Silibinin and SARS-CoV-2: Dual Targeting of Host Cytokine Storm and Virus Replication Machinery for Clinical Management of COVID-19 Patients

 ,
,  , , and
, , and 
Abstract
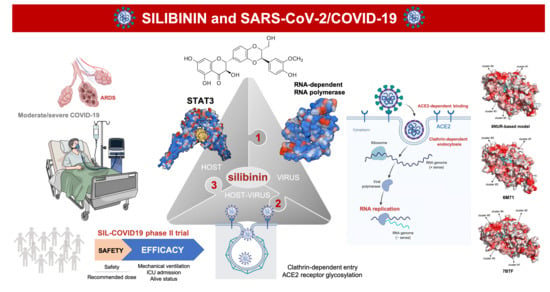
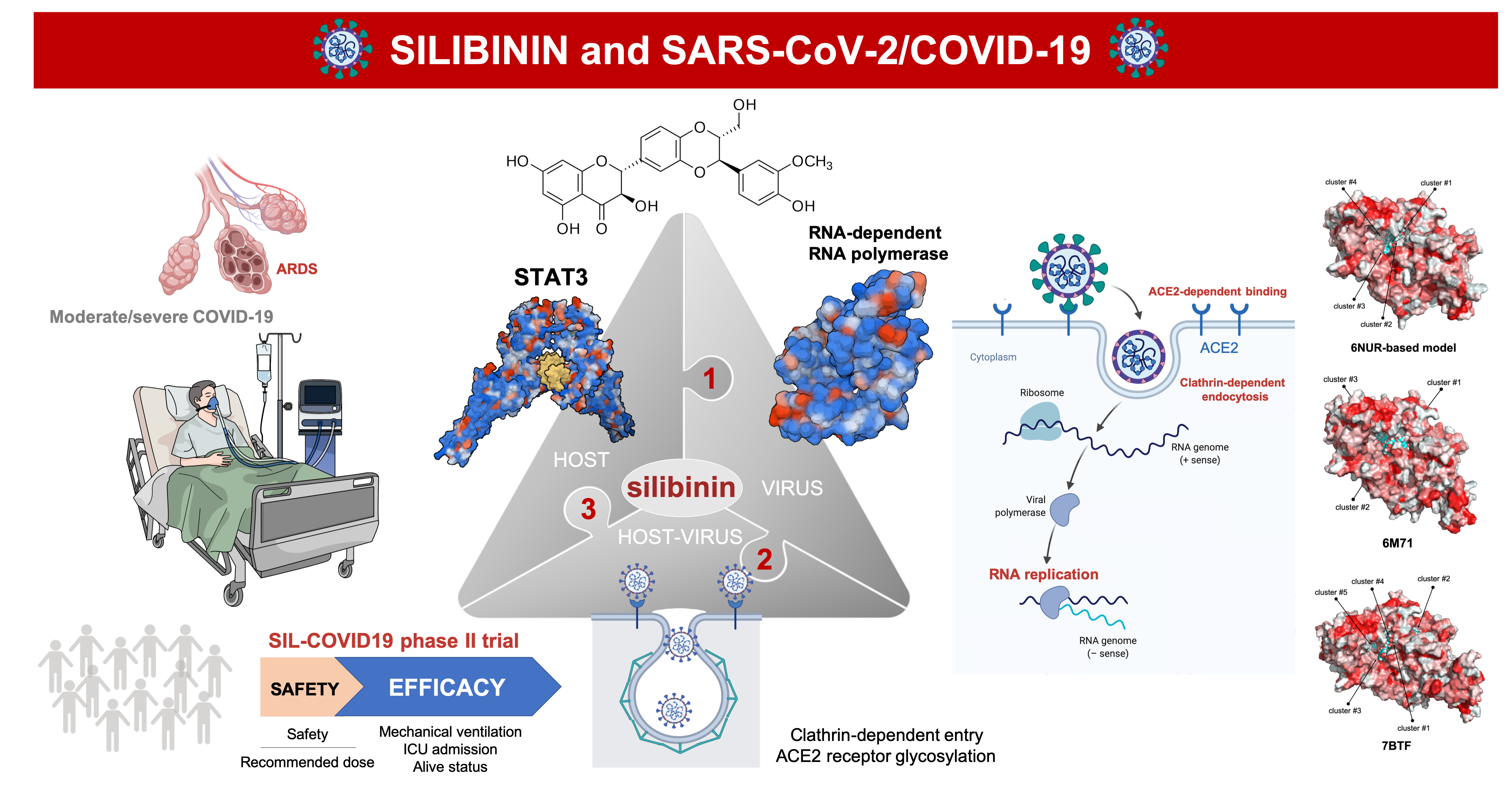
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Silibinin: From an Old Remedy to a Direct STAT3 Inhibitor
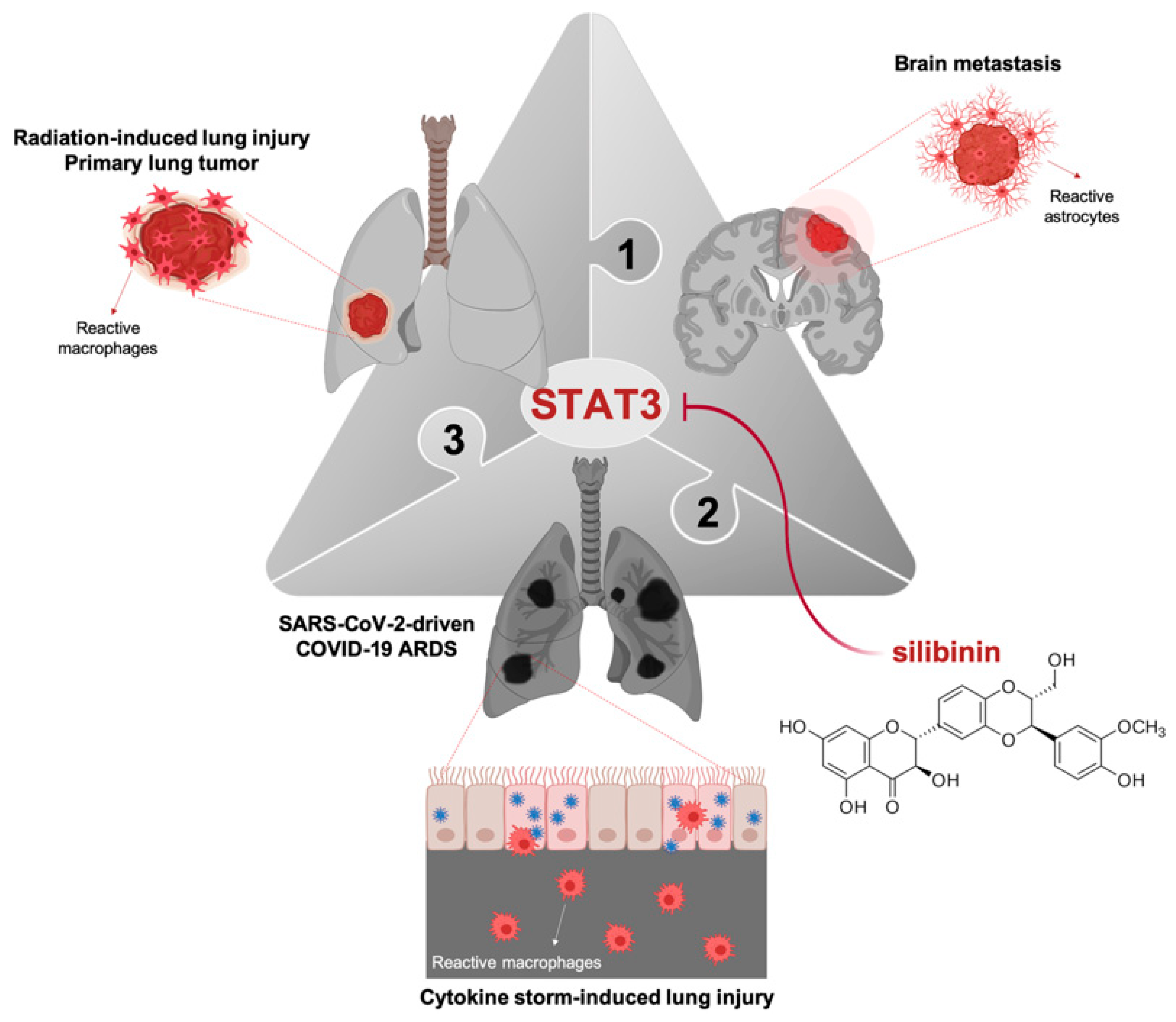
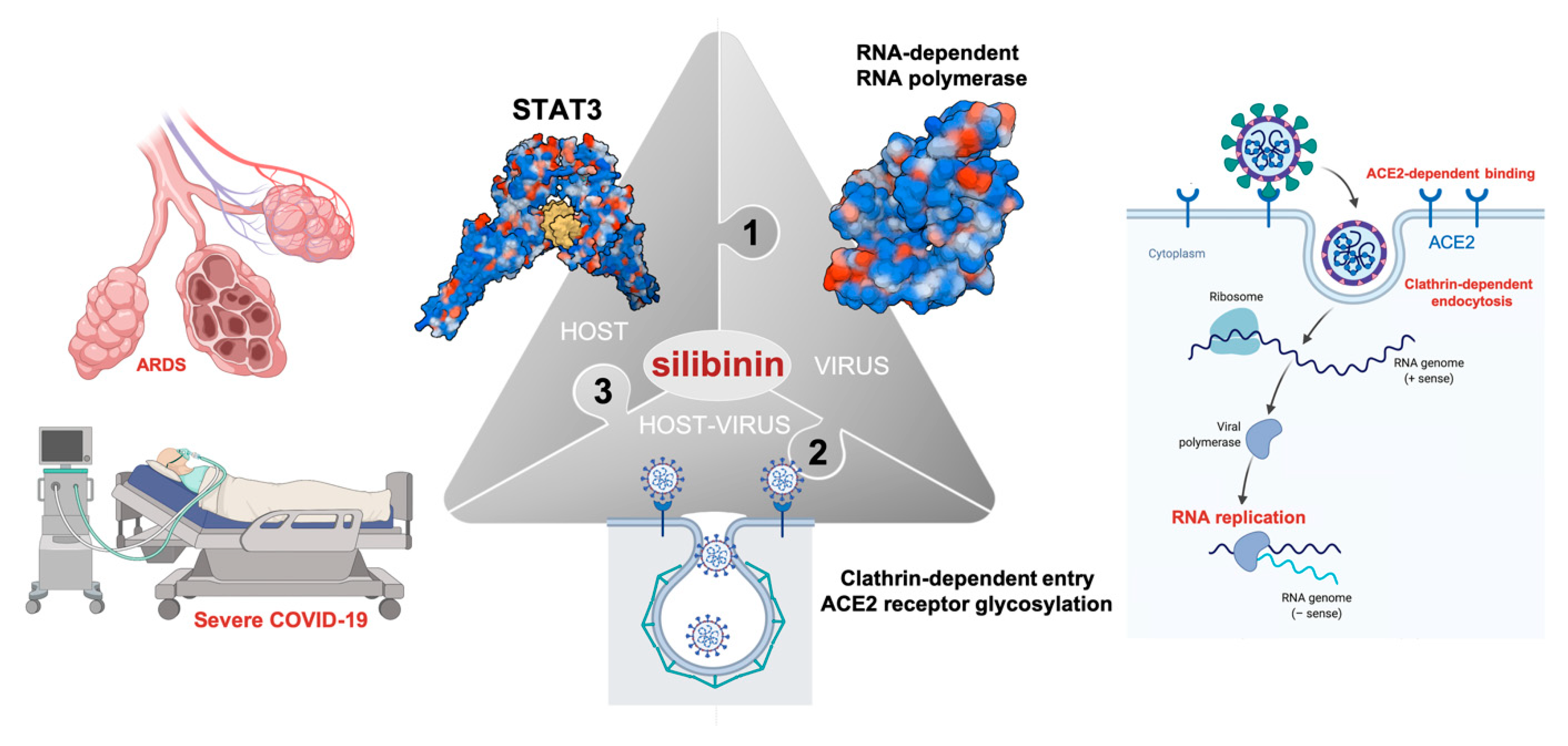
3. Silibinin and STAT3: Targeting the (Reactive) Cytokine Storm
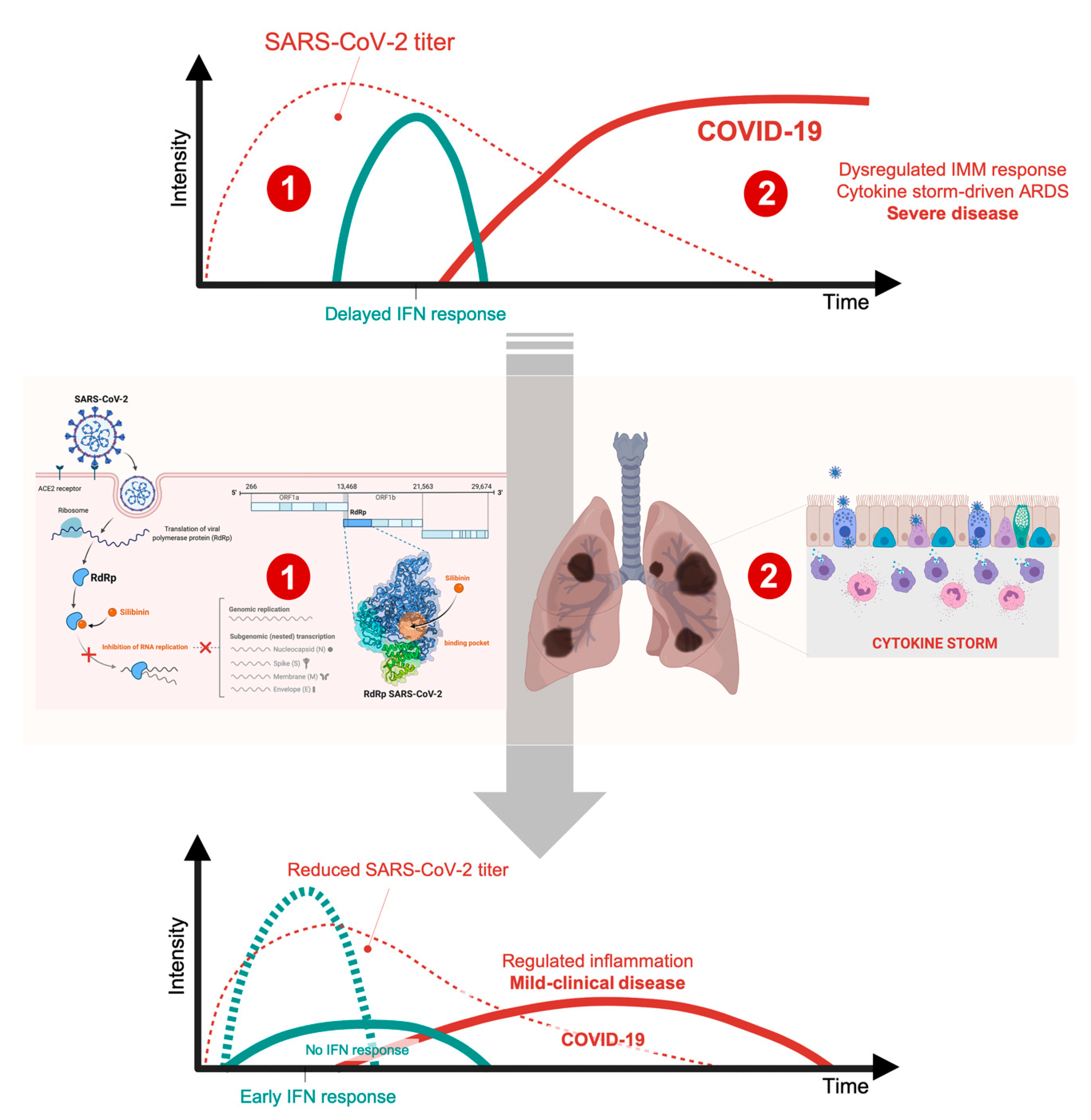
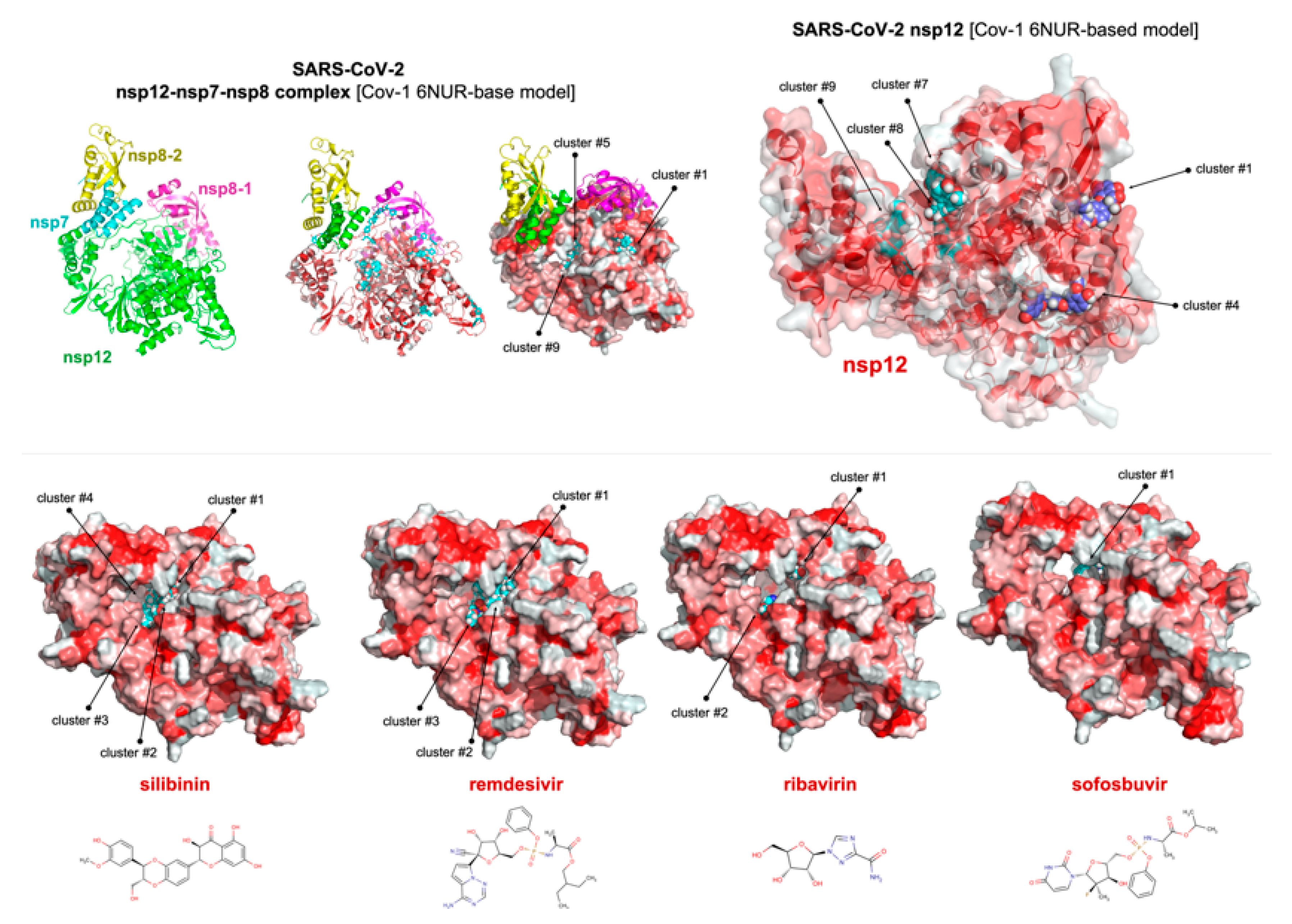
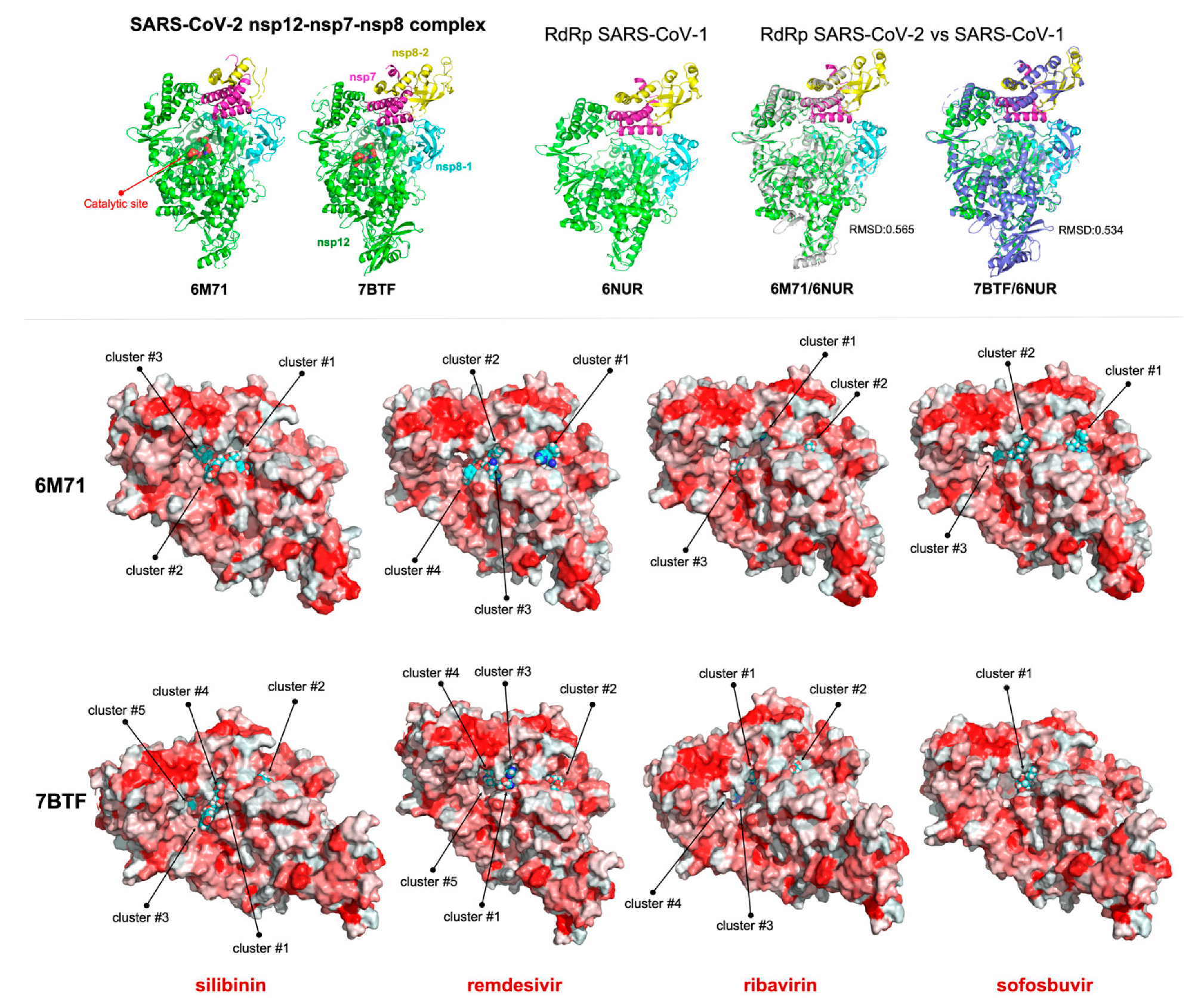
4. Silibinin and the RNA-Dependent RNA Polymerase Complex: Targeting Virus Replication
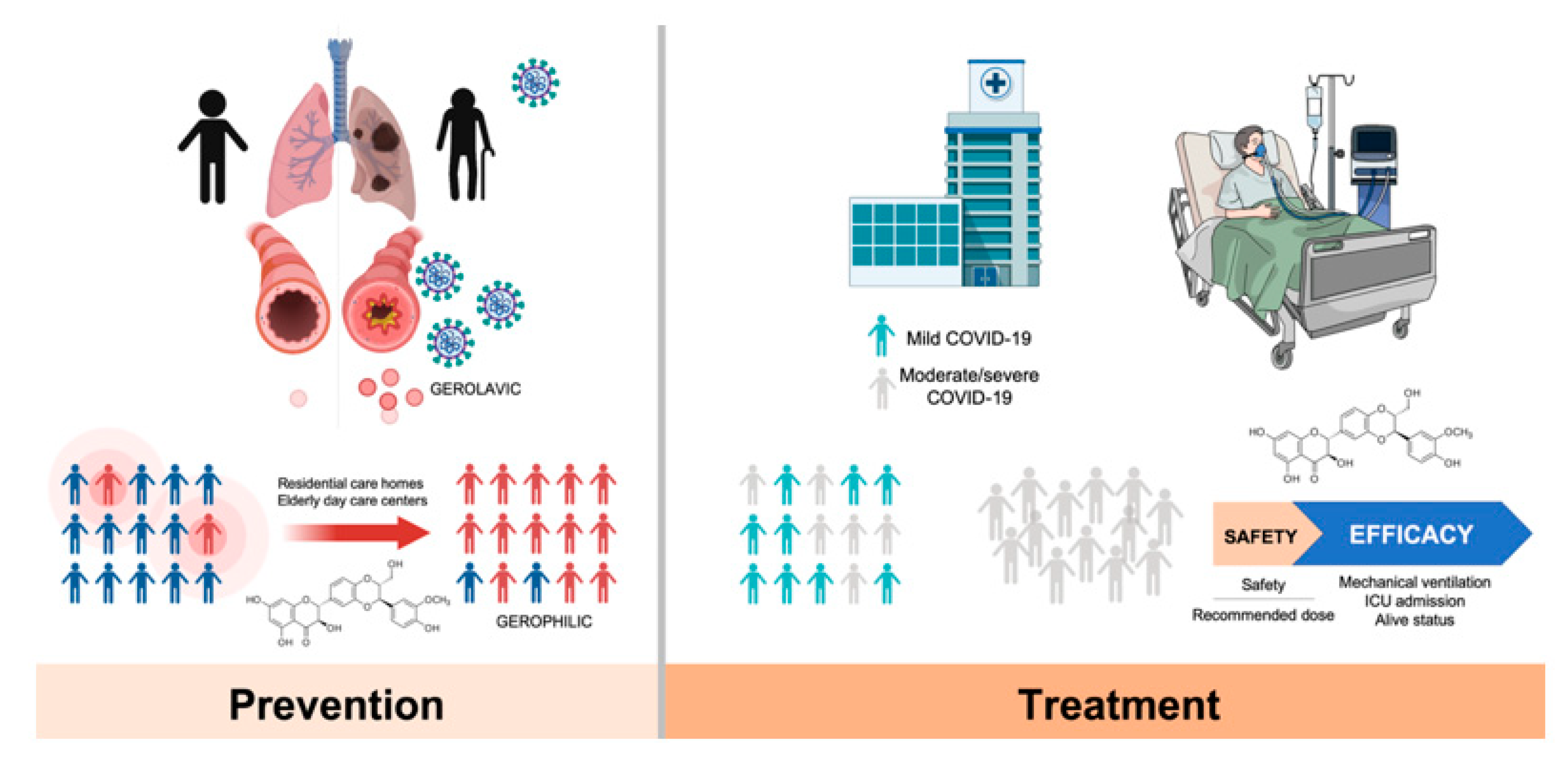
5. Silibinin and COVID-19: An Ongoing Clinical Trial
6. Silibinin and SARS-CoV-2: An Early-Intervention in Older Individuals at Risk of Severe COVID-19?
7. Silibinin Against the Lifecycle of SARS-CoV-2: Beyond Host STAT3 and Viral Replication
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Health Organization. Coronavirus Disease (COVID-19) Outbreak. Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019 (accessed on 28 April 2020).
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Guan, W.J.; Ni, Z.Y.; Hu, Y.; Liang, W.H.; Ou, C.Q.; He, J.X.; Liu, L.; Shan, H.; Lei, C.L.; Hui, D.S.C.; et al. China Medical Treatment Expert Group for Covid-19. Clinical Characteristics of Coronavirus Disease 2019 in China. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Li, T.; Han, M.; Li, X.; Wu, D.; Xu, Y.; Zhu, Y.; Liu, Y.; Wang, X.; Wang, L. Diagnostic Utility of Clinical Laboratory Data Determinations for Patients with the Severe COVID-19. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Wu, D.; Guo, W.; Cao, Y.; Huang, D.; Wang, H.; Wang, T.; Zhang, X.; Chen, H.; Yu, H.; et al. Clinical and immunological features in severe and moderate Coronavirus Disease 2019. J. Clin. Investig. 2020, 130, 2620–2629. [Google Scholar] [CrossRef] [Green Version]
- Pedersen, S.F.; Ho, Y.C. SARS-CoV-2: A Storm is Raging. J. Clin. Investig. 2020, 130, 2202–2205. [Google Scholar] [CrossRef]
- Zhang, C.; Wu, Z.; Li, J.W.; Zhao, H.; Wang, G.Q. The cytokine release syndrome (CRS) of severe COVID-19 and Interleukin-6 receptor (IL-6R) antagonist Tocilizumab may be the key to reduce the mortality. Int. J. Antimicrob. Agents 2020, 55, 105954. [Google Scholar] [CrossRef]
- Michot, J.M.; Albiges, L.; Chaput, N.; Saada, V.; Pommeret, F.; Griscelli, F.; Balleyguier, C.; Besse, B.; Marabelle, A.; Netzer, F.; et al. Tocilizumab, an anti-IL6 receptor antibody, to treat Covid-19-related respiratory failure: A case report. Ann. Oncol. 2020, 7534, 36387. [Google Scholar]
- Ceribelli, A.; Motta, F.; De Santis, M.; Ansari, A.A.; Ridgway, W.M.; Gershwin, M.E.; Selmi, C. Recommendations for coronavirus infection in rheumatic diseases treated with biologic therapy. J. Autoimmun. 2020, 109, 102442. [Google Scholar] [CrossRef]
- Luo, P.; Liu, Y.; Qiu, L.; Liu, X.; Liu, D.; Li, J. Tocilizumab treatment in COVID-19: A single center experience. J. Med. Virol. 2020. [Google Scholar] [CrossRef]
- Stebbing, J.; Phelan, A.; Griffin, I.; Tucker, C.; Oechsle, O.; Smith, D.; Richardson, P. COVID-19: Combining antiviral and anti-inflammatory treatments. Lancet Infect. Dis. 2020, 20, 400–402. [Google Scholar] [CrossRef]
- Richardson, P.; Griffin, I.; Tucker, C.; Smith, D.; Oechsle, O.; Phelan, A.; Stebbing, J. Baricitinib as potential treatment for 2019-nCoV acute respiratory disease. Lancet. 2020, 395, e30–e31. [Google Scholar] [CrossRef] [Green Version]
- Favalli, E.G.; Biggioggero, M.; Maioli, G.; Caporali, R. Baricitinib for COVID-19: A suitable treatment? Lancet Infect. Dis. 2020, 3099, 30262. [Google Scholar] [CrossRef]
