Impact of the Gut Microbiota on Atorvastatin Mediated Effects on Blood Lipids

 ,
, 
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
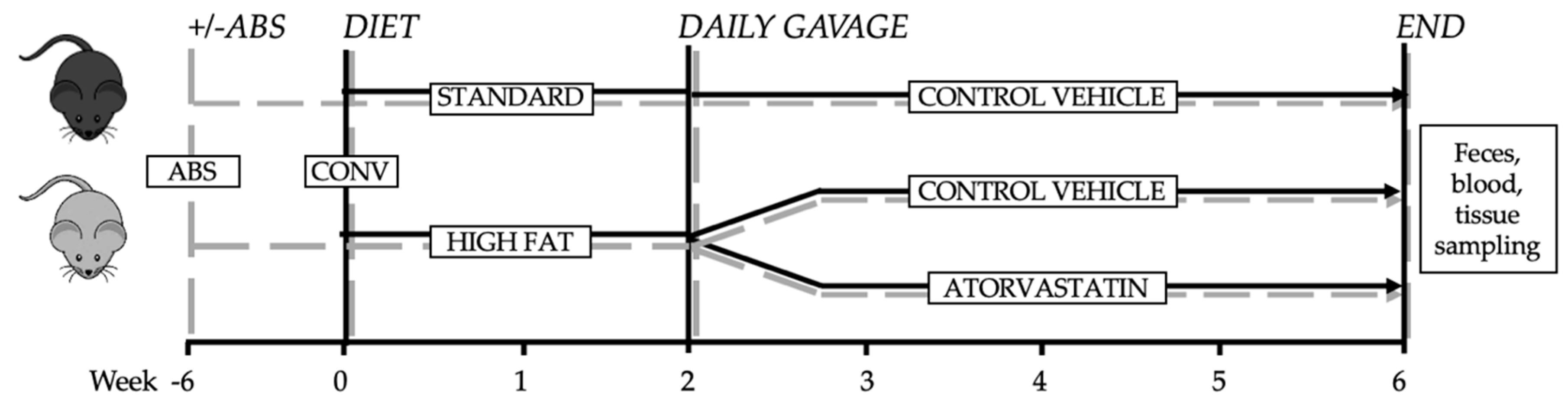
2.1. Mouse Experiments
2.2. Generation of Secondary Abiotic Mice
2.3. Molecular Analysis of Intestinal Microbiota
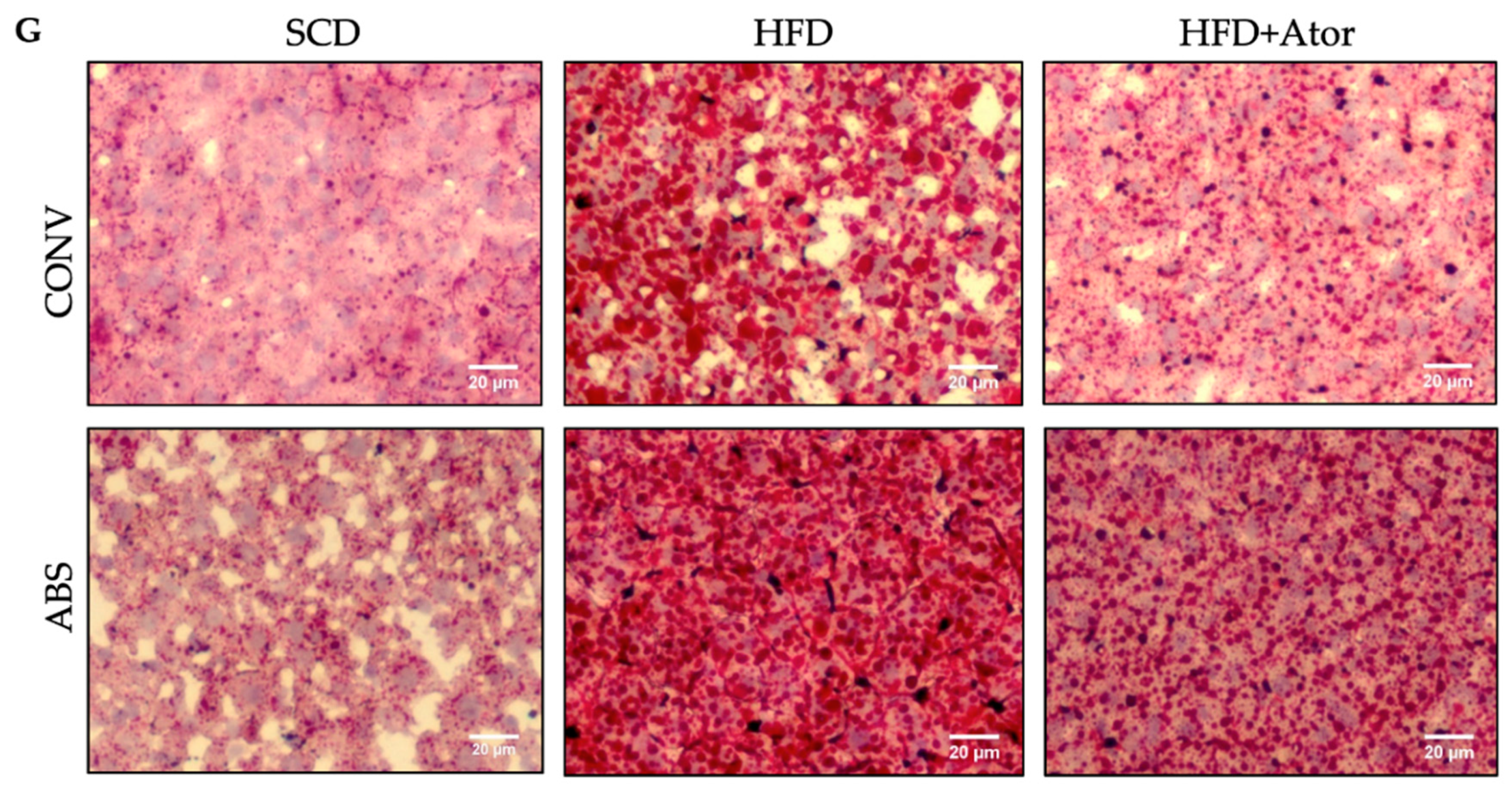
2.4. Histology
2.5. qRT-PCR and Western Blot
2.6. Metabolite Profiling and Lipoprotein Separation
2.7. Statistical Analyses
3. Results
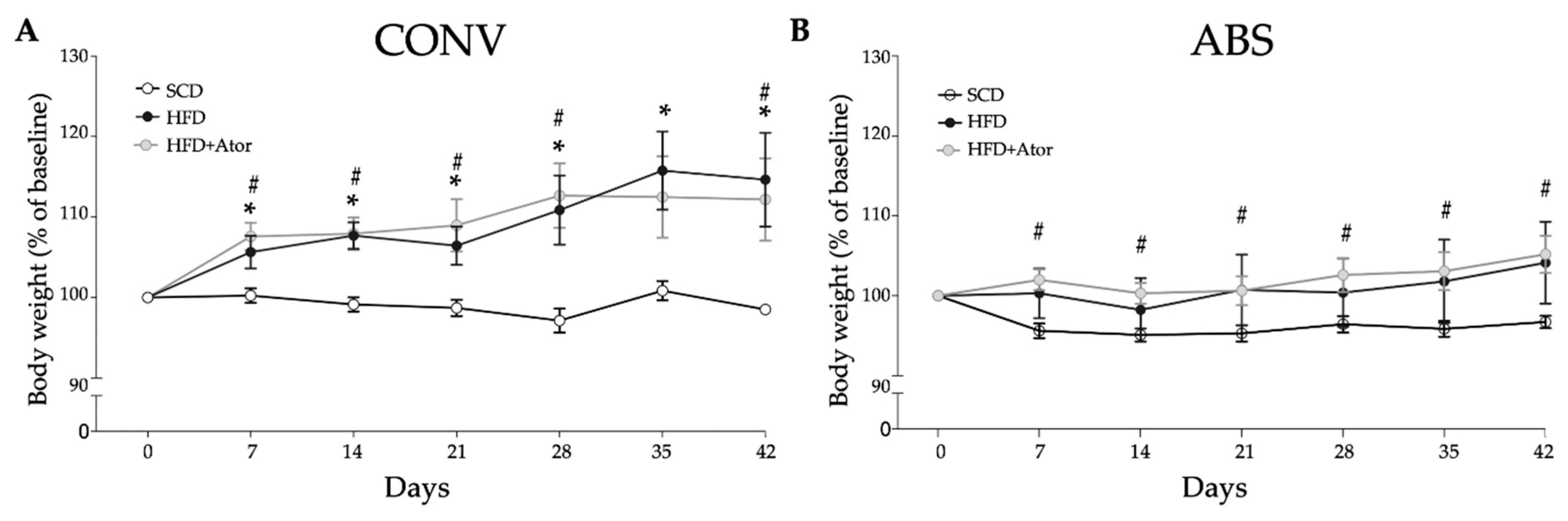
3.1. Differences in Diet-Induced Weight Gain in Conventionally Raised and Abiotic Mice
3.2. Effect of Atorvastatin on Cholesterol and Lipoprotein Levels in Conventionally Raised and Abiotic Mice
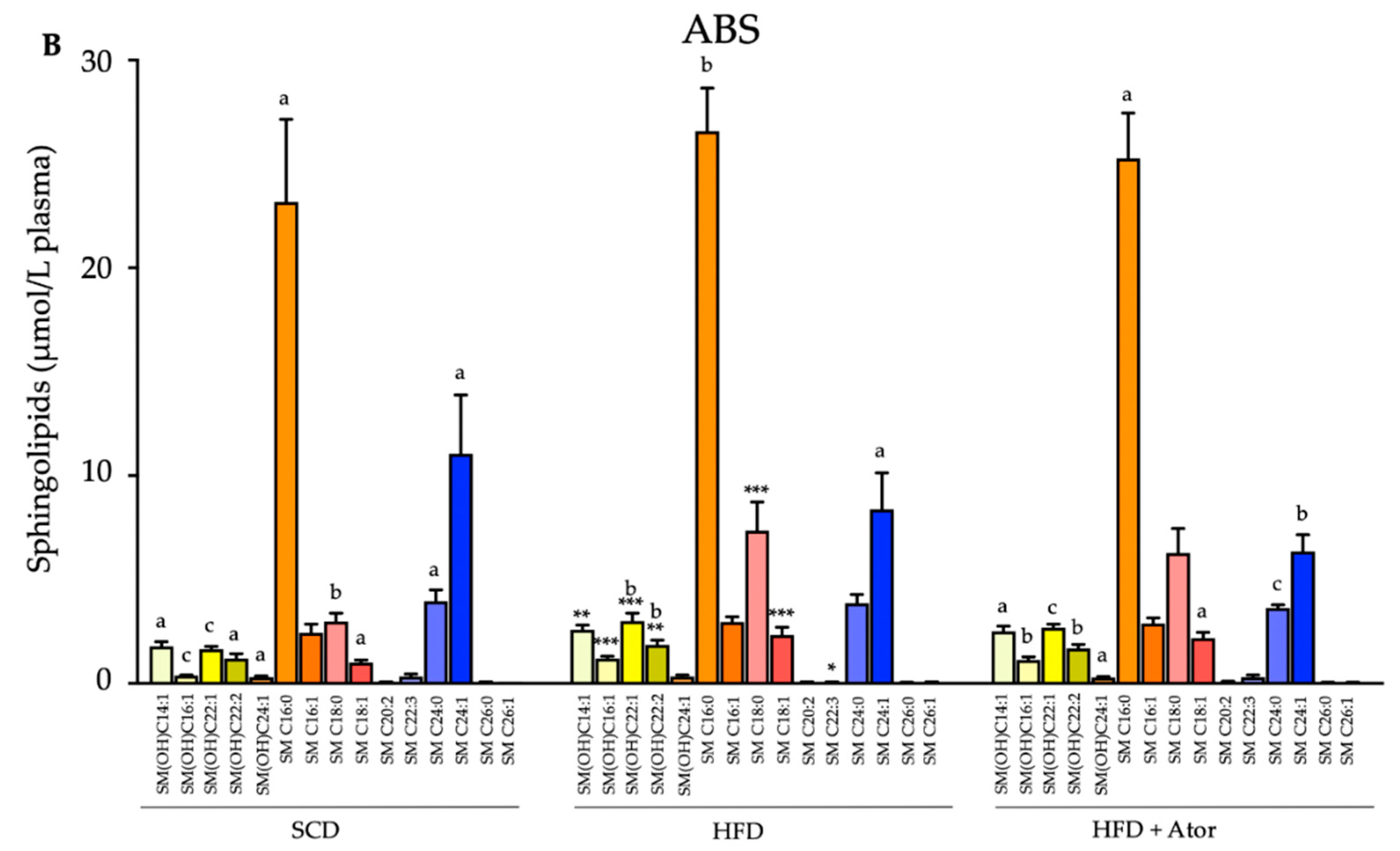
3.3. Regulatory Effect of Atorvastatin on Blood Lipidome in Mice with Intact and Depleted Gut Microbiota under High Fat Diet
3.4. Atorvastatin Alters the of Gut Microbiota Composition
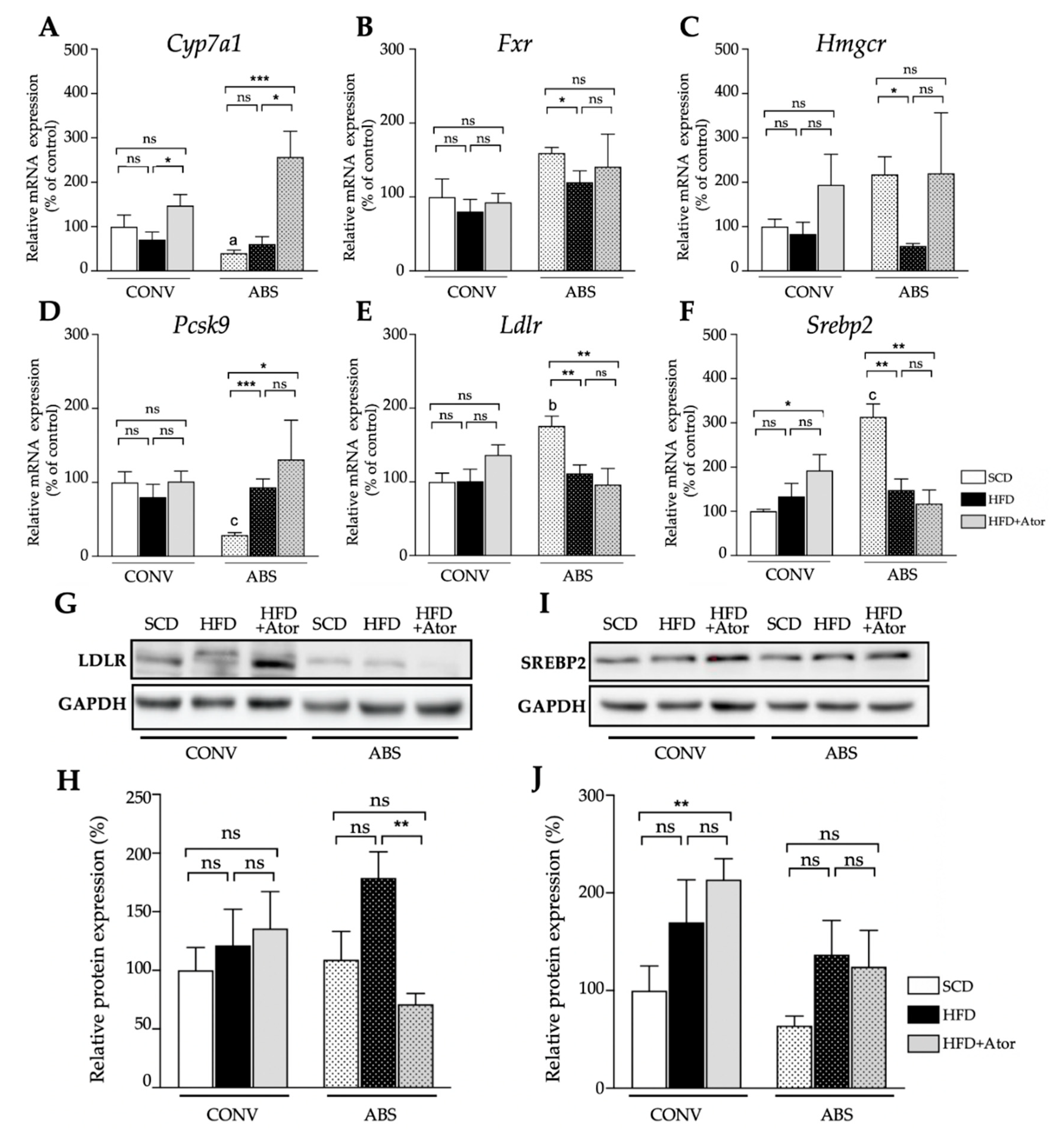
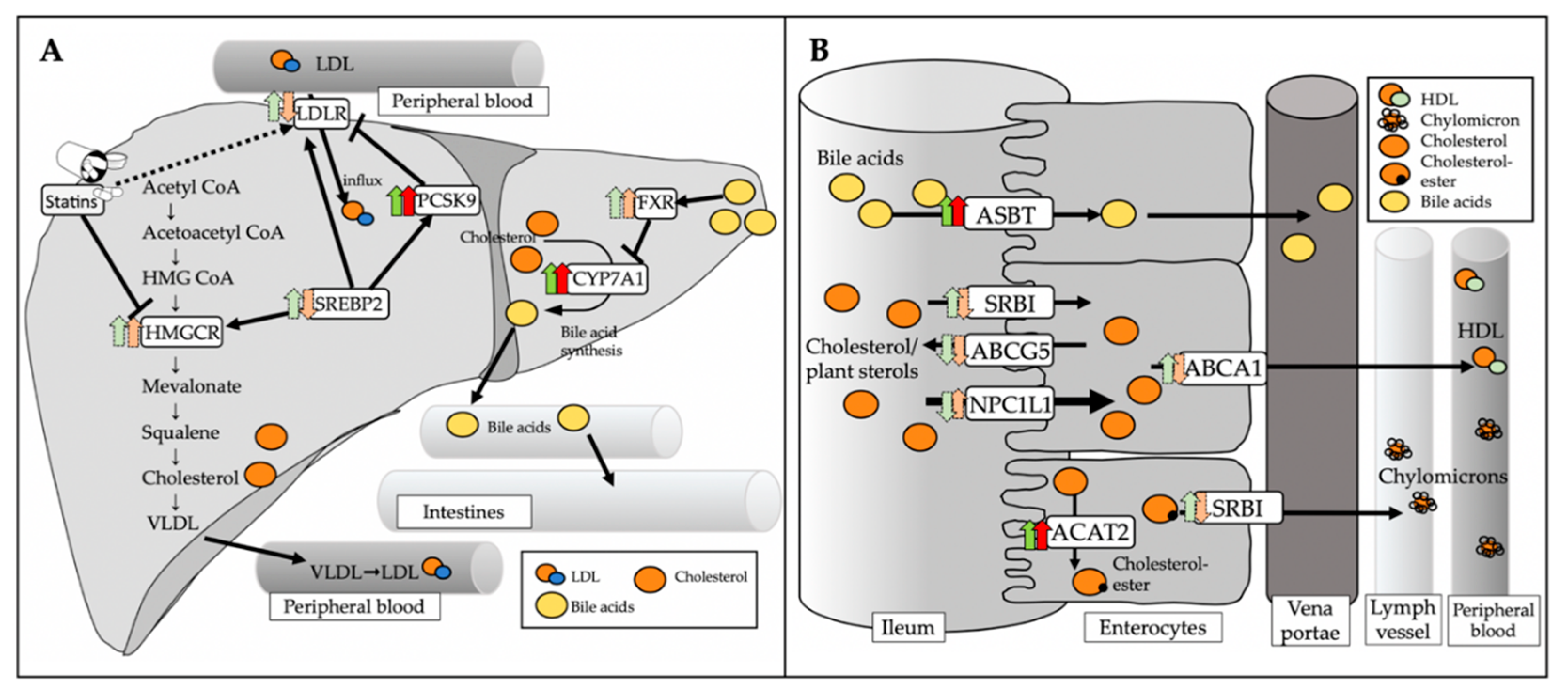
