Immunomodulation of the Natural Killer Cell Phenotype and Response during HCV Infection

 , ,
, ,
Abstract
:1. Introduction
2. Natural Killer Cells
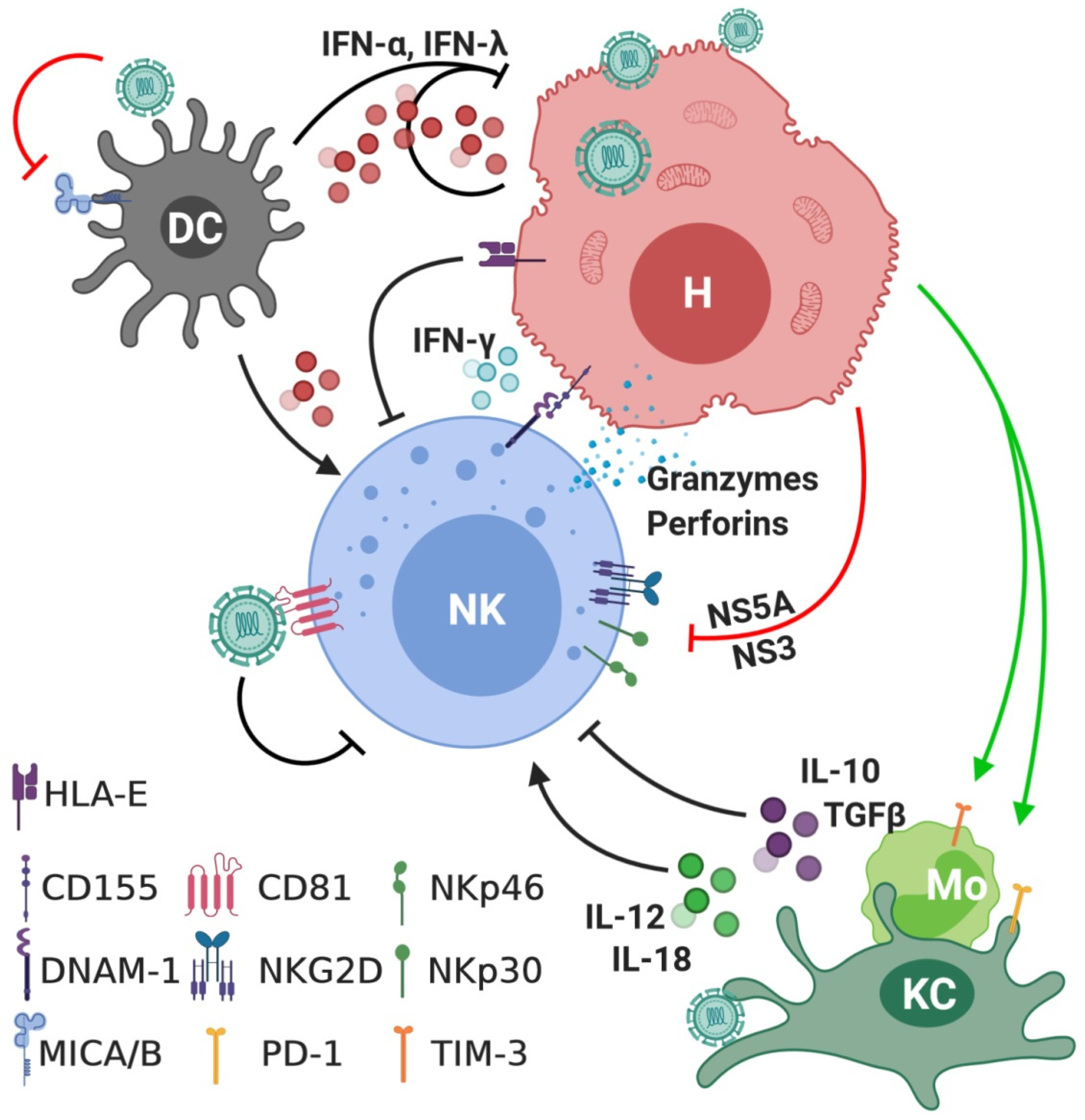
3. Interactions between NK Cells and HCV
4. The Role of NK Cells in Acute Hepatitis C Infection
5. The Role of NK Cells in Chronic Hepatitis C (CHC) Infection
6. NK Cell Memory in HCV
7. The Effect of Anti-HCV Therapy on NK Cell Phenotype and Activity
7.1. Effect of Pegylated IFN-α/Ribavirin-Based Therapy on NK Cells
7.2. Effect of Direct-Acting Antivirals-Based Therapy on NK Cells
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shepard, C.W.; Finelli, L.; Alter, M.J. Global epidemiology of hepatitis C virus infection. Lancet Infect. Dis. 2005, 5, 558–567. [Google Scholar] [CrossRef]
- Lingala, S.; Ghany, M.G. Natural History of Hepatitis C. Gastroenterol. Clin. N. Am. 2015, 44, 717–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Averhoff, F.M.; Glass, N.; Holtzman, D. Global burden of hepatitis C: Considerations for healthcare providers in the United States. Clin. Infect. Dis. 2012, 55 (Suppl. 1), S10–S15. [Google Scholar] [CrossRef]
- Gower, E.; Estes, C.; Blach, S.; Razavi-Shearer, K.; Razavi, H. Global epidemiology and genotype distribution of the hepatitis C virus infection. J. Hepatol. 2014, 61, S45–S57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petruzziello, A.; Marigliano, S.; Loquercio, G.; Cozzolino, A.; Cacciapuoti, C. Global epidemiology of hepatitis C virus infection: An up-date of the distribution and circulation of hepatitis C virus genotypes. World J. Gastroenterol. 2016, 22, 7824–7840. [Google Scholar] [CrossRef]
- Mandal, A.; Viswanathan, C. Natural killer cells: In health and disease. Hematol. Oncol. Stem. Cell Ther. 2015, 8, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Long, E.O.; Kim, H.S.; Liu, D.; Peterson, M.E.; Rajagopalan, S. Controlling natural killer cell responses: Integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013, 31, 227–258. [Google Scholar] [CrossRef] [Green Version]
- Horowitz, A.; Strauss-Albee, D.M.; Leipold, M.; Kubo, J.; Nemat-Gorgani, N.; Dogan, O.C.; Dekker, C.L.; Mackey, S.; Maecker, H.; Swan, G.E.; et al. Genetic and Environmental Determinants of Human NK Cell Diversity Revealed by Mass Cytometry. Sci. Transl. Med. 2013, 5, 208ra145. [Google Scholar] [CrossRef] [Green Version]
- Prager, I.; Watzl, C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J. Leukoc. Biol. 2019, 105, 1319–1329. [Google Scholar] [CrossRef]
- Cosman, D.; Mullberg, J.; Sutherland, C.L.; Chin, W.; Armitage, R.; Fanslow, W.; Kubin, M.; Chalupny, N.J. ULBPs, novel MHC class I-related molecules, bind to CMV glycoprotein UL16 and stimulate NK cytotoxicity through the NKG2D receptor. Immunity 2001, 14, 123–133. [Google Scholar] [CrossRef]
- Bauer, S.; Groh, V.; Wu, J.; Steinle, A.; Phillips, J.H.; Lanier, L.L.; Spies, T. Activation of NK cells and T cells by NKG2D, a receptor for stress-inducible MICA. Science 1999, 285, 727–729. [Google Scholar] [CrossRef]
- Braud, V.M.; Allan, D.S.; O’Callaghan, C.A.; Soderstrom, K.; D’Andrea, A.; Ogg, G.S.; Lazetic, S.; Young, N.T.; Bell, J.I.; Phillips, J.H.; et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature 1998, 391, 795–799. [Google Scholar] [CrossRef]
- Rajagopalan, S.; Long, E.O. A human histocompatibility leukocyte antigen (HLA)-G-specific receptor expressed on all natural killer cells. J. Exp. Med. 1999, 189, 1093–1099. [Google Scholar] [CrossRef] [Green Version]
- Cognet, C.; Farnarier, C.; Gauthier, L.; Frassati, C.; Andre, P.; Magerus-Chatinet, A.; Anfossi, N.; Rieux-Laucat, F.; Vivier, E.; Schleinitz, N. Expression of the HLA-C2-specific activating killer-cell Ig-like receptor KIR2DS1 on NK and T cells. Clin. Immunol. 2010, 135, 26–32. [Google Scholar] [CrossRef]
- Liu, J.X.; Xiao, Z.W.; Ko, H.L.; Shen, M.X.; Ren, E.C. Activating killer cell immunoglobulin-like receptor 2DS2 binds to HLA-A*11. Proc. Natl. Acad. Sci. USA 2014, 111, 2662–2667. [Google Scholar] [CrossRef] [Green Version]
- Sim, M.J.W.; Rajagopalan, S.; Altmann, D.M.; Boyton, R.J.; Sun, P.D.; Long, E.O. Human NK cell receptor KIR2DS4 detects a conserved bacterial epitope presented by HLA-C. Proc. Natl. Acad. Sci. USA 2019, 116, 12964–12973. [Google Scholar] [CrossRef] [Green Version]
- Graef, T.; Moesta, A.K.; Norman, P.J.; Abi-Rached, L.; Vago, L.; Aguilar, A.M.O.; Gleimer, M.; Hammond, J.A.; Guethlein, L.A.; Bushnell, D.A.; et al. KIR2DS4 is a product of gene conversion with KIR3DL2 that introduced specificity for HLA-A*11 while diminishing avidity for HLA-C. J. Exp. Med. 2009, 206, 2557–2572. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Beltran, W.F.; Holzemer, A.; Martrus, G.; Chung, A.W.; Pacheco, Y.; Simoneau, C.R.; Rucevic, M.; Lamothe-Molina, P.A.; Pertel, T.; Kim, T.E.; et al. Open conformers of HLA-F are high-affinity ligands of the activating NK-cell receptor KIR3DS1. Nat. Immunol. 2016, 17, 1067–1074. [Google Scholar] [CrossRef]
- Brandt, C.S.; Baratin, M.; Yi, E.C.; Kennedy, J.; Gao, Z.; Fox, B.; Haldeman, B.; Ostrander, C.D.; Kaifu, T.; Chabannon, C.; et al. The B7 family member B7-H6 is a tumor cell ligand for the activating natural killer cell receptor NKp30 in humans. J. Exp. Med. 2009, 206, 1495–1503. [Google Scholar] [CrossRef] [Green Version]
