronic lymphocytic leukemia. Leukemia 2011, 25, 638–647. [Google Scholar] [CrossRef] [PubMed]
- Chanan-Khan, A.A.; Zaritskey, A.; Egyed, M.; Vokurka, S.; Semochkin, S.; Schuh, A.; Kassis, J.; Simpson, D.; Zhang, J.; Purse, B.; et al. Lenalidomide maintenance therapy in previously treated chronic lymphocytic leukaemia (CONTINUUM): A randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. Haematol. 2017, 4, e534–e543. [Google Scholar] [CrossRef]
- Kessler, T.; Koschmieder, S.; Schliemann, C.; Crysandt, M.; Mikesch, J.-H.; von Stillfried, S.; Stelljes, M.; Pohlen, M.; Lenz, G.; Kirsch, A.; et al. Phase II clinical trial of pazopanib in patients with acute myeloid leukemia (AML), relapsed or refractory or at initial diagnosis without an intensive treatment option (PazoAML). Ann. Hematol. 2019, 98, 1393–1401. [Google Scholar] [CrossRef]
- Forero, A.; Hamadani, M.; Kipps, T.; Fanale, M.; Cuneo, A.; Perez de Oteyza, J.; Gladstone, D.; Andre, M.; Bellam, N.; Goswami, T.; et al. Safety and disease response to MEDI-551, an anti-CD19 antibody, in chronic lymphocytic leukemia patients previously treated with rituximab. J. Immunother. Cancer 2013, 1, 43. [Google Scholar] [CrossRef] [Green Version]
- Fraietta, J.A.; Beckwith, K.A.; Patel, P.R.; Ruella, M.; Zheng, Z.; Barrett, D.M.; Lacey, S.F.; Melenhorst, J.J.; McGettigan, S.E.; Cook, D.R.; et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood 2016, 127, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Cheson, B.D.; Horning, S.J.; Coiffier, B.; Shipp, M.A.; Fisher, R.I.; Connors, J.M.; Andrew Lister, T.; Vose, J.; Grillo-López, A.; Hagenbeek, A.; et al. Report of an International Workshop to Standardize Response Criteria for Non-Hodgkin’s Lymphomas. J. Clin. Oncol. 1999, 17, 1244. [Google Scholar] [CrossRef]
- Advani, R.; Flinn, I.; Popplewell, L.; Forero, A.; Bartlett, N.L.; Ghosh, N.; Kline, J.; Roschewski, M.; LaCasce, A.; Collins, G.P.; et al. CD47 Blockade by Hu5F9-G4 and Rituximab in Non-Hodgkin’s Lymphoma. N. Engl. J. Med. 2018, 379, 1711–1721. [Google Scholar] [CrossRef]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2017, 35, 180–188. [Google Scholar] [CrossRef]
- Kasinski, A.L.; Slack, F.J. miRNA-34 Prevents Cancer Initiation and Progression in a Therapeutically Resistant K-ras and p53-Induced Mouse Model of Lung Adenocarcinoma. Cancer Res. 2012, 72, 5576–5587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zauli, G.; Voltan, R.; di Iasio, M.G.; Bosco, R.; Melloni, E.; Sana, M.E.; Secchiero, P. miR-34a Induces the Downregulation of Both E2F1 and B-Myb Oncogenes in Leukemic Cells. Clin. Cancer Res. 2011, 17, 2712–2724. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hart, M.; Walch-Rückheim, B.; Friedmann, K.S.; Rheinheimer, S.; Tänzer, T.; Glombitza, B.; Sester, M.; Lenhof, H.-P.; Hoth, M.; Schwarz, E.C.; et al. miR-34a: A new player in the regulation of T cell function by modulation of NF-κB signaling. Cell Death Dis. 2019, 10, 46. [Google Scholar] [CrossRef] [Green Version]
- Hart, M.; Walch-Rückheim, B.; Krammes, L.; Kehl, T.; Rheinheimer, S.; Tänzer, T.; Glombitza, B.; Sester, M.; Lenhof, H.-P.; Keller, A.; et al. miR-34a as hub of T cell regulation networks. J. Immunother. Cancer 2019, 7, 187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Li, J.; Dong, K.; Lin, F.; Long, M.; Ouyang, Y.; Wei, J.; Chen, X.; Weng, Y.; He, T.; et al. Tumor suppressor miR-34a targets PD-L1 and functions as a potential immunotherapeutic target in acute myeloid leukemia. Cell Signal. 2015, 27, 443–452. [Google Scholar] [CrossRef]