3.5. Regulatory Effect of Atorvastatin on Hepatic and Intestinal Genes Involved in Cholesterol Metabolism in Mice with Intact and Depleted Gut Microbiome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Yusuf, S.; Dagenais, G.; Liu, L.; Peters, R.J.G.; Lewis, B.S.; McKelvie, R.; Jung, H.; Gao, P.; Bosch, J.; Zhu, J.; et al. Cholesterol Lowering in Intermediate-Risk Persons without Cardiovascular Disease. N. Engl. J. Med. 2016, 374, 2021–2031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cannon, C.P.; Blazing, M.A.; Giugliano, R.P.; McCagg, A.; White, J.A.; Théroux, P.; Darius, H.; Lewis, B.S.; Ophuis, T.O.; Jukema, J.W.; et al. Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes. N. Engl. J. Med. 2015, 372, 2387–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirtori, C.R. The pharmacology of statins. Pharmacol. Res. 2014, 88, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Schonewille, M.; De Boer, J.F.; Mele, L.; Wolters, H.; Bloks, V.W.; Wolters, J.C.; Kuivenhoven, J.A.; Tietge, U.J.F.; Brufau, G.; Groen, A.K. Statins increase hepatic cholesterol synthesis and stimulate fecal cholesterol elimination in mice. J. Lipid Res. 2016, 57, 1455–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Heimesaat, M.M.; Bereswill, S.; Fischer, A.; Fuchs, D.; Struck, D.; Niebergall, J.; Jahn, H.-K.; Dunay, I.R.; Moter, A.; Gescher, D.M.; et al. Gram-negative bacteria aggravate murine small intestinal Th1-type immunopathology following oral infection with Toxoplasma gondii. J. Immunol. 2006, 177, 8785–8795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Klitzing, E.; Ekmekciu, I.; Kühl, A.A.; Bereswill, S.; Heimesaat, M.M. Multidrug-resistant Pseudomonas aeruginosa aggravates inflammatory responses in murine chronic colitis. Sci. Rep. 2018, 8, 6685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haghikia, A.; Missol-Kolka, E.; Tsikas, D.; Venturini, L.; Brundiers, S.; Castoldi, M.; Muckenthaler, M.U.; Eder, M.; Stapel, B.; Thum, T.; et al. Signal transducer and activator of transcription 3-mediated regulation of miR-199a-5p links cardiomyocyte and endothelial cell function in the heart: A key role for ubiquitin-conjugating enzymes. Eur. Heart J. 2010, 32, 1287–1297. [Google Scholar] [CrossRef] [PubMed]