- Arnon, T.I.; Achdout, H.; Levi, O.; Markel, G.; Saleh, N.; Katz, G.; Gazit, R.; Gonen-Gross, T.; Hanna, J.; Nahari, E.; et al. Inhibition of the NKp30 activating receptor by pp65 of human cytomegalovirus. Nat. Immunol. 2005, 6, 515–523. [Google Scholar] [CrossRef]
- Hecht, M.L.; Rosental, B.; Horlacher, T.; Hershkovitz, O.; De Paz, J.L.; Noti, C.; Schauer, S.; Porgador, A.; Seeberger, P.H. Natural Cytotoxicity Receptors NKp30, NKp44 and NKp46 Bind to Different Heparan Sulfate/Heparin Sequences. J. Proteome Res. 2009, 8, 712–720. [Google Scholar] [CrossRef] [PubMed]
- Von Strandmann, E.P.; Simhadri, V.R.; von Tresckow, B.; Sasse, S.; Reiners, K.S.; Hansen, H.P.; Rothe, A.; Boll, B.; Simhadri, V.L.; Borchmann, P.; et al. Human leukocyte antigen-B-associated transcript 3 is released from tumor cells and engages the NKp30 receptor on natural killer cells. Immunity 2007, 27, 965–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandelboim, O.; Lieberman, N.; Lev, M.; Paul, L.; Arnon, T.I.; Bushkin, Y.; Davis, D.M.; Strominger, J.L.; Yewdell, J.W.; Porgador, A. Recognition of haemagglutinins on virus-infected cells by NKp46 activates lysis by human NK cells. Nature 2001, 409, 1055–1060. [Google Scholar] [CrossRef] [PubMed]
- Narni-Mancinelli, E.; Gauthier, L.; Baratin, M.; Guia, S.; Fenis, A.; Deghmane, A.E.; Rossi, B.; Fourquet, P.; Escaliere, B.; Kerdiles, Y.M.; et al. Complement factor P is a ligand for the natural killer cell-activating receptor NKp46. Sci. Immunol. 2017, 2, eaam9628. [Google Scholar] [CrossRef] [Green Version]
- Jarahian, M.; Watzl, C.; Fournier, P.; Arnold, A.; Djandji, D.; Zahedi, S.; Cerwenka, A.; Paschen, A.; Schirrmacher, V.; Momburg, F. Activation of Natural Killer Cells by Newcastle Disease Virus Hemagglutinin-Neuraminidase. J. Virol. 2009, 83, 8108–8121. [Google Scholar] [CrossRef] [Green Version]
- Rosental, B.; Hadad, U.; Brusilovsky, M.; Campbell, K.S.; Porgador, A. A novel mechanism for cancer cells to evade immune attack by NK cells The interaction between NKp44 and proliferating cell nuclear antigen. Oncoimmunology 2012, 1, 572–574. [Google Scholar] [CrossRef] [Green Version]
- Arnon, T.I.; Lev, M.; Katz, G.; Chernobrov, Y.; Porgador, A.; Mandelboim, O. Recognition of viral hemagglutinins by NKp44 but not by NKp30. Eur. J. Immunol. 2001, 31, 2680–2689. [Google Scholar] [CrossRef]
- Bottino, C.; Castriconi, R.; Pende, D.; Rivera, P.; Nanni, M.; Carnemolla, B.; Cantoni, C.; Grassi, J.; Marcenaro, S.; Reymond, N.; et al. Identification of PVR (CD155) and nectin-2 (CD112) as cell surface ligands for the human DNAM-1 (CD226) activating molecule. J. Exp. Med. 2003, 198, 557–567. [Google Scholar] [CrossRef] [Green Version]
- Snyder, G.A.; Brooks, A.G.; Sun, P.D. Crystal structure of the HLA-Cw3 allotype-specific killer cell inhibitory receptor KIR2DL2. Proc. Natl. Acad. Sci. USA 1999, 96, 3864–3869. [Google Scholar] [CrossRef] [Green Version]
- Mandelboim, O.; Reyburn, H.T.; ValesGomez, M.; Pazmany, L.; Colonna, M.; Borsellino, G.; Strominger, J.L. Protection from lysis by natural killer cells of group 1 and 2 specificity is mediated by residue 80 in human histocompatibility leukocyte antigen C alleles and also occurs with empty major histocompatibility complex molecules. J. Exp. Med. 1996, 184, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Brusilovsky, M.; Cordoba, M.; Rosental, B.; Hershkovitz, O.; Andrake, M.D.; Pecherskaya, A.; Einarson, M.B.; Zhou, Y.; Braiman, A.; Campbell, K.S.; et al. Genome-Wide siRNA Screen Reveals a New Cellular Partner of NK Cell Receptor KIR2DL4: Heparan Sulfate Directly Modulates KIR2DL4-Mediated Responses. J. Immunol. 2013, 191, 5256–5267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumperz, J.E.; Litwin, V.; Phillips, J.H.; Lanier, L.L.; Parham, P. The Bw4 Public Epitope of Hla-B Molecules Confers Reactivity with Natural-Killer-Cell Clones That Express Nkb1, a Putative Hla Receptor. J. Exp. Med. 1995, 181, 1133–1144. [Google Scholar] [CrossRef] [PubMed]
- Hansasuta, P.; Dong, T.; Thananchai, H.; Weekes, M.; Willberg, C.; Aldemir, H.; Rowland-Jones, S.; Braud, V.M. Recognition of HLA-A3 and HLA-A11 by KIR3DL2 is peptide-specific. Eur. J. Immunol. 2004, 34, 1673–1679. [Google Scholar] [CrossRef] [PubMed]
- Arnold, V.; Cummings, J.S.; Moreno-Nieves, U.Y.; Didier, C.; Gilbert, A.; Barre-Sinoussi, F.; Scott-Algara, D. S100A9 protein is a novel ligand for the CD85j receptor and its interaction is implicated in the control of HIV-1 replication by NK cells. Retrovirology 2013, 10, 122. [Google Scholar] [CrossRef] [Green Version]
- Chapman, T.L.; Heikema, A.P.; West, A.P.; Bjorkman, P.J. Crystal structure and ligand binding properties of the D1D2 region of the inhibitory receptor LIR-1 (ILT2). Immunity 2000, 13, 727–736. [Google Scholar] [CrossRef] [Green Version]
- Colonna, M.; Navarro, F.; Bellon, T.; Llano, M.; Garcia, P.; Samaridis, J.; Angman, L.; Cella, M.; LopezBotet, M. A common inhibitory receptor for major histocompatibility complex class I molecules on human lymphoid and myelomonocytic cells. J. Exp. Med. 1997, 186, 1809–1818. [Google Scholar] [CrossRef] [Green Version]
- Latchman, Y.; McKay, P.F.; Reiser, H. Cutting edge: Identification of the 2B4 molecule as a counter-receptor for CD48. J. Immunol. 1998, 161, 5809–5812. [Google Scholar]