- Richardson, P.J.; Corbellino, M.; Stebbing, J. Baricitinib for COVID-19: A suitable treatment?—Authors’ reply. Lancet Infect. Dis. 2020, S1473–3099, 30270-X. [Google Scholar] [CrossRef]
- Praveen, D.; Chowdary, P.R.; Aanandhi, M.V. Janus kinase inhibitor baricitinib is not an ideal option for management of COVID-19. Int. J. Antimicrob. Agents. 2020, 55, 105967. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.C.; Graf, T.N.; Sparacino, C.M.; Wani, M.C.; Wall, M.E. Complete isolation and characterization of silybins and isosilybins from milk thistle (Silybum marianum). Org. Biomol. Chem. 2003, 1, 1684–1689. [Google Scholar] [CrossRef]
- Gazák, R.; Walterová, D.; Kren, V. Silybin and silymarin—New and emerging applications in medicine. Curr. Med. Chem. 2007, 14, 315–338. [Google Scholar] [CrossRef]
- Hackett, E.S.; Twedt, D.C.; Gustafson, D.L. Milk thistle and its derivative compounds: A review of opportunities for treatment of liver disease. J. Vet. Intern. Med. 2013, 27, 10–16. [Google Scholar] [CrossRef]
- Abenavoli, L.; Izzo, A.A.; Milić, N.; Cicala, C.; Santini, A.; Capasso, R. Milk thistle (Silybum marianum): A concise overview on its chemistry, pharmacological, and nutraceutical uses in liver diseases. Phytother. Res. 2018, 32, 2202–2213. [Google Scholar] [CrossRef]
- Bijak, M. Silybin, a Major Bioactive Component of Milk Thistle (Silybum marianum L. Gaernt.)—Chemistry, Bioavailability, and Metabolism. Molecules 2017, 22, E1942. [Google Scholar] [CrossRef] [Green Version]
- Rho, J.K.; Choi, Y.J.; Jeon, B.S.; Choi, S.J.; Cheon, G.J.; Woo, S.K.; Kim, H.R.; Kim, C.H.; Choi, C.M.; Lee, J.C. Combined treatment with silibinin and epidermal growth factor receptor tyrosine kinase inhibitors overcomes drug resistance caused by T790M mutation. Mol. Cancer Ther. 2010, 9, 3233–3243. [Google Scholar] [CrossRef] [Green Version]
- Cufí, S.; Bonavia, R.; Vazquez-Martin, A.; Corominas-Faja, B.; Oliveras-Ferraros, C.; Cuyàs, E.; Martin-Castillo, B.; Barrajón-Catalán, E.; Visa, J.; Segura-Carretero, A.; et al. Silibinin meglumine, a water-soluble form of milk thistle silymarin, is an orally active anti-cancer agent that impedes the epithelial-to-mesenchymal transition (EMT) in EGFR-mutant non-small-cell lung carcinoma cells. Food Chem. Toxicol. 2013, 60, 360–368. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Menendez, J.A. Silibinin and STAT3: A natural way of targeting transcription factors for cancer therapy. Cancer Treat. Rev. 2015, 41, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Bosch-Barrera, J.; Sais, E.; Cañete, N.; Marruecos, J.; Cuyàs, E.; Izquierdo, A.; Porta, R.; Haro, M.; Brunet, J.; Pedraza, S.; et al. Response of brain metastasis from lung cancer patients to an oral nutraceutical product containing silibinin. Oncotarget 2016, 7, 32006–32014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belli, V.; Sforza, V.; Cardone, C.; Martinelli, E.; Barra, G.; Matrone, N.; Napolitano, S.; Morgillo, F.; Tuccillo, C.; Federico, A.; et al. Regorafenib in combination with silybin as a novel potential strategy for the treatment of metastatic colorectal cancer. Oncotarget 2017, 8, 68305–68316. [Google Scholar] [CrossRef] [Green Version]
- Bosch-Barrera, J.; Queralt, B.; Menendez, J.A. Targeting STAT3 with silibinin to improve cancer therapeutics. Cancer Treat. Rev. 2017, 58, 61–69. [Google Scholar] [CrossRef]
- Pérez-Sánchez, A.; Cuyàs, E.; Ruiz-Torres, V.; Agulló-Chazarra, L.; Verdura, S.; González-Álvarez, I.; Bermejo, M.; Joven, J.; Micol, V.; Bosch-Barrera, J.; et al. Intestinal Permeability Study of Clinically Relevant Formulations of Silibinin in Caco-2 Cell Monolayers. Int. J. Mol. Sci. 2019, 20, E1606. [Google Scholar] [CrossRef] [Green Version]
- Priego, N.; Zhu, L.; Monteiro, C.; Mulders, M.; Wasilewski, D.; Bindeman, W.; Doglio, L.; Martínez, L.; Martínez-Saez, E.; Ramón, Y.; et al. STAT3 labels a subpopulation of reactive astrocytes required for brain metastasis. Nat. Med. 2018, 24, 1024–1035. [Google Scholar] [CrossRef]
