Tumor Immune Microenvironment Clusters in Localized Prostate Adenocarcinoma: Prognostic Impact of Macrophage Enriched/Plasma Cell Non-Enriched Subtypes

 , ,
, ,
Abstract
:1. Introduction
2. Experimental Section
2.1. Study Design
2.2. Clustering Based on Immune Cell Subpopulations
2.3. Survival Analysis of Clusters
2.4. Retrieving Raw Count Data
2.5. Statistical Analysis
3. Results
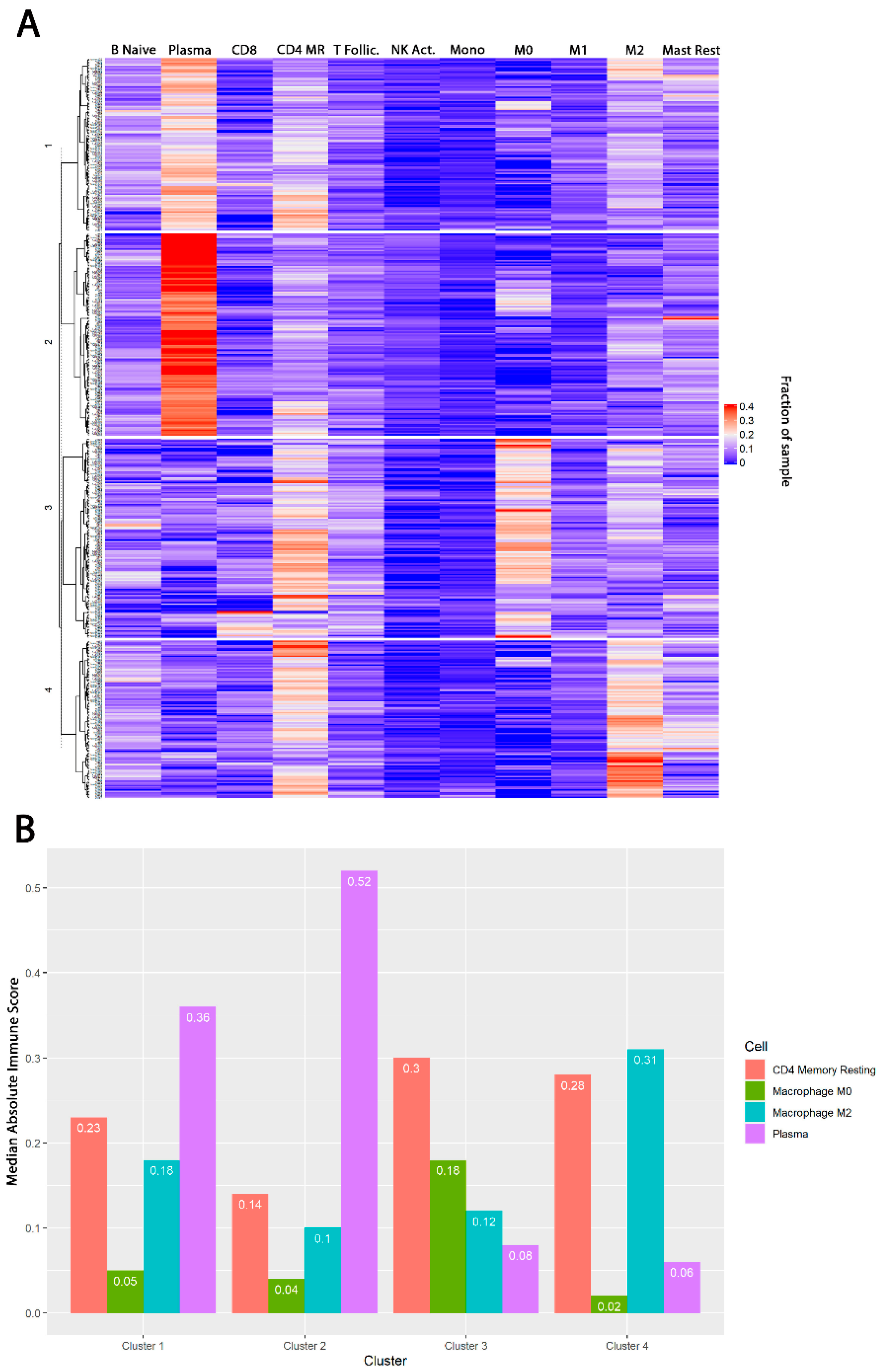
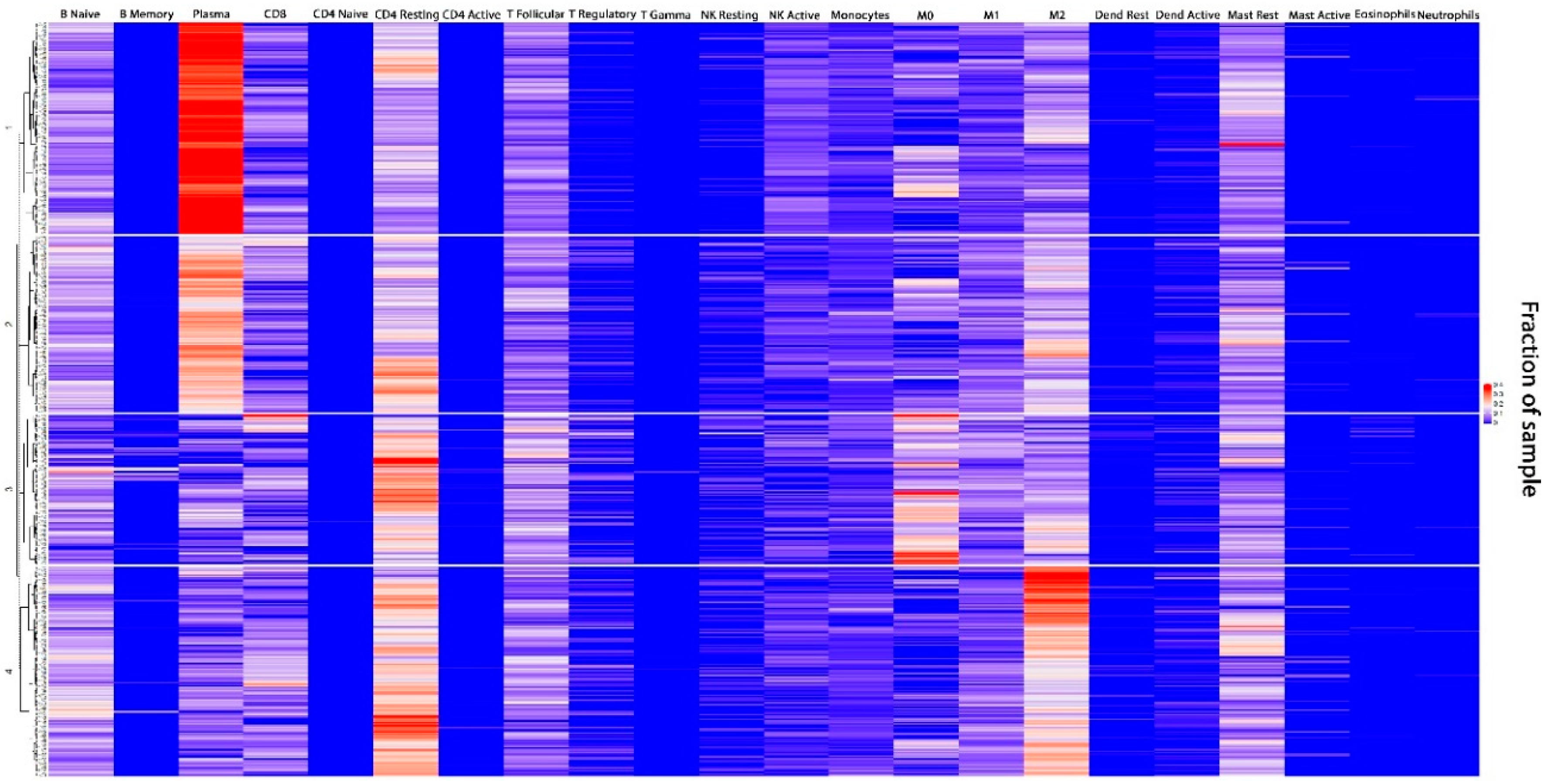
3.1. TIME Clustering
3.2. Baseline Demographic and Treatment Details by TIME Cluster
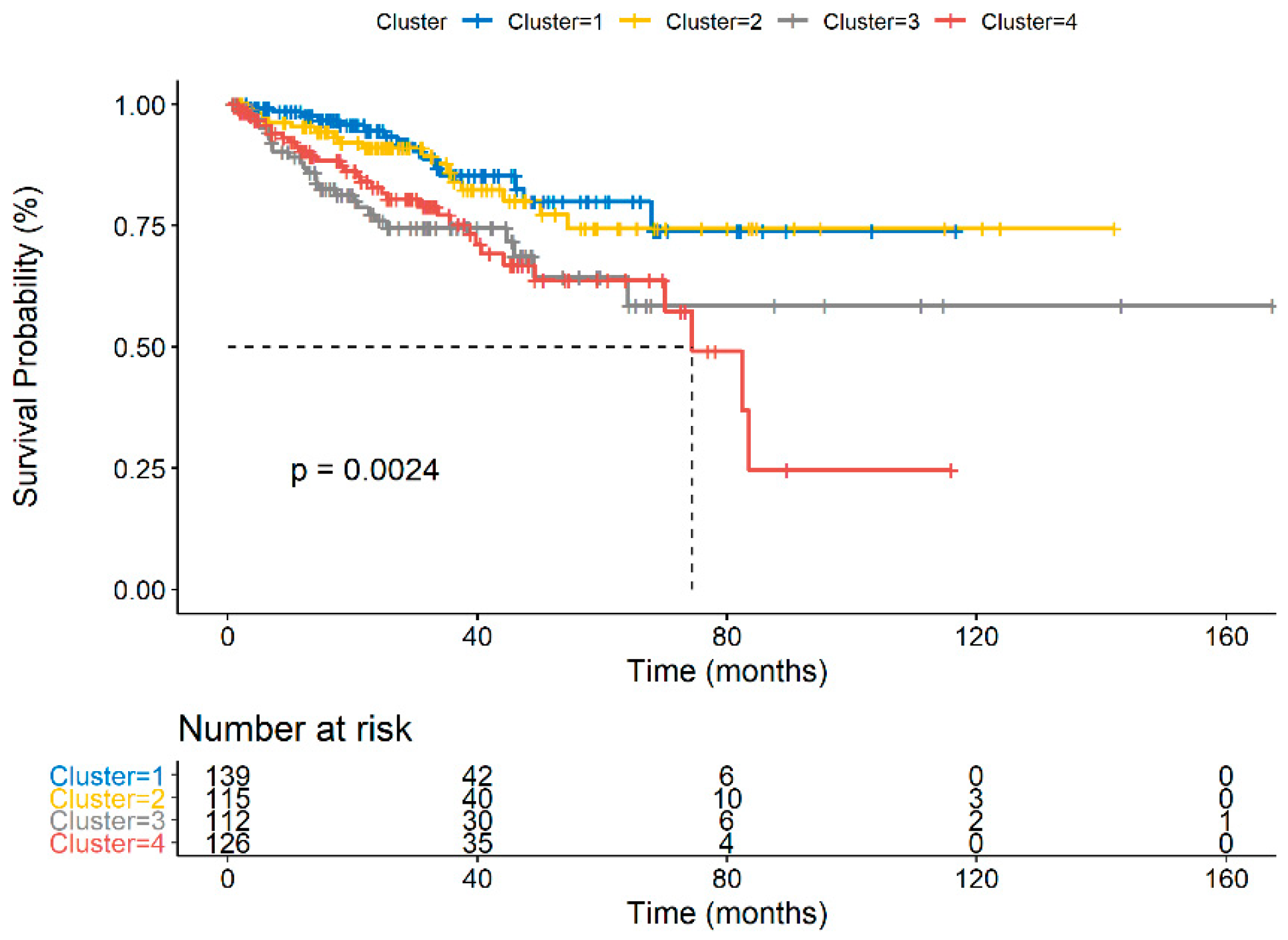
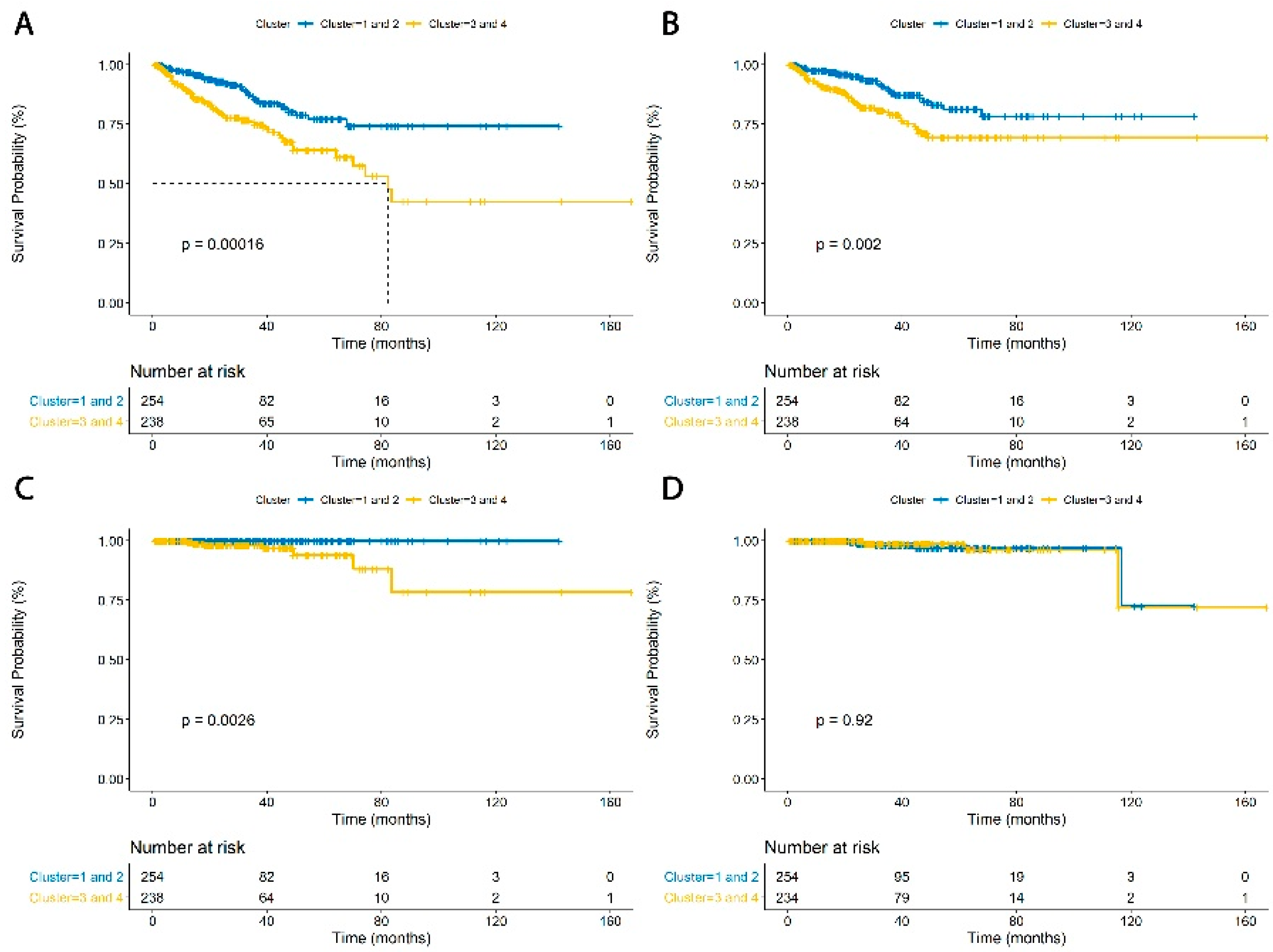
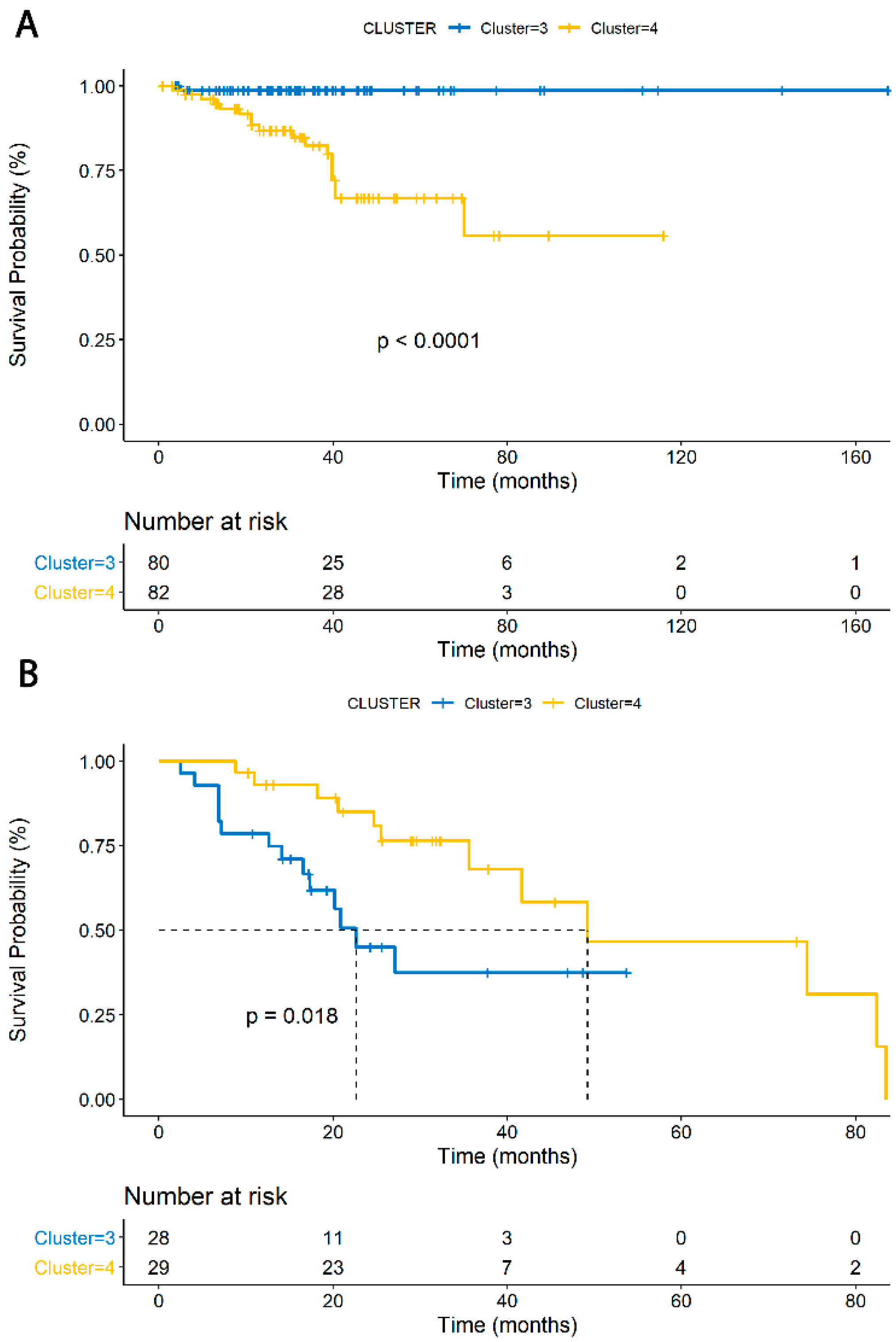
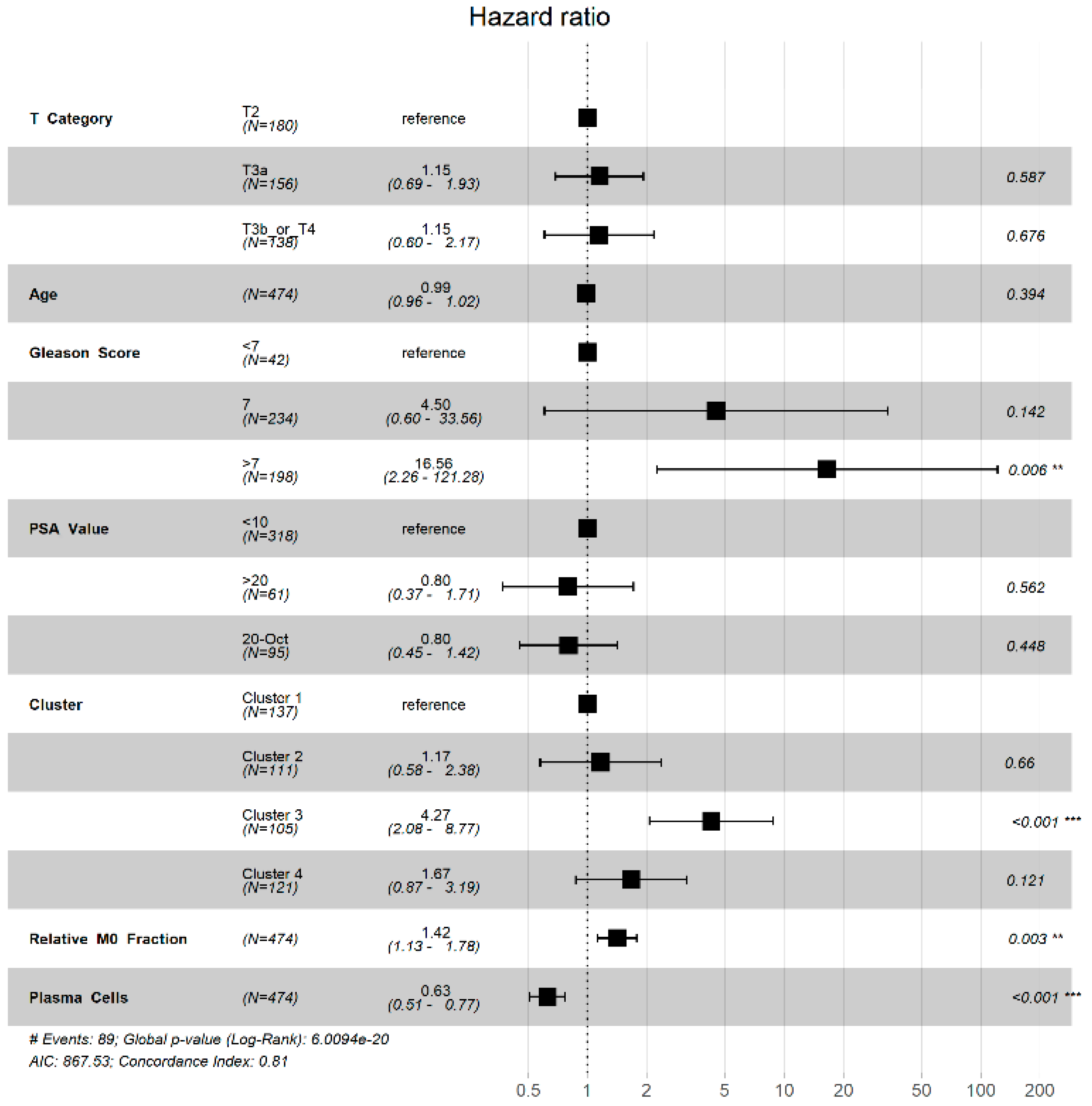