- Aldemir, H.; Prod’homme, V.; Dumaurier, M.J.; Retiere, C.; Poupon, G.; Cazareth, J.; Bih, F.; Braud, V.M. Cutting edge: Lectin-like transcript 1 is a ligand for the CD161 receptor. J. Immunol. 2005, 175, 7791–7795. [Google Scholar] [CrossRef] [Green Version]
- Glassner, A.; Eisenhardt, M.; Kramer, B.; Korner, C.; Coenen, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL- and NKG2D-dependent manner. Lab. Investig. 2012, 92, 967–977. [Google Scholar] [CrossRef] [Green Version]
- Ahlenstiel, G.; Edlich, B.; Hogdal, L.J.; Rotman, Y.; Noureddin, M.; Feld, J.J.; Holz, L.E.; Titerence, R.H.; Liang, T.J.; Rehermann, B. Early changes in natural killer cell function indicate virologic response to interferon therapy for hepatitis C. Gastroenterology 2011, 141, 1231–1239. [Google Scholar] [CrossRef] [Green Version]
- Prager, I.; Liesche, C.; van Ooijen, H.; Urlaub, D.; Verron, Q.; Sandstrom, N.; Fasbender, F.; Claus, M.; Eils, R.; Beaudouin, J.; et al. NK cells switch from granzyme B to death receptor-mediated cytotoxicity during serial killing. J. Exp. Med. 2019, 216, 2113–2127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijaya, R.S.; Read, S.A.; Schibeci, S.; Eslam, M.; Azardaryany, M.K.; El-Khobar, K.; van der Poorten, D.; Lin, R.; Yuen, L.; Lam, V.; et al. KLRG1+ natural killer cells exert a novel antifibrotic function in chronic hepatitis B. J. Hepatol. 2019, 71, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Huang, C.X.; Ye, L.; Wang, X.; Song, L.; Wang, Y.J.; Liang, H.; Huang, X.Y.; Ho, W.Z. Natural killer cells suppress full cycle HCV infection of human hepatocytes. J. Viral Hepat. 2008, 15, 855–864. [Google Scholar] [CrossRef] [Green Version]
- Orr, M.T.; Lanier, L.L. Natural killer cell education and tolerance. Cell 2010, 142, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carretero, M.; Palmieri, G.; Llano, M.; Tullio, V.; Santoni, A.; Geraghty, D.E.; Lopez-Botet, M. Specific engagement of the CD94/NKG2-A killer inhibitory receptor by the HLA-E class Ib molecule induces SHP-1 phosphatase recruitment to tyrosine-phosphorylated NKG2-A: Evidence for receptor function in heterologous transfectants. Eur. J. Immunol. 1998, 28, 1280–1291. [Google Scholar] [CrossRef]
- Moretta, A.; Moretta, L. HLA class I specific inhibitory receptors. Curr. Opin. Immunol. 1997, 9, 694–701. [Google Scholar] [CrossRef]
- Borrego, F.; Kabat, J.; Kim, D.K.; Lieto, L.; Maasho, K.; Pena, J.; Solana, R.; Coligan, J.E. Structure and function of major histocompatibility complex (MHC) class I specific receptors expressed on human natural killer (NK) cells. Mol. Immunol. 2002, 38, 637–660. [Google Scholar] [CrossRef] [Green Version]
- Kang, W.; Shin, E.C. Interferon-induced MHC class I expression is attenuated by Hepatitis C Virus. Hepatology 2012, 56, 700A. [Google Scholar]
- Abel, A.M.; Yang, C.; Thakar, M.S.; Malarkannan, S. Natural Killer Cells: Development, Maturation, and Clinical Utilization. Front. Immunol. 2018, 9, 1869. [Google Scholar] [CrossRef] [Green Version]
- Rolle, A.; Meyer, M.; Calderazzo, S.; Jager, D.; Momburg, F. Distinct HLA-E Peptide Complexes Modify Antibody-Driven Effector Functions of Adaptive NK Cells. Cell Rep. 2018, 24, 1967. [Google Scholar] [CrossRef] [Green Version]
- Melsen, J.E.; Lugthart, G.; Lankester, A.C.; Schilham, M.W. Human Circulating and Tissue-Resident CD56(bright) Natural Killer Cell Populations. Front. Immunol. 2016, 7, 262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, T.; Poli, A.; Cuapio, A.; Briquemont, B.; Iserentant, G.; Ollert, M.; Zimmer, J. Human CD56(bright) NK Cells: An Update. J. Immunol. 2016, 196, 2923–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bjorkstrom, N.K.; Ljunggren, H.G.; Sandberg, J.K. CD56 negative NK cells: Origin, function, and role in chronic viral disease. Trends Immunol. 2010, 31, 401–406. [Google Scholar] [CrossRef]
- Milush, J.M.; Lopez-Verges, S.; York, V.A.; Deeks, S.G.; Martin, J.N.; Hecht, F.M.; Lanier, L.L.; Nixon, D.F. CD56(neg)CD16(+) NK cells are activated mature NK cells with impaired effector function during HIV-1 infection. Retrovirology 2013, 10, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, V.D.; Falconer, K.; Bjorkstrom, N.K.; Blom, K.G.; Weiland, O.; Ljunggren, H.G.; Alaeus, A.; Sandberg, J.K. Expansion of Functionally Skewed CD56-Negative NK Cells in Chronic Hepatitis C Virus Infection: Correlation with Outcome of Pegylated IFN-alpha and Ribavirin Treatment. J. Immunol. 2009, 183, 6612–6618. [Google Scholar] [CrossRef] [Green Version]
- Carrega, P.; Ferlazzo, G. Natural killer cell distribution and trafficking in human tissues. Front. Immunol. 2012, 3, 347. [Google Scholar] [CrossRef] [Green Version]
- Mikulak, J.; Bruni, E.; Oriolo, F.; Di Vito, C.; Mavilio, D. Hepatic Natural Killer Cells: Organ-Specific Sentinels of Liver Immune Homeostasis and Physiopathology. Front. Immunol. 2019, 10, 946. [Google Scholar] [CrossRef] [Green Version]
- Hudspeth, K.; Donadon, M.; Cimino, M.; Pontarini, E.; Tentorio, P.; Preti, M.; Hong, M.; Bertoletti, A.; Bicciato, S.; Invernizzi, P.; et al. Human liver-resident CD56(bright)/CD16(neg) NK cells are retained within hepatic sinusoids via the engagement of CCR5 and CXCR6 pathways. J. Autoimmun. 2016, 66, 40–50. [Google Scholar] [CrossRef] [Green Version]