- Yang, N.; Zhu, S.; Lv, X.; Qiao, Y.; Liu, Y.-J.; Chen, J. MicroRNAs: Pleiotropic Regulators in the Tumor Microenvironment. Front. Immunol. 2018, 9, 2491. [Google Scholar] [CrossRef] [Green Version]
- Bresin, A.; Callegari, E.; D’Abundo, L.; Cattani, C.; Bassi, C.; Zagatti, B.; Grazia Narducci, M.; Caprini, E.; Pekarsky, Y.; Croce, C.M.; et al. miR-181b as a therapeutic agent for chronic lymphocytic leukemia in the Eµ-TCL1 mouse model. Oncotarget 2015, 6, 19807–19818. [Google Scholar] [CrossRef]
- Gallant-Behm, C.L.; Piper, J.; Lynch, J.M.; Seto, A.G.; Hong, S.J.; Mustoe, T.A.; Maari, C.; Pestano, L.A.; Dalby, C.M.; Jackson, A.L.; et al. A MicroRNA-29 Mimic (Remlarsen) Represses Extracellular Matrix Expression and Fibroplasia in the Skin. J. Investig. Dermatol. 2019, 139, 1073–1081. [Google Scholar] [CrossRef]
- Misso, G.; Zarone, M.R.; Lombardi, A.; Grimaldi, A.; Cossu, A.M.; Ferri, C.; Russo, M.; Vuoso, D.C.; Luce, A.; Kawasaki, H.; et al. miR-125b Upregulates miR-34a and Sequentially Activates Stress Adaption and Cell Death Mechanisms in Multiple Myeloma. Mol. Ther. Nucleic Acids 2019, 16, 391–406. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Liu, Y.; Zhang, C.; Duan, C. Multiple Roles of Exosomal Long Noncoding RNAs in Cancers. Biomed Res. Int. 2019, 2019, 1460572. [Google Scholar] [CrossRef]
- Crompot, E.; Van Damme, M.; Pieters, K.; Vermeersch, M.; Perez-Morga, D.; Mineur, P.; Maerevoet, M.; Meuleman, N.; Bron, D.; Lagneaux, L.; et al. Extracellular vesicles of bone marrow stromal cells rescue chronic lymphocytic leukemia B cells from apoptosis, enhance their migration and induce gene expression modifications. Haematologica 2017, 102, 1594–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armstrong, J.P.K.; Stevens, M.M. Strategic design of extracellular vesicle drug delivery systems. Adv. Drug Deliv. Rev. 2018, 130, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Li, I.; Nabet, B.Y. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol. Cancer 2019, 18, 32. [Google Scholar] [CrossRef] [PubMed]
- Steinbichler, T.B.; Dudás, J.; Skvortsov, S.; Ganswindt, U.; Riechelmann, H.; Skvortsova, I.-I. Therapy resistance mediated by exosomes. Mol. Cancer 2019, 18, 58. [Google Scholar] [CrossRef]
- Fuster-Matanzo, A.; Gessler, F.; Leonardi, T.; Iraci, N.; Pluchino, S. Acellular approaches for regenerative medicine: On the verge of clinical trials with extracellular membrane vesicles? Stem. Cell Res. Ther. 2015, 6, 227. [Google Scholar] [CrossRef] [Green Version]
- Yamada, N.O.; Heishima, K.; Akao, Y.; Senda, T. Extracellular Vesicles Containing MicroRNA-92a-3p Facilitate Partial Endothelial-Mesenchymal Transition and Angiogenesis in Endothelial Cells. Int. J. Mol. Sci. 2019, 20, 4406. [Google Scholar] [CrossRef] [Green Version]
- Palma, L.M.A.; Gehrke, I.; Kreuzer, K.-A. Angiogenic factors in chronic lymphocytic leukaemia (CLL): Where do we stand? Crit. Rev. Oncol. Hematol. 2015, 93, 225–236. [Google Scholar] [CrossRef]
- Brander, D.; Rizzieri, D.; Gockerman, J.; Diehl, L.; Shea, T.C.; Decastro, C.; Moore, J.O.; Beaven, A. Phase II open label study of the oral vascular endothelial growth factor-receptor inhibitor PTK787/ZK222584 (vatalanib) in adult patients with refractory or relapsed diffuse large B-cell lymphoma. Leuk Lymphoma 2013, 54, 2627–2630. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