3.3. TIME Cluster Prognostic Effect
4. Discussion
Author Contributions
Funding
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| M2 Macrophages | M0 Macrophages | ||||||
| Cluster | 1 | 2 | 3 | Cluster | 1 | 2 | 3 |
| 2 | <0.0001 * | NA | NA | 2 | 0.937 | NA | NA |
| 3 | <0.0001 * | 0.075 * | NA | 3 | <0.0001 * | <0.0001 * | NA |
| 4 | <0.0001 * | <0.0001 * | <0.0001 * | 4 | 0.047 * | 0.045 * | <0.0001 * |
| Plasma Cells | CD4 Memory Resting T Cells | ||||||
| Cluster | 1 | 2 | 3 | Cluster | 1 | 2 | 3 |
| 2 | <0.0001 * | NA | NA | 2 | <0.0001 * | NA | NA |
| 3 | <0.0001 * | <0.0001 * | NA | 3 | <0.0001 * | <0.0001 * | NA |
| 4 | <0.0001 * | <0.0001 * | 0.018 * | 4 | <0.0001 * | <0.0001 * | 0.46 |
References
- Global Cancer Observatory. Cancer Today Data Table. Available online: http://gco.iarc.fr/today/online-analysistable?v=2018&mode=cancer&mode_population=continents&population=900&populations=900&key=asr&sex=0&cancer=39&type=0&statistic=5&prevalence=0&population_group=0&ages_group%5B%5D=0&ages_group%5B%5D=17&group_cancer=1&include_nmsc=1&include_nmsc_other=1 (accessed on 1 May 2020).