- Cuff, A.O.; Robertson, F.P.; Stegmann, K.A.; Pallett, L.J.; Maini, M.K.; Davidson, B.R.; Male, V. Eomeshi NK Cells in Human Liver Are Long-Lived and Do Not Recirculate but Can Be Replenished from the Circulation. J. Immunol. 2016, 197, 4283–4291. [Google Scholar] [CrossRef] [Green Version]
- Yoon, J.C.; Yang, C.M.; Song, Y.; Lee, J.M. Natural killer cells in hepatitis C: Current progress. World J. Gastroenterol. 2016, 22, 1449–1460. [Google Scholar] [CrossRef]
- Marra, F.; Tacke, F. Roles for Chemokines in Liver Disease. Gastroenterology 2014, 147, 577. [Google Scholar] [CrossRef] [PubMed]
- Saiman, Y.; Friedman, S.L. The role of chemokines in acute liver injury. Front. Physiol. 2012, 3, 213. [Google Scholar] [CrossRef] [Green Version]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stegmann, K.A.; Bjorkstrom, N.K.; Ciesek, S.; Lunemann, S.; Jaroszewicz, J.; Wiegand, J.; Malinski, P.; Dustin, L.B.; Rice, C.M.; Manns, M.P.; et al. Interferon alpha-Stimulated Natural Killer Cells From Patients With Acute Hepatitis C Virus (HCV) Infection Recognize HCV-Infected and Uninfected Hepatoma Cells via DNAX accessory molecule-1. J. Infect. Dis. 2012, 205, 1351–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.C.; Lim, J.B.; Park, J.H.; Lee, J.M. Cell-to-Cell Contact with Hepatitis C Virus-Infected Cells Reduces Functional Capacity of Natural Killer Cells. J. Virol. 2011, 85, 12557–12569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holder, K.A.; Stapleton, S.N.; Gallant, M.E.; Russell, R.S.; Grant, M.D. Hepatitis C Virus-Infected Cells Downregulate NKp30 and Inhibit Ex Vivo NK Cell Functions. J. Immunol. 2013, 191, 3308–3318. [Google Scholar] [CrossRef] [Green Version]
- Sene, D.; Levasseur, F.; Abel, M.; Camous, X.; Rosenbereg, A.R.; Marche, P.N.; Cacoub, P.; Caillat-Zucman, S. Hepatitis C Virus (Hcv) Evades Nkg2d-Dependent NK Cell Responses through Ns5a-Mediated Imbalance of Inflammatory Cytokines. Hepatology 2010, 52, 740a. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.M.; Yoon, J.C.; Park, J.H.; Lee, J.M. Hepatitis C virus impairs natural killer cell activity via viral serine protease NS3. PLoS ONE 2017, 12, e0175793. [Google Scholar] [CrossRef] [Green Version]
- Sene, D.; Levasseur, F.; Abel, M.; Lambert, M.; Camous, X.; Hernandez, C.; Pene, V.; Rosenberg, A.R.; Jouvin-Marche, E.; Marche, P.N.; et al. Hepatitis C Virus (HCV) Evades NKG2D-Dependent NK Cell Responses through NS5A-Mediated Imbalance of Inflammatory Cytokines. PLoS Pathog. 2010, 6, e1001184. [Google Scholar] [CrossRef] [Green Version]
- Tseng, C.T.K.; Klimpel, G.R. Binding of the hepatitis C virus envelope protein E2 to CD81 inhibits natural killer cell functions. J. Exp. Med. 2002, 195, 43–49. [Google Scholar] [CrossRef] [Green Version]
- Nattermann, J.; Nischalke, H.D.; Hofmeister, V.; Ahlenstiel, G.; Zimmermann, H.; Leifeld, L.; Weiss, E.H.; Sauerbruch, T.; Spengler, U. The HLA-A2 restricted T cell epitope HCV core 35-44 stabilizes HLA-E expression and inhibits cytolysis mediated by natural killer cells. Am. J. Pathol. 2005, 166, 443–453. [Google Scholar] [CrossRef]
- Schulte, D.; Vogel, M.; Langhans, B.; Kramer, B.; Korner, C.; Nischalke, H.D.; Steinberg, V.; Michalk, M.; Berg, T.; Rockstroh, J.K.; et al. The HLA-E(R)/HLA-E(R) genotype affects the natural course of hepatitis C virus (HCV) infection and is associated with HLA-E-restricted recognition of an HCV-derived peptide by interferon-gamma-secreting human CD8(+) T cells. J. Infect. Dis. 2009, 200, 1397–1401. [Google Scholar] [CrossRef] [Green Version]
- Irshad, M.; Khushboo, I.; Singh, S.; Singh, S. Hepatitis C Virus (HCV): A Review of Immunological Aspects. Int. Rev. Immunol. 2008, 27, 497–517. [Google Scholar] [CrossRef]
- Takahashi, K.; Asabe, S.; Wieland, S.; Garaigorta, U.; Gastaminza, P.; Isogawa, M.; Chisari, F.V. Plasmacytoid dendritic cells sense hepatitis C virus-infected cells, produce interferon, and inhibit infection. Proc. Natl. Acad. Sci. USA 2010, 107, 7431–7436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Kanto, T.; Groh, V.; Spies, T.; Suzuki, T.; Miyagi, T.; Hayashi, N. Autocrine/paracrine IL-15 that is required for type I IFN-mediated dendritic cell expression of MHC class I-related chain A and B is impaired in hepatitis C virus infection. J. Immunol. 2003, 171, 5423–5429. [Google Scholar] [CrossRef] [PubMed]
- Lucas, M.; Schachterle, W.; Oberle, K.; Aichele, P.; Diefenbach, A. Dendritic cells prime natural killer cells by trans-presenting interleukin 15. Immunity 2007, 26, 503–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, C.J.; Ni, L.; Zhang, Y.; Zhang, C.L.; Wu, X.Y.; Atia, A.N.; Thayer, P.; Moorman, J.P.; Yao, Z.Q. PD-1 negatively regulates interleukin-12 expression by limiting STAT-1 phosphorylation in monocytes/macrophages duringchronic hepatitis C virus infection. Immunology 2011, 132, 421–431. [Google Scholar] [CrossRef]
- Wang, J.M.; Shi, L.; Ma, C.J.; Ji, X.J.; Ying, R.S.; Wu, X.Y.; Wang, K.S.; Li, G.; Moorman, J.P.; Yao, Z.Q. Differential regulation of interleukin-12 (IL-12)/IL-23 by Tim-3 drives T(H)17 cell development during hepatitis C virus infection. J. Virol. 2013, 87, 4372–4383. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Ma, C.J.; Wang, J.M.; Ji, X.J.; Wu, X.Y.; Jia, Z.S.; Moorman, J.P.; Yao, Z.Q. Tim-3 negatively regulates IL-12 expression by monocytes in HCV infection. PLoS ONE 2011, 6, e19664. [Google Scholar] [CrossRef] [Green Version]