- Bäckhed, F.; Manchester, J.K.; Semenkovich, C.F.; Gordon, J.I. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. USA 2007, 104, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akyea, R.K.; Kai, J.; Qureshi, N.; Iyen, B.; Weng, S.F. Sub-optimal cholesterol response to initiation of statins and future risk of cardiovascular disease. Heart 2019, 105, 975–981. [Google Scholar] [CrossRef] [PubMed]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Pedersen, H.K.; et al. Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef] [PubMed]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The mechanisms of action of metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.-Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Altmann, S.W.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.N.; Maguire, M.; Golovko, A.; Wang, L.; Murgolo, N.; Graziano, M.P.; et al. Niemann-Pick C1 Like 1 Protein Is Critical for Intestinal Cholesterol Absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2017, 19, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Gérard, P.; Lepercq, P.; Leclerc, M.; Gavini, F.; Raibaud, P.; Juste, C. Bacteroides sp. Strain D8, the First Cholesterol-Reducing Bacterium Isolated from Human Feces. Appl. Environ. Microbiol. 2007, 73, 5742–5749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mihalik, S.J.; Goodpaster, B.H.; Kelley, D.E.; Chace, D.H.; Vockley, J.; Toledo, F.G.; Delany, J.P. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 2010, 18, 1695–1700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaddurah-Daouk, R.; Baillie, R.; Zhu, H.; Zeng, Z.-B.; Wiest, M.M.; Nguyen, U.T.; Wojnoonski, K.; Watkins, S.M.; Trupp, M.; Krauss, R.M. Enteric Microbiome Metabolites Correlate with Response to Simvastatin Treatment. PLoS ONE 2011, 6, e25482. [Google Scholar] [CrossRef] [PubMed] [Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zimmermann, F.; Roessler, J.; Schmidt, D.; Jasina, A.; Schumann, P.; Gast, M.; Poller, W.; Leistner, D.; Giral, H.; Kränkel, N.; et al. Impact of the Gut Microbiota on Atorvastatin Mediated Effects on Blood Lipids. J. Clin. Med. 2020, 9, 1596. https://doi.org/10.3390/jcm9051596