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, S.S.; Stockert, J.A.; Hackert, V.; Yadav, K.K.; Tewari, A.K. Intratumor heterogeneity in prostate cancer. Urol. Oncol. 2018, 36, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Beerenwinkel, N.; Antal, T.; Dingli, D.; Traulsen, A.; Kinzler, K.W.; Velculescu, V.E.; Vogelstein, B.; Nowak, M.A. Genetic progression and the waiting time to cancer. PLoS Comput. Biol. 2007, 3, e225. [Google Scholar] [CrossRef] [PubMed]
- Liu, A.Y.; Roudier, M.P.; True, L.D. Heterogeneity in primary and metastatic prostate cancer as defined by cell surface CD profile. Am. J. Pathol. 2004, 165, 1543–1556. [Google Scholar] [CrossRef] [Green Version]
- Ross, A.E.; D’Amico, A.V.; Freedland, S.J. Which, when and why? Rational use of tissue-based molecular testing in localized prostate cancer. Prostate Cancer Prostatic Dis. 2016, 19, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Moschini, M.; Spahn, M.; Mattei, A.; Cheville, J.; Karnes, R.J. Incorporation of tissue-based genomic biomarkers into localized prostate cancer clinics. BMC Med. 2016, 14, 67. [Google Scholar] [CrossRef] [Green Version]
- Nagarsheth, N.; Wicha, M.S.; Zou, W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017, 17, 559–572. [Google Scholar] [CrossRef] [Green Version]
- Seager, R.J.; Hajal, C.; Spill, F.; Kamm, R.D.; Zaman, M.H. Dynamic interplay between tumour, stroma and immune system can drive or prevent tumour progression. Converg. Sci. Phys. Oncol. 2017, 3. [Google Scholar] [CrossRef]
- Roma-Rodrigues, C.; Mendes, R.; Baptista, P.V.; Fernandes, A.R. Targeting Tumor Microenvironment for Cancer Therapy. Int. J. Mol. Sci. 2019, 20, 840. [Google Scholar] [CrossRef] [Green Version]
- Stanta, G.; Bonin, S. Overview on Clinical Relevance of Intra-Tumor Heterogeneity. Front. Med. (Lausanne) 2018, 5, 85. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef] [PubMed]
- Netea-Maier, R.T.; Smit, J.W.A.; Netea, M.G. Metabolic changes in tumor cells and tumor-associated macrophages: A mutual relationship. Cancer Lett. 2018, 413, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Chen, Y. Tumor microenvironment: Sanctuary of the devil. Cancer Lett. 2015, 368, 7–13. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- Nishikawa, H.; Sakaguchi, S. Regulatory T cells in cancer immunotherapy. Curr. Opin. Immunol. 2014, 27, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, S.G.; Lehrer, J.; Chang, S.L.; Das, R.; Erho, N.; Liu, Y.; Sjöström, M.; Den, R.B.; Freedland, S.J.; Klein, E.A.; et al. The Immune Landscape of Prostate Cancer and Nomination of PD-L2 as a Potential Therapeutic Target. JNCI J. Natl. Cancer Inst. 2019, 111, 301–310. [Google Scholar] [CrossRef]
- Newman, A.M.; Steen, C.B.; Liu, C.L.; Gentles, A.J.; Chaudhuri, A.A.; Scherer, F.; Khodadoust, M.S.; Esfahani, M.S.; Luca, B.A.; Steiner, D.; et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 2019, 37, 773–782. [Google Scholar] [CrossRef]
- Cerami, E.; Gao, J.; Dogrusoz, U.; Gross, B.E.; Sumer, S.O.; Aksoy, B.A.; Jacobsen, A.; Byrne, C.J.; Heuer, M.L.; Larsson, E.; et al. The cBio cancer genomics portal: An open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012, 2, 401–404. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [Green Version]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef] [Green Version]
- TCGA Firebrowse. Available online: http://firebrowse.org/ (accessed on 16 February 2020).
- Kassambara, A.; Kosinski, M.; Biecek, P.; Fabian, S. Drawing Survival Curves Using ’Ggplot2′. R Package. 2016, 4, 2. [Google Scholar]
- Mounir, M.; Lucchetta, M.; Silva, T.C.; Olsen, C.; Bontempi, G.; Chen, X.; Noushmehr, H.; Colaprico, A.; Papaleo, E. New functionalities in the TCGAbiolinks package for the study and integration of cancer data from GDC and GTEx. PLoS Comput. Biol. 2019, 15, e1006701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silva, C.T.; Colaprico, A.; Olsen, C.; D’Angelo, F.; Bontempi, G.; Ceccarelli, M.; Noushmehr, H. TCGA Workflow: Analyze cancer genomics and epigenomics data using Bioconductor packages. Research 2016, 5, 1542. [Google Scholar]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Qiu, S.-Q.; Waaijer, S.J.H.; Zwager, M.C.; de Vries, E.G.E.; van der Vegt, B.; Schröder, C.P. Tumor-associated macrophages in breast cancer: Innocent bystander or important player? Cancer Treat. Rev. 2018, 70, 178–189. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Baer, C.; Squadrito, M.L.; Laoui, D.; Thompson, D.; Hansen, S.K.; Kiialainen, A.; Hoves, S.; Ries, C.H.; Ooi, C.-H.; De Palma, M. Suppression of microRNA activity amplifies IFN-γ-induced macrophage activation and promotes anti-tumour immunity. Nat. Cell Biol. 2016, 18, 790–802. [Google Scholar] [CrossRef]