- Khakoo, S.I.; Thio, C.L.; Martin, M.P.; Brooks, C.R.; Gao, X.; Astemborski, J.; Cheng, J.; Goedert, J.J.; Vlahov, D.; Hilgartner, M.; et al. HLA and NK cell inhibitory receptor genes in resolving hepatitis C virus infection. Science 2004, 305, 872–874. [Google Scholar] [CrossRef]
- Romero, V.; Azocar, J.; Zuniga, J.; Clavijo, O.P.; Terreros, D.; Gu, X.; Husain, Z.; Chung, R.T.; Amos, C.; Yunis, E.J. Interaction of NK inhibitory receptor genes with HLA-C and MHC class II alleles in Hepatitis C virus infection outcome. Mol. Immunol. 2008, 45, 2429–2436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moesta, A.K.; Norman, P.J.; Yawata, M.; Yawata, N.; Gleimer, M.; Parham, P. Synergistic polymorphism at two positions distal to the ligand-binding site makes KIR2DL2 a stronger receptor for HLA-C than KIR2DL3. J. Immunol. 2008, 180, 3969–3979. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, C.M. NK cell function and receptor diversity in the context of HCV infection. Front. Microbiol. 2015, 6, 1061. [Google Scholar] [CrossRef] [PubMed]
- Dring, M.M.; Morrison, M.H.; McSharry, B.P.; Guinan, K.J.; Hagan, R.; Irish, H.C.V.R.C.; O’Farrelly, C.; Gardiner, C.M. Innate immune genes synergize to predict increased risk of chronic disease in hepatitis C virus infection. Proc. Natl. Acad. Sci. USA 2011, 108, 5736–5741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strong, R.K.; Holmes, M.A.; Morris, D.L.; Braun, L.; Lee, N.; Geraghty, D.E. HLA-E allelic variants: Correlating differential expression, peptide affinities, crystal structures and thermal stabilities. Tissue Antigens 2002, 59, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amadei, B.; Urbani, S.; Cazaly, A.; Fisicaro, P.; Zerbini, A.; Ahmed, P.; Missale, G.; Ferrari, C.; Khakoo, S.I. Activation of natural killer cells during acute infection with hepatitis C virus. Gastroenterology 2010, 138, 1536–1545. [Google Scholar] [CrossRef] [Green Version]
- Alter, G.; Jost, S.; Rihn, S.; Reyor, L.L.; Nolan, B.E.; Ghebremichael, M.; Bosch, R.; Altfeld, M.; Lauer, G.M. Reduced frequencies of NKp30+NKp46+, CD161+, and NKG2D+NK cells in acute HCV infection may predict viral clearance. J. Hepatol. 2011, 55, 278–288. [Google Scholar] [CrossRef] [Green Version]
- Kokordelis, P.; Kramer, B.; Korner, C.; Boesecke, C.; Voigt, E.; Ingiliz, P.; Glassner, A.; Eisenhardt, M.; Wolter, F.; Kaczmarek, D.; et al. An Effective Interferon-Gamma-Mediated Inhibition of Hepatitis C Virus Replication by Natural Killer Cells Is Associated With Spontaneous Clearance of Acute Hepatitis C in Human Immunodeficiency Virus-Positive Patients. Hepatology 2014, 59, 814–827. [Google Scholar] [CrossRef]
- Golden-Mason, L.; Madrigal-Estebas, L.; McGrath, E.; Conroy, M.J.; Ryan, E.J.; Hegarty, J.E.; O’Farrelly, C.; Doherty, D.G. Altered natural killer cell subset distributions in resolved and persistent hepatitis C virus infection following single source exposure. Gut 2008, 57, 1121–1128. [Google Scholar] [CrossRef]
- Dessouki, O.; Kamiya, Y.; Nagahama, H.; Tanaka, M.; Suzu, S.; Sasaki, Y.; Okada, S. Chronic hepatitis C viral infection reduces NK cell frequency and suppresses cytokine secretion: Reversion by anti-viral treatment. Biochem. Biophys. Res. Commun. 2010, 393, 331–337. [Google Scholar] [CrossRef]
- Ahlenstiel, G.; Titerence, R.H.; Koh, C.; Edlich, B.; Feld, J.J.; Rotman, Y.; Ghany, M.G.; Hoofnagle, J.H.; Liang, T.J.; Heller, T.; et al. Natural Killer Cells Are Polarized Toward Cytotoxicity in Chronic Hepatitis C in an Interferon-Alfa-Dependent Manner. Gastroenterology 2010, 138, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliviero, B.; Varchetta, S.; Paudice, E.; Michelone, G.; Zaramella, M.; Mavilio, D.; De Filippi, F.; Bruno, S.; Mondelli, M.U. Natural killer cell functional dichotomy in chronic hepatitis B and chronic hepatitis C virus infections. Gastroenterology 2009, 137, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Gil, M.P.; Wang, X.; Louten, J.; Chu, W.M.; Biron, C.A. High basal STAT4 balanced by STAT1 induction to control type 1 interferon effects in natural killer cells. J. Exp. Med. 2007, 204, 2383–2396. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, T.; Takehara, T.; Nishio, K.; Shimizu, S.; Kohga, K.; Li, W.; Tatsumi, T.; Hiramatsu, N.; Kanto, T.; Hayashi, N. Altered interferon-alpha-signaling in natural killer cells from patients with chronic hepatitis C virus infection. J. Hepatol. 2010, 53, 424–430. [Google Scholar] [CrossRef]
- Edlich, B.; Ahlenstiel, G.; Azpiroz, A.Z.; Stoltzfus, J.; Noureddin, M.; Serti, E.; Feld, J.J.; Liang, T.J.; Rotman, Y.; Rehermann, B. Early changes in interferon signaling define natural killer cell response and refractoriness to interferon-based therapy of hepatitis C patients. Hepatology 2012, 55, 39–48. [Google Scholar] [CrossRef] [Green Version]
- Pembroke, T.; Christian, A.; Jones, E.; Hills, R.K.; Wang, E.C.; Gallimore, A.M.; Godkin, A. The paradox of NKp46+ natural killer cells: Drivers of severe hepatitis C virus-induced pathology but in-vivo resistance to interferon alpha treatment. Gut 2014, 63, 515–524. [Google Scholar] [CrossRef] [Green Version]