- Agarwal, C.; Tyagi, A.; Kaur, M.; Agarwal, R. Silibinin inhibits constitutive activation of Stat3, and causes caspase activation and apoptotic death of human prostate carcinoma DU145 cells. Carcinogenesis 2007, 28, 1463–1470. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Singh, R.P.; Ramasamy, K.; Raina, K.; Redente, E.F.; Dwyer-Nield, L.D.; Radcliffe, R.A.; Malkinson, A.M.; Agarwal, R. Growth inhibition and regression of lung tumors by silibinin: Modulation of angiogenesis by macrophage-associated cytokines and nuclear factor-kappaB and signal transducers and activators of transcription 3. Cancer Prev. Res. 2009, 2, 74–83. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.P.; Raina, K.; Deep, G.; Chan, D.; Agarwal, R. Silibinin suppresses growth of human prostate carcinoma PC-3 orthotopic xenograft via activation of extracellular signal-regulated kinase 1/2 and inhibition of signal transducers and activators of transcription signaling. Clin. Cancer Res. 2009, 15, 613–621. [Google Scholar] [CrossRef] [Green Version]
- Tyagi, A.; Agarwal, C.; Dwyer-Nield, L.D.; Singh, R.P.; Malkinson, A.M.; Agarwal, R. Silibinin modulates TNF-α and IFN-γ mediated signaling to regulate COX2 and iNOS expression in tumorigenic mouse lung epithelial LM2 cells. Mol Carcinog. 2012, 51, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Pérez-Sánchez, A.; Micol, V.; Menendez, J.A.; Bosch-Barrera, J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle. 2016, 15, 3413–3418. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Zhou, Q.; Gao, S.; Li, W.; Li, X.; Liu, Z.; Jin, P.; Jiang, J. Silibinin inhibits endometrial carcinoma via blocking pathways of STAT3 activation and SREBP1-mediated lipid accumulation. Life Sci. 2019, 217, 70–80. [Google Scholar] [CrossRef]
- Zheng, R.; Ma, J.; Wang, D.; Dong, W.; Wang, S.; Liu, T.; Xie, R.; Liu, L.; Wang, B.; Cao, H. Chemopreventive Effects of Silibinin on Colitis-Associated Tumorigenesis by Inhibiting IL-6/STAT3 Signaling Pathway. Mediat. Inflamm. 2018, 2018, 1562010. [Google Scholar] [CrossRef] [PubMed]
- Verdura, S.; Cuyàs, E.; Llorach-Parés, L.; Pérez-Sánchez, A.; Micol, V.; Nonell-Canals, A.; Joven, J.; Valiente, M.; Sánchez-Martínez, M.; Bosch-Barrera, J.; et al. Silibinin is a direct inhibitor of STAT3. Food Chem. Toxicol. 2018, 116, 161–172. [Google Scholar] [CrossRef]
- Gao, H.; Ward, P.A. STAT3 and suppressor of cytokine signaling 3: Potential targets in lung inflammatory responses. Expert Opin. Ther. Targets 2007, 11, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Carnesecchi, S.; Dunand-Sauthier, I.; Zanetti, F.; Singovski, G.; Deffert, C.; Donati, Y.; Cagarelli, T.; Pache, J.C.; Krause, K.H.; Reith, W.; et al. NOX1 is responsible for cell death through STAT3 activation in hyperoxia and is associated with the pathogenesis of acute respiratory distress syndrome. Int. J. Clin. Exp. Pathol. 2014, 7, 537–551. [Google Scholar]
- Mizushina, Y.; Shirasuna, K.; Usui, F.; Karasawa, T.; Kawashima, A.; Kimura, H.; Kobayashi, M.; Komada, T.; Inoue, Y.; Mato, N.; et al. NLRP3 protein deficiency exacerbates hyperoxia-induced lethality through Stat3 protein signaling independent of interleukin-1β. J. Biol. Chem. 2015, 290, 5065–5077. [Google Scholar] [CrossRef] [Green Version]