| Standard FDA Approved Approaches for CLL Treatment | Type of Therapy/ Clinical Trial Code | Reference |
|---|---|---|
| Acalabrutinib | Targeted therapy (BTK) NCT02029443 | [94] |
| BR (bendamustine, rituximab) | Chemo-Immunotherapy NCT02381899 | [196] |
| CG (Chlorambucil, obinutuzumab) | Chemo-Immunotherapy Approved for first-line therapy NCT01010061 | [197] |
| FCR (fludarabine, cyclophosphamide, rituximab) | Chemo-Immunotherapy NCT00090051 | [132] |
| FR (fludarabine, rituximab) | Chemo-Immunotherapy Approved for first-line therapy NCT00860457 | [198] |
| Ibrutinib | Targeted therapy (BTK) Approved for first-line therapy NCT02801578 | [91] |
| Ibrutinib/obinutuzumab | Chemo-Immunotherapy NCT02537613 | [199] |
| Ofatumumab/chlorambucil | Chemo-Immunotherapy NCT00748189 | [138] |
| PCR (pentostatin, cyclophosphamide, and rituximab) | Chemo-Immunotherapy Approved for first-line therapy NCT00049413 | [200] |
| Rituximab/chlorambucil | Chemo-Immunotherapy Approved for first-line therapy NCT00532129 | [201] |
| Rituximab/human hyaluronidase | Chemo-Immunotherapy NCT03467867 | [202] |
| Venetoclax | Targeted therapy (Bcl2) NCT01328626 | [118] |
| Clinical Trial ID | Treatment | Phase | Status | Date of Start | Reference for Results |
|---|---|---|---|---|---|
| NCT00060372 | Ipilimumab | I | Completed | 04/2003 | |
| NCT00108108 | Lucatumumab | I/II | Terminated | 04/2005 | [157] |
| NCT00285103 | SPC2996 | I/II | Completed | 06/2005 | [203] |
| NCT00511043 | PTK787 (vatalanib) | II | Terminated | 11/2005 | |
| NCT00602459 | Lenalidomide combined with fludarabine and rituximab | II | Completed | 01/2008 | [192] |
| NCT00738829 | Lenalidomide combined with fludarabine and rituximab (dose escalation) | I/II | Completed | 10/2008 | [193] |
| NCT00774345 | Lenalidomide as maintenance therapy for CLL | III | Active | 01/2009 | [204] |
| NCT01029366 | Autologous CART19 | I | Completed | 03/2010 | [171] |
| NCT01161511 | XmAb5574 | I | Completed | 10/2010 | [142] |
| NCT01188681 | Otlertuzumab in combination with bendamustine | I/II | Completed | 10/2010 | [151] |
| NCT01361334 | Pazopanib | II | Completed | 06/2011 | [205] |
| NCT01400685 | Lenalidomide as first line with bendamustine and rituximab | I | Completed | 07/2011 | |
| NCT01466153 | Inebilizumab in combination with bendamustine or rituximab | II | Completed | 02/2012 | [206] |
| NCT01569295 | Idelalisib in Combination With Bendamustine and Rituximab | III | Completed | 06/2012 | [106] |
| NCT01699152 | TG02 | I | Completed | 09/2012 | |
| NCT01747486 | Autologous CART19 (dose optimization) | II | Completed | 02/2013 | [207] |
| NCT01829971 | miR-RX34 liposomal injection | I | Terminated | 04/2013 | |
| NCT02005289 | MOR00208 in combination with lenalidomide | II | Active | 12/2013 | |
| NCT02137889 | Ianalumab | I | Terminated | 07/2012 | |
| NCT02222688 | Cirmtuzumab | I | Completed | 10/2014 | |
| NCT02242942 | Obinutuzumab in combination with venetoclax, and obinutuzumab and chlorambucil | III | Active | 12/2014 | [133] |
| NCT02254772 | Ipilimumab with SD-101 and radiation therapy | I/II | Completed | 09/2014 | [208] |
| NCT02329847 | Nivolumab with ibrutinib | I/II | Active | 03/2015 | [178] |