- Kramer, B.; Korner, C.; Kebschull, M.; Glassner, A.; Eisenhardt, M.; Nischalke, H.D.; Alexander, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. Natural killer p46High expression defines a natural killer cell subset that is potentially involved in control of hepatitis C virus replication and modulation of liver fibrosis. Hepatology 2012, 56, 1201–1213. [Google Scholar] [CrossRef]
- Jinushi, M.; Takehara, T.; Tatsumi, T.; Kanto, T.; Miyagi, T.; Suzuki, T.; Kanazawa, Y.; Hiramatsu, N.; Hayashi, N. Negative regulation of NK cell activities by inhibitory receptor CD94/NKG2A leads to altered NK cell-induced modulation of dendritic cell functions in chronic hepatitis C virus infection. J. Immunol. 2004, 173, 6072–6081. [Google Scholar] [CrossRef]
- Szereday, L.; Meggyes, M.; Halasz, M.; Szekeres-Bartho, J.; Par, A.; Par, G. Immunological changes in different patient populations with chronic hepatitis C virus infection. World J. Gastroenterol. 2016, 22, 4848–4859. [Google Scholar] [CrossRef]
- Bozzano, F.; Picciotto, A.; Costa, P.; Marras, F.; Fazio, V.; Hirsch, I.; Olive, D.; Moretta, L.; De Maria, A. Activating NK cell receptor expression/function (NKp30, NKp46, DNAM-1) during chronic viraemic HCV infection is associated with the outcome of combined treatment. Eur. J. Immunol. 2011, 41, 2905–2914. [Google Scholar] [CrossRef]
- Beziat, V.; Dalgard, O.; Asselah, T.; Halfon, P.; Bedossa, P.; Boudifa, A.; Hervier, B.; Theodorou, I.; Martinot, M.; Debre, P.; et al. CMV drives clonal expansion of NKG2C(+) NK cells expressing self-specific KIRs in chronic hepatitis patients. Eur. J. Immunol. 2012, 42, 447–457. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Wang, X.M.; Li, S.R.; Twelkmeyer, T.; Wang, W.H.; Zhang, S.Y.; Wang, S.F.; Chen, J.Z.; Jin, X.; Wu, Y.Z.; et al. NKG2A is a NK cell exhaustion checkpoint for HCV persistence. Nat. Commun. 2019, 10, 1507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cosgrove, C.; Berger, C.T.; Kroy, D.C.; Cheney, P.C.; Ghebremichael, M.; Aneja, J.; Tomlinson, M.; Kim, A.Y.; Lauer, G.M.; Alter, G. Chronic HCV Infection Affects the NK Cell Phenotype in the Blood More than in the Liver. PLoS ONE 2014, 9, e0105950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rivero-Juarez, A.; Gonzalez, R.; Camacho, A.; Manzanares-Martin, B.; Caruz, A.; Martinez-Peinado, A.; Torre-Cisneros, J.; Pineda, J.A.; Pena, J.; Rivero, A. Natural Killer KIR3DS1 Is Closely Associated with HCV Viral Clearance and Sustained Virological Response in HIV/HCV Patients. PLoS ONE 2013, 8, e0061992. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Vazquez, A.; Rodrigo, L.; Martinez-Borra, J.; Perez, R.; Rodriguez, M.; Fdez-Morera, J.L.; Fuentes, D.; Rodriguez-Rodero, S.; Gonzalez, S.; Lopez-Larrea, C. Protective effect of the HLA-Bw4I80 epitope and the killer cell immunoglobulin-like receptor 3DS1 gene against the development of hepatocellular carcinoma in patients with hepatitis C virus infection. J. Infect. Dis. 2005, 192, 162–165. [Google Scholar] [CrossRef] [Green Version]
- Lunemann, S.; Schobel, A.; Kah, J.; Fittje, P.; Holzemer, A.; Langeneckert, A.E.; Hess, L.U.; Poch, T.; Martrus, G.; Garcia-Beltran, W.F.; et al. Interactions Between KIR3DS1 and HLA-F Activate Natural Killer Cells to Control HCV Replication in Cell Culture. Gastroenterology 2018, 155, 1366. [Google Scholar] [CrossRef]
- De Groen, R.A.; Groothuismink, Z.M.A.; van Oord, G.; Kootstra, N.A.; Janssen, H.L.A.; Prins, M.; Schinkel, J.; Boonstra, A. NK cells in self-limited HCV infection exhibit a more extensively differentiated, but not memory-like, repertoire. J. Viral Hepat. 2017, 24, 917–926. [Google Scholar] [CrossRef]
- Krueger, P.D.; Narayanan, S.; Surette, F.A.; Brown, M.G.; Sung, S.J.; Hahn, Y.S. Murine liver-resident group 1 innate lymphoid cells regulate optimal priming of anti-viral CD8+ T cells. J. Leukoc. Biol. 2017, 101, 329–338. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Tian, Z. Natural Killer Cell Memory: Progress and Implications. Front. Immunol. 2017, 8, 1143. [Google Scholar] [CrossRef] [Green Version]
- Bjorkstrom, N.K.; Lindgren, T.; Stoltz, M.; Fauriat, C.; Braun, M.; Evander, M.; Michaelsson, J.; Malmberg, K.J.; Klingstrom, J.; Ahlm, C.; et al. Rapid expansion and long-term persistence of elevated NK cell numbers in humans infected with hantavirus. J. Exp. Med. 2011, 208, 13–21. [Google Scholar] [CrossRef]
- Reeves, R.K.; Li, H.; Jost, S.; Blass, E.; Li, H.; Schafer, J.L.; Varner, V.; Manickam, C.; Eslamizar, L.; Altfeld, M.; et al. Antigen-specific NK cell memory in rhesus macaques. Nat. Immunol. 2015, 16, 927–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, Q.; Ruckert, T.; Borst, E.M.; Dunst, J.; Haubner, A.; Durek, P.; Heinrich, F.; Gasparoni, G.; Babic, M.; Tomic, A.; et al. Peptide-specific recognition of human cytomegalovirus strains controls adaptive natural killer cells. Nat. Immunol. 2018, 19, 453–463. [Google Scholar] [CrossRef]