- Bingle, L.; Brown, N.J.; Lewis, C.E. The role of tumour-associated macrophages in tumour progression: Implications for new anticancer therapies. J. Pathol. 2002, 196, 254–265. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Kaneda, M.M.; Messer, K.S.; Ralainirina, N.; Li, H.; Leem, C.J.; Gorjestani, S.; Woo, G.; Nguyen, A.V.; Figueiredo, C.C.; Foubert, P.; et al. PI3Kγ is a molecular switch that controls immune suppression. Nature 2016, 539, 437–442. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Schioppa, T.; Porta, C.; Allavena, P.; Sica, A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006, 25, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.-Z.; Pollard, J.W. Macrophage diversity enhances tumor progression and metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef] [Green Version]
- Lissbrant, I.F.; Stattin, P.; Wikstrom, P.; Damber, J.E.; Egevad, L.; Bergh, A. Tumor associated macrophages in human prostate cancer: Relation to clinicopathological variables and survival. Int. J. Oncol. 2000, 17, 445–451. [Google Scholar] [CrossRef]
- Izumi, K.; Fang, L.-Y.; Mizokami, A.; Namiki, M.; Li, L.; Lin, W.-J.; Chang, C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO Mol. Med. 2013, 5, 1383–1401. [Google Scholar] [CrossRef]
- Lin, T.-H.; Izumi, K.; Lee, S.O.; Lin, W.-J.; Yeh, S.; Chang, C. Anti-androgen receptor ASC-J9 versus anti-androgens MDV3100 (Enzalutamide) or Casodex (Bicalutamide) leads to opposite effects on prostate cancer metastasis via differential modulation of macrophage infiltration and STAT3-CCL2 signaling. Cell Death Dis. 2013, 4, e764. [Google Scholar] [CrossRef]
- Maolake, A.; Izumi, K.; Shigehara, K.; Natsagdorj, A.; Iwamoto, H.; Kadomoto, S.; Takezawa, Y.; Machioka, K.; Narimoto, K.; Namiki, M.; et al. Tumor-associated macrophages promote prostate cancer migration through activation of the CCL22-CCR4 axis. Oncotarget 2017, 8, 9739–9751. [Google Scholar] [CrossRef] [Green Version]
- Pienta, K.J.; Machiels, J.-P.; Schrijvers, D.; Alekseev, B.; Shkolnik, M.; Crabb, S.J.; Li, S.; Seetharam, S.; Puchalski, T.A.; Takimoto, C.; et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Investig. New Drugs 2013, 31, 760–768. [Google Scholar] [CrossRef] [PubMed]
- Ries, C.H.; Cannarile, M.A.; Hoves, S.; Benz, J.; Wartha, K.; Runza, V.; Rey-Giraud, F.; Pradel, L.P.; Feuerhake, F.; Klaman, I.; et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014, 25, 846–859. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Wouters, M.C.A.; Nelson, B.H. Prognostic Significance of Tumor-Infiltrating B Cells and Plasma Cells in Human Cancer. Clin. Cancer Res. 2018, 24, 6125–6135. [Google Scholar] [CrossRef] [Green Version]
- Gentles, A.J.; Newman, A.M.; Liu, C.L.; Bratman, S.V.; Feng, W.; Kim, D.; Nair, V.S.; Xu, Y.; Khuong, A.; Hoang, C.D.; et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat. Med. 2015, 21, 938–945. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Shiao, S.L. The role of macrophage phenotype in regulating the response to radiation therapy. Transl. Res. 2018, 191, 64–80. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-S.; Chen, F.-H.; Wang, C.-C.; Huang, H.-L.; Jung, S.-M.; Wu, C.-J.; Lee, C.-C.; McBride, W.H.; Chiang, C.-S.; Hong, J.-H. Macrophages from irradiated tumors express higher levels of iNOS, arginase-I and COX-2, and promote tumor growth. Int. J. Radiat. Oncol. Biol. Phys. 2007, 68, 499–507. [Google Scholar] [CrossRef]
- Milas, L. Tumor bed effect in murine tumors: Relationship to tumor take and tumor macrophage content. Radiat. Res. 1990, 123, 232–236. [Google Scholar] [CrossRef]
- De Palma, M.; Lewis, C.E. Macrophage regulation of tumor responses to anticancer therapies. Cancer Cell 2013, 23, 277–286. [Google Scholar] [CrossRef] [Green Version]