- Kwok, H.H.; Poon, P.Y.; Fok, S.P.; Ying-Kit Yue, P.; Mak, N.K.; Chan, M.C.; Peiris, J.S.; Wong, R.N. Anti-inflammatory effects of indirubin derivatives on influenza A virus-infected human pulmonary microvascular endothelial cells. Sci. Rep. 2016, 6, 18941. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; Hu, X.; Sun, R.; Tu, Y.; Gong, F.; Ni, Y. Resolution acute respiratory distress syndrome through reversing the imbalance of Treg/Th17 by targeting the cAMP signaling pathway. Mol. Med. Rep. 2016, 14, 343–348. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Yu, H.; Liu, Y.; Gibson, S.A.; Yan, Z.; Xu, X.; Gaggar, A.; Li, P.K.; Li, C.; Wei, S.; et al. Protective effect of suppressing STAT3 activity in LPS-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L868–L880. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Ciechanowicz, A.K.; Kaplan, A.R.; Wang, L.; Zhang, P.X.; Lu, Y.C.; Tobin, R.E.; Tobin, B.A.; Cohn, L.; Zeiss, C.J.; et al. Surfactant protein C dampens inflammation by decreasing JAK/STAT activation during lung repair. Am. J. Physiol. Lung Cell Mol. Physiol. 2018, 314, L882–L892. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Yang, N.; Pan, G.; Jin, B.; Wang, S.; Ji, W. Elevated IL-33 promotes expression of MMP2 and MMP9 via activating STAT3 in alveolar macrophages during LPS-induced acute lung injury. Cell Mol. Biol. Lett. 2018, 23, 52. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Luo, Y.; Wang, X.; Zhu, J.; Li, Q.; Feng, J.; He, D.; Zhong, Z.; Zheng, X.; Lu, J.; et al. Global transcriptional regulation of STAT3- and MYC-mediated sepsis-induced ARDS. Ther. Adv. Respir. Dis. 2019, 13, 1753466619879840. [Google Scholar] [CrossRef]
- Li, S.W.; Wang, C.Y.; Jou, Y.J.; Yang, T.C.; Huang, S.H.; Wan, L.; Lin, Y.J.; Lin, C.W. SARS coronavirus papain-like protease induces Egr-1-dependent up-regulation of TGF-β1 via ROS/p38 MAPK/STAT3 pathway. Sci. Rep. 2016, 6, 25754. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Wang, B.; Cao, S.; Wang, Y.; Wu, D. Silybin attenuates LPS-induced lung injury in mice by inhibiting NF-κB signaling and NLRP3 activation. Int. J. Mol. Med. 2017, 39, 1111–1118. [Google Scholar] [CrossRef]
- Tian, L.; Li, W.; Wang, T. Therapeutic effects of silibinin on LPS-induced acute lung injury by inhibiting NLRP3 and NF-κB signaling pathways. Microb. Pathog. 2017, 108, 104–108. [Google Scholar] [CrossRef]
- Son, Y.; Lee, H.J.; Rho, J.K.; Chung, S.Y.; Lee, C.G.; Yang, K.; Kim, S.H.; Lee, M.; Shin, I.S.; Kim, J.S. The ameliorative effect of silibinin against radiation-induced lung injury: Protection of normal tissue without decreasing therapeutic efficacy in lung cancer. BMC Pulm. Med. 2015, 15, 68. [Google Scholar] [CrossRef] [Green Version]
- Channappanavar, R.; Fehr, A.R.; Vijay, R.; Mack, M.; Zhao, J.; Meyerholz, D.K.; Perlman, S. Dysregulated Type I Interferon and Inflammatory Monocyte-Macrophage Responses Cause Lethal Pneumonia in SARS-CoV-Infected Mice. Cell Host Microbe 2016, 19, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Kindler, E.; Thiel, V. SARS-CoV and IFN: Too Little, Too Late. Cell Host Microbe 2016, 19, 139–141. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Guo, R.; Lei, L.; Liu, H.; Wang, Y.; Wang, Y.; Dai, T.; Zhang, T.; Lai, Y.; Wang, J.; et al. COVID-19 infection induces readily detectable morphological and inflammation-related phenotypic changes in peripheral blood monocytes, the severity of which correlate with patient outcome. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Liao, M.; Liu, Y.; Yuan, J.; Wen, Y.; Xu, G.; Zhao, J.; Chen, L.; Li, J.; Wang, X.; Wang, F.; et al. The landscape of lung bronchoalveolar immune cells in COVID-19 revealed by single-cell RNA sequencing. medRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Chan, J.F.; Kok, K.H.; Zhu, Z.; Chu, H.; To, K.K.; Yuan, S.; Yuen, K.Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. Methods Mol. Biol. 2015, 1282, 1–23. [Google Scholar]
- Totura, A.L.; Bavari, S. Broad-spectrum coronavirus antiviral drug discovery. Expert Opin. Drug Discov. 2019, 14, 397–412. [Google Scholar] [CrossRef] [Green Version]