- Foley, B.; Cooley, S.; Verneris, M.R.; Curtsinger, J.; Luo, X.; Waller, E.K.; Anasetti, C.; Weisdorf, D.; Miller, J.S. Human cytomegalovirus (CMV)-induced memory-like NKG2C(+) NK cells are transplantable and expand in vivo in response to recipient CMV antigen. J. Immunol. 2012, 189, 5082–5088. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paust, S.; Gill, H.S.; Wang, B.Z.; Flynn, M.P.; Moseman, E.A.; Senman, B.; Szczepanik, M.; Telenti, A.; Askenase, P.W.; Compans, R.W.; et al. Critical role for the chemokine receptor CXCR6 in NK cell-mediated antigen-specific memory of haptens and viruses. Nat. Immunol. 2010, 11, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Abdul-Careem, M.F.; Lee, A.J.; Pek, E.A.; Gill, N.; Gillgrass, A.E.; Chew, M.V.; Reid, S.; Ashkar, A.A. Genital HSV-2 infection induces short-term NK cell memory. PLoS ONE 2012, 7, e32821. [Google Scholar] [CrossRef] [PubMed]
- Wijaya, R.S.; Read, S.A.; Truong, N.R.; Han, S.; Chen, D.; Shahidipour, H.; Fewings, N.L.; Schibeci, S.; Azardaryany, M.K.; Parnell, G.P.; et al. HBV vaccination and HBV infection induces HBV-specific natural killer cell memory. Gut 2020. [Google Scholar] [CrossRef] [PubMed]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.H.; Albrecht, J.K.; et al. Peginterferon alfa-2b plus ribavirin compared with interferon alfa-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef]
- Poordad, F.; McCone, J., Jr.; Bacon, B.R.; Bruno, S.; Manns, M.P.; Sulkowski, M.S.; Jacobson, I.M.; Reddy, K.R.; Goodman, Z.D.; Boparai, N.; et al. Boceprevir for untreated chronic HCV genotype 1 infection. N. Engl. J. Med. 2011, 364, 1195–1206. [Google Scholar] [CrossRef] [Green Version]
- Jacobson, I.M.; McHutchison, J.G.; Dusheiko, G.; Di Bisceglie, A.M.; Reddy, K.R.; Bzowej, N.H.; Marcellin, P.; Muir, A.J.; Ferenci, P.; Flisiak, R.; et al. Telaprevir for previously untreated chronic hepatitis C virus infection. N. Engl. J. Med. 2011, 364, 2405–2416. [Google Scholar] [CrossRef]
- Bourliere, M.; Gordon, S.C.; Flamm, S.L.; Cooper, C.L.; Ramji, A.; Tong, M.; Ravendhran, N.; Vierling, J.M.; Tran, T.T.; Pianko, S.; et al. Sofosbuvir, Velpatasvir, and Voxilaprevir for Previously Treated HCV Infection. N. Engl. J. Med. 2017, 376, 2134–2146. [Google Scholar] [CrossRef]
- Zeuzem, S.; Foster, G.R.; Wang, S.; Asatryan, A.; Gane, E.; Feld, J.J.; Asselah, T.; Bourliere, M.; Ruane, P.J.; Wedemeyer, H.; et al. Glecaprevir-Pibrentasvir for 8 or 12 Weeks in HCV Genotype 1 or 3 Infection. N. Engl. J. Med. 2018, 378, 354–369. [Google Scholar] [CrossRef] [PubMed]
- Ascierto, M.L.; Bozzano, F.; Bedognetti, D.; Marras, F.; Schechterly, C.; Matsuura, K.; Picciotto, A.; Marenco, S.; Zhao, Y.D.; DeGiorgi, V.; et al. Inherent transcriptional signatures of NK cells are associated with response to IFN alpha plus rivabirin therapy in patients with Hepatitis C Virus. J. Transl. Med. 2015, 13, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, X.L.; Jiang, Y.F.; Li, X.R.; Gao, Y.H.; Guo, X.L.; Chi, X.M.; Yan, H.Q.; Feng, J.Y.; Zhong, J.; Sun, B.; et al. Long-Term Effect on Natural Killer Cells by Interferon-alpha Therapy on the Outcomes of HCV Infection. J. Interferon Cytokine Res. 2014, 34, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarasin-Filipowicz, M.; Oakeley, E.J.; Duong, F.H.; Christen, V.; Terracciano, L.; Filipowicz, W.; Heim, M.H. Interferon signaling and treatment outcome in chronic hepatitis C. Proc. Natl. Acad. Sci. USA 2008, 105, 7034–7039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conry, S.J.; Meng, Q.L.; Hardy, G.; Yonkers, N.L.; Sugalski, J.M.; Hirsch, A.; Davitkov, P.; Compan, A.; Falck-Ytter, Y.; Blanton, R.E.; et al. Genetically Associated CD16(+)56(-) Natural Killer Cell Interferon (IFN)-alpha R Expression Regulates Signaling and Is Implicated in IFN-alpha-Induced Hepatitis C Virus Decline. J. Infect. Dis. 2012, 205, 1131–1141. [Google Scholar] [CrossRef] [Green Version]
- Vidal-Castineira, J.R.; Lopez-Vazquez, A.; Diaz-Pena, R.; Alonso-Arias, R.; Martinez-Borra, J.; Perez, R.; Fernandez-Suarez, J.; Melon, S.; Prieto, J.; Rodrigo, L.; et al. Effect of killer immunoglobulin-like receptors in the response to combined treatment in patients with chronic hepatitis C virus infection. J. Virol. 2010, 84, 475–481. [Google Scholar] [CrossRef] [Green Version]
- Suppiah, V.; Gaudieri, S.; Armstrong, N.J.; O’Connor, K.S.; Berg, T.; Weltman, M.; Abate, M.L.; Spengler, U.; Bassendine, M.; Dore, G.J.; et al. IL28B, HLA-C, and KIR variants additively predict response to therapy in chronic hepatitis C virus infection in a European Cohort: A cross-sectional study. PLoS Med. 2011, 8, e1001092. [Google Scholar] [CrossRef]
- Oliviero, B.; Mele, D.; Degasperi, E.; Aghemo, A.; Cremonesi, E.; Rumi, M.G.; Tinelli, C.; Varchetta, S.; Mantovani, S.; Colombo, M.; et al. Natural killer cell dynamic profile is associated with treatment outcome in patients with chronic HCV infection. J. Hepatol. 2013, 59, 38–44. [Google Scholar] [CrossRef]
- Werner, J.M.; Serti, E.; Chepa-Lotrea, X.; Stoltzfus, J.; Ahlenstiel, G.; Noureddin, M.; Feld, J.J.; Liang, T.J.; Rotman, Y.; Rehermann, B. Ribavirin improves the IFN-γ response of natural killer cells to IFN-based therapy of hepatitis C virus infection. Hepatology 2014, 60, 1160–1169. [Google Scholar] [CrossRef] [Green Version]
- Falade-Nwulia, O.; Suarez-Cuervo, C.; Nelson, D.R.; Fried, M.W.; Segal, J.B.; Sulkowski, M.S. Oral Direct-Acting Agent Therapy for Hepatitis C Virus Infection: A Systematic Review. Ann. Intern. Med. 2017, 166, 637–648. [Google Scholar] [CrossRef] [Green Version]