| NCT02332980 | Pembrolizumab in combination with idelalisib or ibrutinib | II | Recruiting | 02/2015 | [176] |
| NCT02406742 | CC-122 combined with ibrutinib and obinutuzumab | I/II | Active | 09/2015 | |
| NCT02420912 | Nivolumab and ibrutinib | II | Active | 06/2015 | |
| NCT02500407 | BTCT4465A (Mosunetuzumab) as a single agent and combined with Atezolizumab | I | Recruiting | ||
| NCT02535286 | Ublituximab in combination with umbralisib | I/II | Recruiting | 09/2015 | |
| NCT02580552 | MRG-106 | I | Recruiting | 02/2016 | |
| NCT02640209 | CART19 with ibrutinib | --- | Active | 12/2015 | |
| NCT02706392 | ROR1-specific CART-cells | I | Recruiting | 03/2016 | |
| NTC02733042 | Durvalumab in combinations with lenalidomide, rituximab, ibrutinib, and bendamustine | I/II | Active | 03/2016 | |
| NCT02742090 | Duvelisib | II | Active | 04/2016 | [109] |
| NCT02846623 | Atezolizumab in combination with obinutuzumab and venetoclax | II | Recruiting | 01/2017 | |
| NCT02910583 | Ibrutinib plus venetoclax | II | Active | 10/2016 | |
| NCT02953509 | Hu5F9-G4 in Combination with Rituximab | I/II | Recruiting | 11/2016 | [209] |
| NCT02968563 | Tirabrutinib and Idelalisib with and Without Obinutuzumab | II | Active | 12/2016 | |
| NCT03037645 | Vecabrutinib | I/II | Recruiting | 04/2017 | |
| NCT03056339 | CAR-NK | I/II | Recruiting | 06/2017 | |
| NCT03088878 | Cirmtuzumab in combination with ibrutinib | I/II | Recruiting | 01/2018 | |
| NCT03162536 | ARQ-531 | I/II | Recruiting | 07/2017 | |
| NCT03218683 | AZD5991 with or without venetoclax | I | Recruiting | 10/2017 | [127] |
| NCT03336333 | BGB-3111 (zanubrutinib)with Bendamustine plus Rituximab | III | Recruiting | 11/2017 | |
| NCT03400176 | Ianalumab with ibrutinib | I | Recruiting | 04/2018 | |
| NCT03447808 | Daratumumab with ibrutinib | I | Recruiting | 07/2018 | |
| NCT03454165 | BNC105P in combination with ibrutinib | I | Recruiting | 03/2018 | |
| NCT03572634 | TP-0903 | I/II | Recruiting | 06/2019 | |
| NCT03734016 | Zanubrutinib (BGB-3111) versus Ibrutinib | III | Active | 11/2018 | |
| NCT03739554 | CYC065 and venetoclax | I | Recruiting | 01/2019 | |
| NCT03740529 | LOXO-305 | I/II | Recruiting | 11/2018 | |
| NCT03823365 | Blinatumomab | I | Recruiting | 12/2018 | |
| NCT03824483 | Zanubrutinib, obinutuzumab, and venetoclax | II | Recruiting | 02/2019 | |
| NCT04116437 | Zanubrutinib (BGB-3111) | II | Recruiting | 10/2019 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pepe, F.; Balatti, V. Role of Non-Coding RNAs in the Development of Targeted Therapy and Immunotherapy Approaches for Chronic Lymphocytic Leukemia. J. Clin. Med. 2020, 9, 593. https://doi.org/10.3390/jcm9020593
Pepe F, Balatti V. Role of Non-Coding RNAs in the Development of Targeted Therapy and Immunotherapy Approaches for Chronic Lymphocytic Leukemia. Journal of Clinical Medicine. 2020; 9(2):593. https://doi.org/10.3390/jcm9020593
Chicago/Turabian StylePepe, Felice, and Veronica Balatti. 2020. "Role of Non-Coding RNAs in the Development of Targeted Therapy and Immunotherapy Approaches for Chronic Lymphocytic Leukemia" Journal of Clinical Medicine 9, no. 2: 593. https://doi.org/10.3390/jcm9020593
APA StylePepe, F., & Balatti, V. (2020). Role of Non-Coding RNAs in the Development of Targeted Therapy and Immunotherapy Approaches for Chronic Lymphocytic Leukemia. Journal of Clinical Medicine, 9(2), 593. https://doi.org/10.3390/jcm9020593