- Sheahan, T.P.; Sims, A.C.; Graham, R.L.; Menachery, V.D.; Gralinski, L.E.; Case, J.B.; Leist, S.R.; Pyrc, K.; Feng, J.Y.; Trantcheva, I.; et al. Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 2017, 9, eaal3653. [Google Scholar] [CrossRef] [Green Version]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93, e01815–e01818. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Zhou, Y.; Vedantham, P.; Lu, K.; Agudelo, J.; Carrion, R., Jr.; Nunneley, J.W.; Barnard, D.; Pöhlmann, S.; McKerrow, J.H.; Renslo, A.R.; et al. Protease inhibitors targeting coronavirus and filovirus entry. Antiviral Res. 2015, 116, 76–84. [Google Scholar] [CrossRef]
- Yamamoto, M.; Matsuyama, S.; Li, X.; Takeda, M.; Kawaguchi, Y.; Inoue, J.I.; Matsuda, Z. Identification of Nafamostat as a Potent Inhibitor of Middle East Respiratory Syndrome Coronavirus S Protein-Mediated Membrane Fusion Using the Split-Protein-Based Cell-Cell Fusion Assay. Antimicrob. Agents Chemother. 2016, 60, 6532–6539. [Google Scholar] [CrossRef] [Green Version]
- Sisk, J.M.; Frieman, M.B.; Machamer, C.E. Coronavirus S protein-induced fusion is blocked prior to hemifusion by Abl kinase inhibitors. J. Gen. Virol. 2018, 99, 619–630. [Google Scholar] [CrossRef] [PubMed]
- Coleman, C.M.; Sisk, J.M.; Mingo, R.M.; Nelson, E.A.; White, J.M.; Frieman, M.B. Abelson Kinase Inhibitors Are Potent Inhibitors of Severe Acute Respiratory Syndrome Coronavirus and Middle East Respiratory Syndrome Coronavirus Fusion. J. Virol. 2016, 90, 8924–8933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, S.; Zhu, Y.; Liu, M.; Lan, Q.; Xu, W.; Wu, Y.; Ying, T.; Liu, S.; Shi, Z.; Jiang, S.; et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Báez-Santos, Y.M.; St John, S.E.; Mesecar, A.D. The SARS-coronavirus papain-like protease: Structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015, 115, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Ju, J.; Kumar, S.; Li, X.; Jockusch, S.; Russo, J.J. Nucleotide analogues as inhibitors of viral polymerases. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wu, C.; Liu, Y.; Yang, Y.; Zhang, P.; Zhong, W.; Wang, Y.; Wang, Q.; Xu, Y.; Li, M.; Li, X.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sin. B 2020. [Google Scholar] [CrossRef]
- Ahn, D.G.; Choi, J.K.; Taylor, D.R.; Oh, J.W. Biochemical characterization of a recombinant SARS coronavirus nsp12 RNA-dependent RNA polymerase capable of copying viral RNA templates. Arch. Virol. 2012, 157, 2095–2104. [Google Scholar] [CrossRef] [Green Version]
- Subissi, L.; Posthuma, C.C.; Collet, A.; Zevenhoven-Dobbe, J.C.; Gorbalenya, A.E.; Decroly, E.; Snijder, E.J.; Canard, B.; Imbert, I. One severe acute respiratory syndrome coronavirus protein complex integrates processive RNA polymerase and exonuclease activities. Proc. Natl. Acad. Sci. USA 2014, 111, E3900–E3909. [Google Scholar] [CrossRef] [Green Version]
- Zhai, Y.; Sun, F.; Li, X.; Pang, H.; Xu, X.; Bartlam, M.; Rao, Z. Insights into SARS-CoV transcription and replication from the structure of the nsp7-nsp8 hexadecamer. Nat. Struct. Mol. Biol. 2005, 12, 980–986. [Google Scholar] [CrossRef]
- Hillen, H.S.; Kokic, G.; Farnung, L.; Dienemann, C.; Tegunov, D.; Cramer, P. Structure of replicating SARS-CoV-2 polymerase. Nature 2020. [Google Scholar] [CrossRef] [PubMed]
- McDonald, S.M. RNA synthetic mechanisms employed by diverse families of RNA viruses. Wiley Interdiscip. Rev. RNA 2013, 4, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Kirchdoerfer, R.N.; Ward, A.B. Structure of the SARS-CoV nsp12 polymerase bound to nsp7 and nsp8 co-factors. Nat. Commun. 2019, 10, 2342. [Google Scholar] [CrossRef] [Green Version]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020. [Google Scholar] [CrossRef] [Green Version]