Zimmermann F, Roessler J, Schmidt D, Jasina A, Schumann P, Gast M, Poller W, Leistner D, Giral H, Kränkel N, et al. Impact of the Gut Microbiota on Atorvastatin Mediated Effects on Blood Lipids. Journal of Clinical Medicine. 2020; 9(5):1596. https://doi.org/10.3390/jcm9051596
Chicago/Turabian StyleZimmermann, Friederike, Johann Roessler, David Schmidt, Andrzej Jasina, Paul Schumann, Martina Gast, Wolfgang Poller, David Leistner, Hector Giral, Nicolle Kränkel, and et al. 2020. "Impact of the Gut Microbiota on Atorvastatin Mediated Effects on Blood Lipids" Journal of Clinical Medicine 9, no. 5: 1596. https://doi.org/10.3390/jcm9051596
APA StyleZimmermann, F., Roessler, J., Schmidt, D., Jasina, A., Schumann, P., Gast, M., Poller, W., Leistner, D., Giral, H., Kränkel, N., Kratzer, A., Schuchardt, S., Heimesaat, M. M., Landmesser, U., & Haghikia, A. (2020). Impact of the Gut Microbiota on Atorvastatin Mediated Effects on Blood Lipids. Journal of Clinical Medicine, 9(5), 1596. https://doi.org/10.3390/jcm9051596