- Hughes, R.; Qian, B.-Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 Macrophages Stimulate Tumor Relapse after Chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Escamilla, J.; Mok, S.; David, J.; Priceman, S.; West, B.; Bollag, G.; McBride, W.; Wu, L. CSF1R signaling blockade stanches tumor-infiltrating myeloid cells and improves the efficacy of radiotherapy in prostate cancer. Cancer Res. 2013, 73, 2782–2794. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Lee, W.R.; Koontz, B.F.; Moul, J.W.; Mundy, K.; Creel, P.; Wood, S.; Davis, K.; et al. A phase 2 multimodality trial of docetaxel/prednisone with sunitinib followed by salvage radiation therapy in men with PSA recurrent prostate cancer after radical prostatectomy. Prostate Cancer Prostatic Dis. 2016, 19, 100–106. [Google Scholar] [CrossRef]





| Cluster 1 (n = 139) | Cluster 2 (n = 115) | Cluster 3 (n = 112) | Cluster 4 (n = 126) | |
|---|---|---|---|---|
| Median Age (years) | 59.9 | 61.1 | 62.5 | 64.7 |
| Median PSA (IQR) | 7.2 (5.0–11.6) | 7.6 (5.6–9.9) | 7.5 (4.8–12.1) | 7.5 (5.0–11.3) |
| Gleason Score | ||||
| ≤6 | 20 (14.4%) | 10 (8.7%) | 6 (5.4%) | 8 (6.3%) |
| 7 | 84 (60.4%) | 72 (62.6%) | 46 (41.4%) | 41 (32.5%) |
| 8-10 | 35 (25.2%) | 33 (28.7%) | 58 (51.8%) | 75 (59.5%) |
| Pathologic T category | ||||
| pT2 | 53 (38.1%) | 46 (40.0%) | 38 (33.9%) | 46 (36.5%) |
| pT3a | 53 (38.1%) | 36 (31.3%) | 31 (27.7%) | 38 (30.2%) |
| pT3b | 28 (20.1%) | 32 (27.8%) | 35 (31.3%) | 35 (27.8%) |
| pT4 | 4 (2.9%) | 0 | 4 (3.6%) | 2 (1.6%) |
| Risk Group | ||||
| Low | 14 (10.1%) | 5 (4.3%) | 3 (2.7%) | 7 (5.6%) |
| Intermediate | 74 (53.2%) | 66 (57.4%) | 42 (37.5%) | 38 (30.2%) |
| High | 42 (30.2%) | 39 (33.9%) | 51 (45.5%) | 60 (47.6%) |
| Zone of Origin | ||||
| Peripheral | 26 (18.7%) | 37 (32.2%) | 30 (26.8%) | 42 (33.3%) |
| Central | 1 (0.7%) | 0 | 1 (0.9%) | 2 (1.6%) |
| Transition | 2 (1.4%) | 2 (1.7%) | 3 (2.7%) | 1 (0.8%) |
| Multiple | 32 (23.0%) | 22 (19.1%) | 32 (28.6%) | 41 (32.5%) |
| TMPRSS2:ERG Fusion | ||||
| Wild-Type | 91 (65.5%) | 66 (57.4%) | 71 (63.4%) | 84 (66.7%) |
| Fusion | 47 (33.8%) | 48 (41.7%) | 40 (35.7%) | 42 (33.3%) |
| Median Follow Up (months) | 28 | 34.3 | 31.2 | 30.8 |
| Recurrence | ||||
| BCR | 10 (7.2%) | 15 (13.0%) | 22 (19.6%) | 21 (16.7%) |
| LRR | 3 (2.2%) | 0 | 1 (0.9%) | 3 (2.4%) |
| Metastasis | 0 | 0 | 2 (1.8%) | 5 (4.0%) |
| Death | 2 (1.4%) | 3 (2.6%) | 3 (2.7%) | 1 (0.8%) |
| Radiation | ||||
| Adjuvant | 11 (7.9%) | 12 (10.4%) | 19 (17.0%) | 21 (16.7%) |
| Salvage | 7 (5.0%) | 5 (4.3%) | 9 (8.0%) | 8 (6.3%) |
| Median Dose (Gy) | 66.6 | 67.5 | 70 | 66 |
| Median Fractions | 33.5 | 35 | 35 | 34 |
| Salvage Systemic Therapy | 13 (9.3%) | 16 (13.9%) | 25 (22.3%) | 25 (19.9%) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jairath, N.K.; Farha, M.W.; Srinivasan, S.; Jairath, R.; Green, M.D.; Dess, R.T.; Jackson, W.C.; Weiner, A.B.; Schaeffer, E.M.; Zhao, S.G.; et al. Tumor Immune Microenvironment Clusters in Localized Prostate Adenocarcinoma: Prognostic Impact of Macrophage Enriched/Plasma Cell Non-Enriched Subtypes. J. Clin. Med. 2020, 9, 1973. https://doi.org/10.3390/jcm9061973
Jairath NK, Farha MW, Srinivasan S, Jairath R, Green MD, Dess RT, Jackson WC, Weiner AB, Schaeffer EM, Zhao SG, et al. Tumor Immune Microenvironment Clusters in Localized Prostate Adenocarcinoma: Prognostic Impact of Macrophage Enriched/Plasma Cell Non-Enriched Subtypes. Journal of Clinical Medicine. 2020; 9(6):1973. https://doi.org/10.3390/jcm9061973
Chicago/Turabian StyleJairath, Neil K., Mark W. Farha, Sudharsan Srinivasan, Ruple Jairath, Michael D. Green, Robert T. Dess, William C. Jackson, Adam B. Weiner, Edward M. Schaeffer, Shuang G. Zhao, and et al. 2020. "Tumor Immune Microenvironment Clusters in Localized Prostate Adenocarcinoma: Prognostic Impact of Macrophage Enriched/Plasma Cell Non-Enriched Subtypes" Journal of Clinical Medicine 9, no. 6: 1973. https://doi.org/10.3390/jcm9061973
APA StyleJairath, N. K., Farha, M. W., Srinivasan, S., Jairath, R., Green, M. D., Dess, R. T., Jackson, W. C., Weiner, A. B., Schaeffer, E. M., Zhao, S. G., Feng, F. Y., El Naqa, I., & Spratt, D. E. (2020). Tumor Immune Microenvironment Clusters in Localized Prostate Adenocarcinoma: Prognostic Impact of Macrophage Enriched/Plasma Cell Non-Enriched Subtypes. Journal of Clinical Medicine, 9(6), 1973. https://doi.org/10.3390/jcm9061973