- Grein, J.; Ohmagari, N.; Shin, D.; Diaz, G.; Asperges, E.; Castagna, A.; Feldt, T.; Green, G.; Green, M.L.; Lescure, F.X.; et al. Compassionate Use of Remdesivir for Patients with Severe Covid-19. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fleming, S.B. Viral Inhibition of the IFN-Induced JAK/STAT Signalling Pathway: Development of Live Attenuated Vaccines by Mutation of Viral-Encoded IFN-Antagonists. Vaccines (Basel) 2016, 4, E23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, C.G.K.; Allon, S.J.; Nyquist, S.K.; Mbano, I.M.; Miao, V.N.; Tzouanas, C.N.; Cao, Y.; Yousif, A.S.; Bals, J.; Hauser, B.M.; et al. HCA Lung Biological Network. SARS-CoV-2 receptor ACE2 is an interferon-stimulated gene in human airway epithelial cells and is detected in specific cell subsets across tissues. Cell 2020. [Google Scholar] [CrossRef]
- Choy, E.H.S.; Miceli-Richard, C.; González-Gay, M.A.; Sinigaglia, L.; Schlichting, D.E.; Meszaros, G.; de la Torre, I.; Schulze-Koops, H. The effect of JAK1/JAK2 inhibition in rheumatoid arthritis: efficacy and safety of baricitinib. Clin. Exp. Rheumatol. 2019, 37, 694–704. [Google Scholar]
- Honda, S.; Harigai, M. The safety of baricitinib in patients with rheumatoid arthritis. Expert Opin. Drug Saf. 2020, 19, 545–551. [Google Scholar] [CrossRef]
- Bechman, K.; Yates, M.; Galloway, J.B. The new entries in the therapeutic armamentarium: The small molecule JAK inhibitors. Pharmacol. Res. 2019, 147, 104392. [Google Scholar] [CrossRef]
- Gadina, M.; Le, M.T.; Schwartz, D.M.; Silvennoinen, O.; Nakayamada, S.; Yamaoka, K.; O’Shea, J.J. Janus kinases to jakinibs: From basic insights to clinical practice. Rheumatology (Oxford) 2019, 58, i4–i16. [Google Scholar] [CrossRef] [PubMed]
- Segler, M.H.S.; Preuss, M.; Waller, M.P. Planning chemical syntheses with deep neural networks and symbolic AI. Nature 2018, 555, 604–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.H. Randomized phase II trials with a prospective control. Stat. Med. 2008, 27, 568–583. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhu, F.; Xie, L.; Wang, C.; Wang, J.; Chen, R.; Jia, P.; Guan, H.Q.; Peng, L.; Chen, Y.; et al. Clinical characteristics of COVID-19-infected cancer patients: A retrospective case study in three hospitals within Wuhan, China. Ann. Oncol. 2020. [Google Scholar] [CrossRef]
- Moltó, J.; Valle, M.; Miranda, C.; Cedeño, S.; Negredo, E.; Clotet, B. Effect of milk thistle on the pharmacokinetics of darunavir-ritonavir in HIV-infected patients. Antimicrob. Agents Chemother. 2012, 56, 2837–2841. [Google Scholar] [CrossRef] [Green Version]
- Zhavoronkov, A. Geroprotective and senoremediative strategies to reduce the comorbidity, infection rates, severity, and lethality in gerophilic and gerolavic infections. Aging (Albany NY) 2020, 12. [Google Scholar] [CrossRef]
- Chen, J.; Lau, Y.F.; Lamirande, E.W.; Paddock, C.D.; Bartlett, J.H.; Zaki, S.R.; Subbarao, K. Cellular immune responses to severe acute respiratory syndrome coronavirus (SARS-CoV) infection in senescent BALB/c mice: CD4+ T cells are important in control of SARS-CoV infection. J. Virol. 2010, 84, 1289–1301. [Google Scholar] [CrossRef] [Green Version]
- Baas, T.; Roberts, A.; Teal, T.H.; Vogel, L.; Chen, J.; Tumpey, T.M.; Katze, M.G.; Subbarao, K. Genomic analysis reveals age-dependent innate immune responses to severe acute respiratory syndrome coronavirus. J. Virol. 2008, 82, 9465–9476. [Google Scholar] [CrossRef] [Green Version]
- Parikh, P.; Wicher, S.; Khandalavala, K.; Pabelick, C.M.; Britt, R.D., Jr.; Prakash, Y.S. Cellular senescence in the lung across the age spectrum. Am. J. Physiol. Lung Cell Mol. Physiol. 2019, 316, L826–L842. [Google Scholar] [CrossRef]
- Wang, Z.N.; Su, R.N.; Yang, B.Y.; Yang, K.X.; Yang, L.F.; Yan, Y.; Chen, Z.G. Potential Role of Cellular Senescence in Asthma. Front. Cell Dev. Biol. 2020, 8, 59. [Google Scholar] [CrossRef]
- Sargiacomo, C.; Sotgia, F.; Lisanti, M.P. COVID-19 and chronological aging: Senolytics and other anti-aging drugs for the treatment or prevention of corona virus infection? Aging (Albany NY) 2020. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Inoue, T.; Kunimoto, H.; Nakajima, K. IL-6-STAT3 signaling and premature senescence. JAKSTAT 2013, 2, e25763. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Kunimoto, H.; Inoue, T.; Nakajima, K. The STAT3-IGFBP5 axis is critical for IL-6/gp130-induced premature senescence in human fibroblasts. Cell Cycle 2012, 11, 730–739. [Google Scholar] [CrossRef] [PubMed]