- Martin, B.; Hennecke, N.; Lohmann, V.; Kayser, A.; Neumann-Haefelin, C.; Kukolj, G.; Böcher, W.O.; Thimme, R. Restoration of HCV-specific CD8+ T cell function by interferon-free therapy. J. Hepatol. 2014, 61, 538–543. [Google Scholar] [CrossRef]
- Serti, E.; Chepa-Lotrea, X.; Kim, Y.J.; Keane, M.; Fryzek, N.; Liang, T.J.; Ghany, M.; Rehermann, B. Successful Interferon-Free Therapy of Chronic Hepatitis C Virus Infection Normalizes Natural Killer Cell Function. Gastroenterology 2015, 149, 190–200.e192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spaan, M.; van Oord, G.; Kreefft, K.; Hou, J.; Hansen, B.E.; Janssen, H.L.; de Knegt, R.J.; Boonstra, A. Immunological Analysis During Interferon-Free Therapy for Chronic Hepatitis C Virus Infection Reveals Modulation of the Natural Killer Cell Compartment. J. Infect. Dis. 2016, 213, 216–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golden-Mason, L.; McMahan, R.H.; Kriss, M.S.; Kilgore, A.L.; Cheng, L.; Dran, R.J.; Wieland, A.; Rosen, H.R. Early and late changes in natural killer cells in response to ledipasvir/sofosbuvir treatment. Hepatol. Commun. 2018, 2, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Alao, H.; Cam, M.; Keembiyehetty, C.; Zhang, F.; Serti, E.; Suarez, D.; Park, H.; Fourie, N.H.; Wright, E.C.; Henderson, W.A.; et al. Baseline Intrahepatic and Peripheral Innate Immunity are Associated with Hepatitis C Virus Clearance During Direct-Acting Antiviral Therapy. Hepatology 2018, 68, 2078–2088. [Google Scholar] [CrossRef] [Green Version]
- Strunz, B.; Hengst, J.; Deterding, K.; Manns, M.P.; Cornberg, M.; Ljunggren, H.G.; Wedemeyer, H.; Björkström, N.K. Chronic hepatitis C virus infection irreversibly impacts human natural killer cell repertoire diversity. Nat. Commun. 2018, 9, 2275. [Google Scholar] [CrossRef]
- Santangelo, L.; Bordoni, V.; Montaldo, C.; Cimini, E.; Zingoni, A.; Battistelli, C.; D’Offizi, G.; Capobianchi, M.R.; Santoni, A.; Tripodi, M.; et al. Hepatitis C virus direct-acting antivirals therapy impacts on extracellular vesicles microRNAs content and on their immunomodulating properties. Liver Int. 2018, 38, 1741–1750. [Google Scholar] [CrossRef]
- Chu, P.S.; Nakamoto, N.; Taniki, N.; Ojiro, K.; Amiya, T.; Makita, Y.; Murata, H.; Yamaguchi, A.; Shiba, S.; Miyake, R.; et al. On-treatment decrease of NKG2D correlates to early emergence of clinically evident hepatocellular carcinoma after interferon-free therapy for chronic hepatitis C. PLoS ONE 2017, 12, e0179096. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
| Types | Receptor | Ligand | Ref (Ligand) | Acute | Chronic | In Vitro | Ref (Expression) |
|---|---|---|---|---|---|---|---|
| Activating | NKG2D | MICA/B, ULBP1-6 | [10,11] | ↔ | ↓ | ↓ | Yoon et al., 2011; Sene et al., 2010; Alter et al., 2011 |
| Receptor | CD94-NKG2C | HLA-E | [12] | ↑ | Szereday et al., 2016 | ||
| KIR2DL4 | HLA-G | [13] | |||||
| KIR2DS1 | HLA-C2 | [14] | ↔ | Cosgrove et al., 2014 | |||
| KIR2DS2 | HLA-A11 | [15] | |||||
| KIR2DS3 | Unknown | ||||||
| KIR2DS4 | HLA-A11, HLA-C05:01 | [16,17] | |||||
| KIR2DS5 | Unknown | ||||||
| KIR3DS1 | HLA-F | [18] | |||||
| NKp30 | B7H6, BAT3, HCMV pp65, HS | [19,20,21,22] | ↓ | ↓ | ↓ | Yoon et al., 2016; Holder et al., 2013; Alter et al., 2011 | |
| NKp46 | Heparin, viral HA and HN, CFP | [21,23,24,25] | ↓ | ↓ | ↓ | Sene et al., 2010; Alter et al., 2011 | |
| NKp44 | Viral HA and HN, PCNA, HS | [21,25,26,27] | ↔ | ↔ | Alter et al., 2011 | ||
| DNAM-1 | CD112, CD155 | [28] | ↓ | ↓ | Yoon et al., 2016; Bozzano et al., 2011 | ||
| Inhibiting | KIR2DL1 | HLA-C2 | [14] | ↔ | ↔ | Alter et al., 2011; Cosgrove et al., 2014 | |
| Receptor | KIR2DL2 | HLA-C1 | [29] | ↔ | Cosgrove et al., 2014 | ||
| KIR2DL3 | HLA-C1 | [30] | ↑ | Szereday et al., 2016 | |||
| KIR2DL4 | HS | [31] | |||||
| KIR3DL1 | HLA-Bw4 | [32] | ↓ | Oliviero et al., 2009; Szereday et al., 2016 | |||
| KIR3DL2 | HLA-A3-A11 | [33] | |||||
| CD94-NKG2A | HLA-E | [12] | ↔ | ↔ | ↔ | Holder et al., 2013; Alter et al., 2011 | |
| ILT2 (CD85j) | MHC-I, HCMV UL18, S100A9 | [34,35,36] | ↔ | Oliviero et al., 2009; Szereday et al., 2016 | |||
| CD244 (2B4) | CD48 | [37] | ↔ | ↔ | Yoon et al., 2016; Cosgrove et al., 2014 | ||
| CD161 (KLRB1) | LLT1 | [38] | ↓ | ↓ | Alter et al., 2011; Cosgrove et al., 2014 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Njiomegnie, G.F.; Read, S.A.; Fewings, N.; George, J.; McKay, F.; Ahlenstiel, G. Immunomodulation of the Natural Killer Cell Phenotype and Response during HCV Infection. J. Clin. Med. 2020, 9, 1030. https://doi.org/10.3390/jcm9041030
Njiomegnie GF, Read SA, Fewings N, George J, McKay F, Ahlenstiel G. Immunomodulation of the Natural Killer Cell Phenotype and Response during HCV Infection. Journal of Clinical Medicine. 2020; 9(4):1030. https://doi.org/10.3390/jcm9041030
Chicago/Turabian StyleNjiomegnie, Gaitan Fabrice, Scott A. Read, Nicole Fewings, Jacob George, Fiona McKay, and Golo Ahlenstiel. 2020. "Immunomodulation of the Natural Killer Cell Phenotype and Response during HCV Infection" Journal of Clinical Medicine 9, no. 4: 1030. https://doi.org/10.3390/jcm9041030
APA StyleNjiomegnie, G. F., Read, S. A., Fewings, N., George, J., McKay, F., & Ahlenstiel, G. (2020). Immunomodulation of the Natural Killer Cell Phenotype and Response during HCV Infection. Journal of Clinical Medicine, 9(4), 1030. https://doi.org/10.3390/jcm9041030