- Adnot, S.; Lipskaia, L.; Bernard, D. The STATus of STAT3 in Lung Cell Senescence? Am. J. Respir. Cell Mol. Biol. 2019, 61, 5–6. [Google Scholar] [CrossRef] [PubMed]
- Waters, D.W.; Blokland, K.E.C.; Pathinayake, P.S.; Wei, L.; Schuliga, M.; Jaffar, J.; Westall, G.P.; Hansbro, P.M.; Prele, C.M.; Mutsaers, S.E.; et al. STAT3 Regulates the Onset of Oxidant-induced Senescence in Lung Fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 61, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malavolta, M.; Bracci, M.; Santarelli, L.; Sayeed, M.A.; Pierpaoli, E.; Giacconi, R.; Costarelli, L.; Piacenza, F.; Basso, A.; Cardelli, M.; et al. Inducers of Senescence, Toxic Compounds, and Senolytics: The Multiple Faces of Nrf2-Activating Phytochemicals in Cancer Adjuvant Therapy. Mediat. Inflamm. 2018, 2018, 4159013. [Google Scholar] [CrossRef] [Green Version]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef] [Green Version]
- Somerville, V.S.; Braakhuis, A.J.; Hopkins, W.G. Effect of Flavonoids on Upper Respiratory Tract Infections and Immune Function: A Systematic Review and Meta-Analysis. Adv. Nutr. 2016, 7, 488–497. [Google Scholar] [CrossRef] [Green Version]
- Roca Suarez, A.A.; Van Renne, N.; Baumert, T.F.; Lupberger, J. Viral manipulation of STAT3: Evade, exploit, and injure. PLoS Pathog. 2018, 14, e1006839. [Google Scholar] [CrossRef] [Green Version]
- Kuchipudi, S.V. The Complex Role of STAT3 in Viral Infections. J. Immunol. Res. 2015, 2015, 272359. [Google Scholar] [CrossRef] [Green Version]
- Tsai, M.H.; Pai, L.M.; Lee, C.K. Fine-Tuning of Type I Interferon Response by STAT3. Front. Immunol. 2019, 10, 1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, T.; Murakami, M. COVID-19: A New Virus, but a Familiar Receptor and Cytokine Release Syndrome. Immunity 2020, 52, 731–733. [Google Scholar] [CrossRef] [PubMed]
- Ferenci, P.; Scherzer, T.M.; Kerschner, H.; Rutter, K.; Beinhardt, S.; Hofer, H.; Schöniger-Hekele, M.; Holzmann, H.; Steindl-Munda, P. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology 2008, 135, 1561–1567. [Google Scholar] [CrossRef] [PubMed]
- Payer, B.A.; Reiberger, T.; Rutter, K.; Beinhardt, S.; Staettermayer, A.F.; Peck-Radosavljevic, M.; Ferenci, P. Successful HCV eradication and inhibition of HIV replication by intravenous silibinin in an HIV-HCV coinfected patient. J. Clin. Virol. 2010, 49, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Rutter, K.; Scherzer, T.M.; Beinhardt, S.; Kerschner, H.; Stättermayer, A.F.; Hofer, H.; Popow-Kraupp, T.; Steindl-Munda, P.; Ferenci, P. Intravenous silibinin as ‘rescue treatment’ for on-treatment non-responders to pegylated interferon/ribavirin combination therapy. Antivir. Ther. 2011, 16, 1327–1333. [Google Scholar] [CrossRef] [Green Version]
- Biermer, M.; Schlosser, B.; Fülöp, B.; van Bömmel, F.; Brodzinski, A.; Heyne, R.; Keller, K.; Sarrazin, C.; Berg, T. High-dose silibinin rescue treatment for HCV-infected patients showing suboptimal virologic response to standard combination therapy. J. Viral Hepat. 2012, 19, 547–553. [Google Scholar] [CrossRef]
- Knapstein, J.; Wörns, M.A.; Galle, P.R.; Zimmermann, T. Combination therapy with silibinin, pegylated interferon and ribavirin in a patient with hepatitis C virus genotype 3 reinfection after liver transplantation: A case report. J. Med. Case Rep. 2014, 8, 257. [Google Scholar] [CrossRef] [Green Version]
- Rendina, M.; D’Amato, M.; Castellaneta, A.; Castellaneta, N.M.; Brambilla, N.; Giacovelli, G.; Rovati, L.; Rizzi, S.F.; Zappimbulso, M.; Bringiotti, R.S.; et al. Antiviral activity and safety profile of silibinin in HCV patients with advanced fibrosis after liver transplantation: A randomized clinical trial. Transpl. Int. 2014, 27, 696–704. [Google Scholar] [CrossRef]
- Braun, D.L.; Rauch, A.; Aouri, M.; Durisch, N.; Eberhard, N.; Anagnostopoulos, A.; Ledergerber, B.; Müllhaupt, B.; Metzner, K.J.; Decosterd, L.; et al. Swiss HIV Cohort Study. A Lead-In with Silibinin Prior to Triple-Therapy Translates into Favorable Treatment Outcomes in Difficult-To-Treat HIV/Hepatitis C Coinfected Patients. PLoS ONE 2015, 10, e0133028. [Google Scholar] [CrossRef] [Green Version]
- Malaguarnera, M.; Motta, M.; Vacante, M.; Malaguarnera, G.; Caraci, F.; Nunnari, G.; Gagliano, C.; Greco, C.; Chisari, G.; Drago, F.; et al. Silybin-vitamin E-phospholipids complex reduces liver fibrosis in patients with chronic hepatitis C treated with pegylated interferon α and ribavirin. Am. J. Transl. Res. 2015, 7, 2510–2518. [Google Scholar]
- Liu, C.H.; Jassey, A.; Hsu, H.Y.; Lin, L.T. Antiviral Activities of Silymarin and Derivatives. Molecules 2019, 24, E1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Shen, H.M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-dependent entry of severe acute respiratory syndrome coronavirus into target cells expressing ACE2 with the cytoplasmic tail deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blaising, J.; Lévy, P.L.; Gondeau, C.; Phelip, C.; Varbanov, M.; Teissier, E.; Ruggiero, F.; Polyak, S.J.; Oberlies, N.H.; Ivanovic, T.; et al. Silibinin inhibits hepatitis C virus entry into hepatocytes by hindering clathrin-dependent trafficking. Cell Microbiol. 2013, 15, 1866–1882. [Google Scholar] [CrossRef]
- Verdura, S.; Cuyàs, E.; Cortada, E.; Brunet, J.; Lopez-Bonet, E.; Martin-Castillo, B.; Bosch-Barrera, J.; Encinar, J.A.; Menendez, J.A. Resveratrol targets PD-L1 glycosylation and dimerization to enhance antitumor T-cell immunity. Aging (Albany NY) 2020, 12, 8–34. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Guo, F.; Comunale, M.A.; Mehta, A.; Sehgal, M.; Jain, P.; Cuconati, A.; Lin, H.; Block, T.M.; Chang, J.; et al. Inhibition of endoplasmic reticulum-resident glucosidases impairs severe acute respiratory syndrome coronavirus and human coronavirus NL63 spike protein-mediated entry by altering the glycan processing of angiotensin I-converting enzyme 2. Antimicrob. Agents Chemother. 2015, 59, 206–216. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Block, T.M.; Guo, J.T. Antiviral therapies targeting host ER alpha-glucosidases: Current status and future directions. Antiviral Res. 2013, 99, 251–260. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bosch-Barrera, J.; Martin-Castillo, B.; Buxó, M.; Brunet, J.; Encinar, J.A.; Menendez, J.A. Silibinin and SARS-CoV-2: Dual Targeting of Host Cytokine Storm and Virus Replication Machinery for Clinical Management of COVID-19 Patients. J. Clin. Med. 2020, 9, 1770. https://doi.org/10.3390/jcm9061770
Bosch-Barrera J, Martin-Castillo B, Buxó M, Brunet J, Encinar JA, Menendez JA. Silibinin and SARS-CoV-2: Dual Targeting of Host Cytokine Storm and Virus Replication Machinery for Clinical Management of COVID-19 Patients. Journal of Clinical Medicine. 2020; 9(6):1770. https://doi.org/10.3390/jcm9061770
Chicago/Turabian StyleBosch-Barrera, Joaquim, Begoña Martin-Castillo, Maria Buxó, Joan Brunet, José Antonio Encinar, and Javier A. Menendez. 2020. "Silibinin and SARS-CoV-2: Dual Targeting of Host Cytokine Storm and Virus Replication Machinery for Clinical Management of COVID-19 Patients" Journal of Clinical Medicine 9, no. 6: 1770. https://doi.org/10.3390/jcm9061770
APA StyleBosch-Barrera, J., Martin-Castillo, B., Buxó, M., Brunet, J., Encinar, J. A., & Menendez, J. A. (2020). Silibinin and SARS-CoV-2: Dual Targeting of Host Cytokine Storm and Virus Replication Machinery for Clinical Management of COVID-19 Patients. Journal of Clinical Medicine, 9(6), 1770. https://doi.org/10.3390/jcm9061770