Genetic Variation in CCL18 Gene Influences CCL18 Expression and Correlates with Survival in Idiopathic Pulmonary Fibrosis—Part B

 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Experimental Section
2.1. Patients and Clinical Data
2.2. Serum Sampling
2.3. Serum CC18 Measurement by Enzyme-Linked Immunosorbent Assay (ELISA)
2.4. Single Nucleotide Polymorphism (SNP) Genotyping
2.5. Statistical Analysis
3. Results
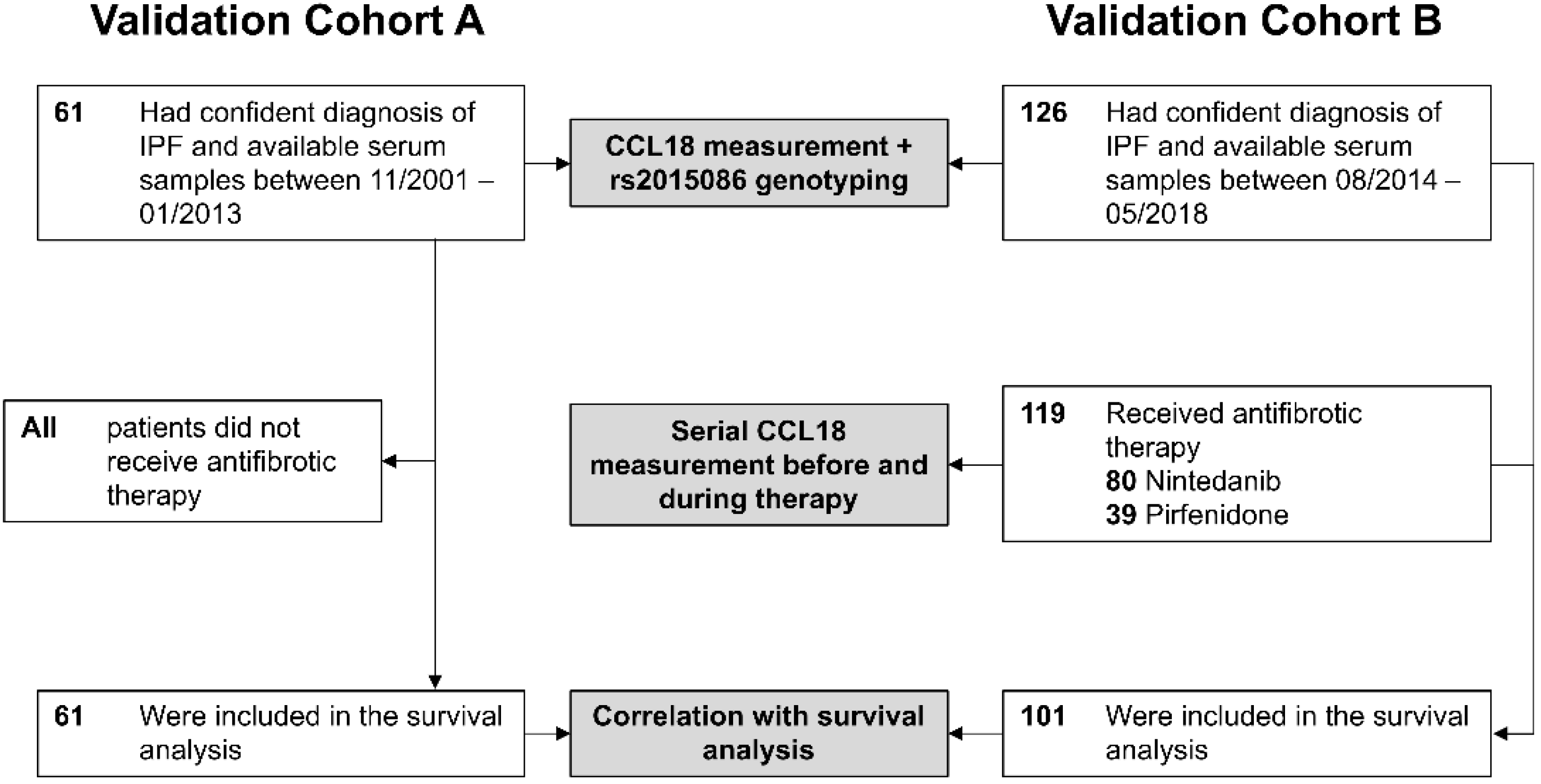
3.1. Validation Cohorts and Patient Baseline Characteristics
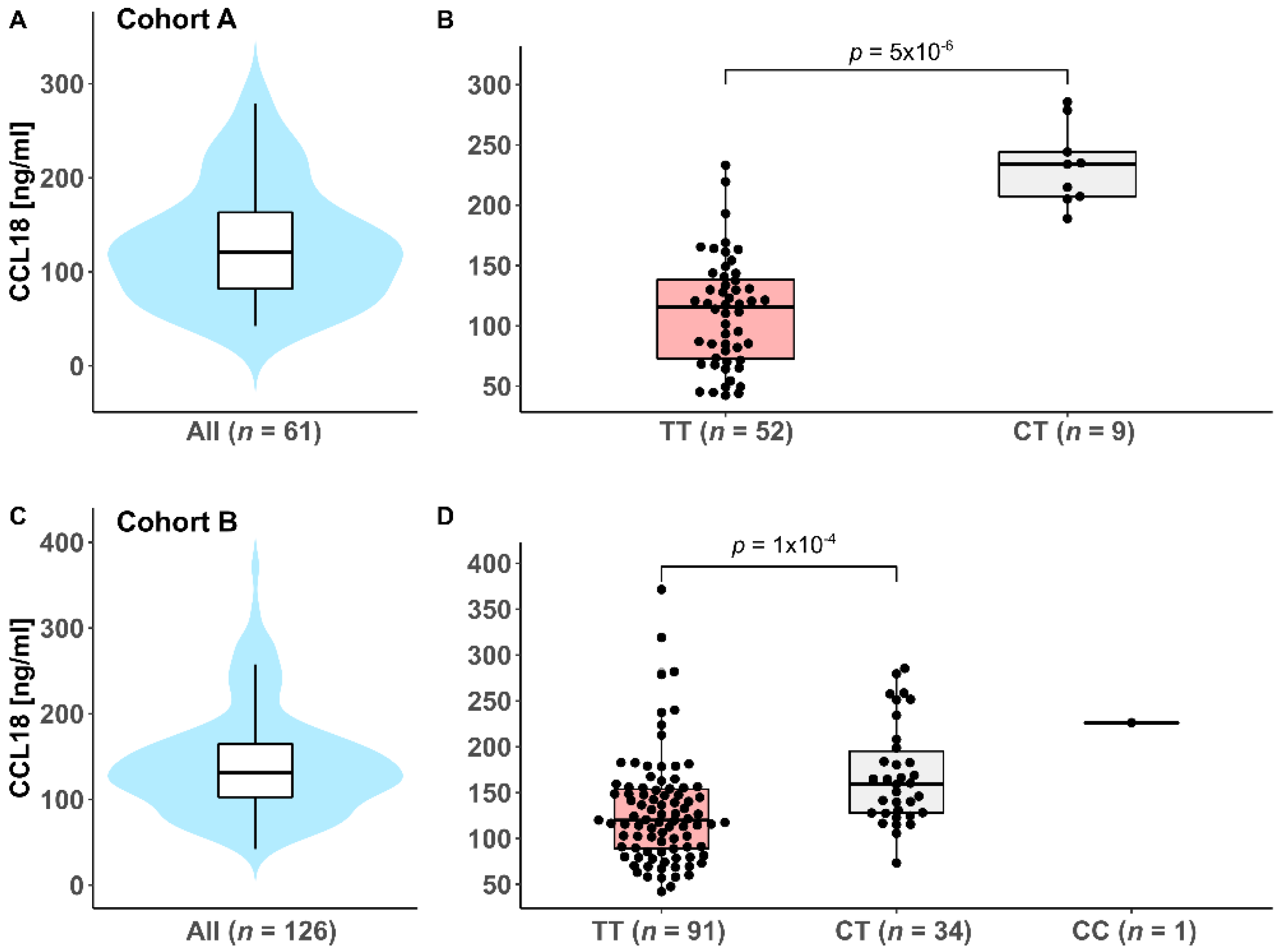
3.2. Genotyping of the CCL18 rs2015086 SNP and Serum CCL18 Levels
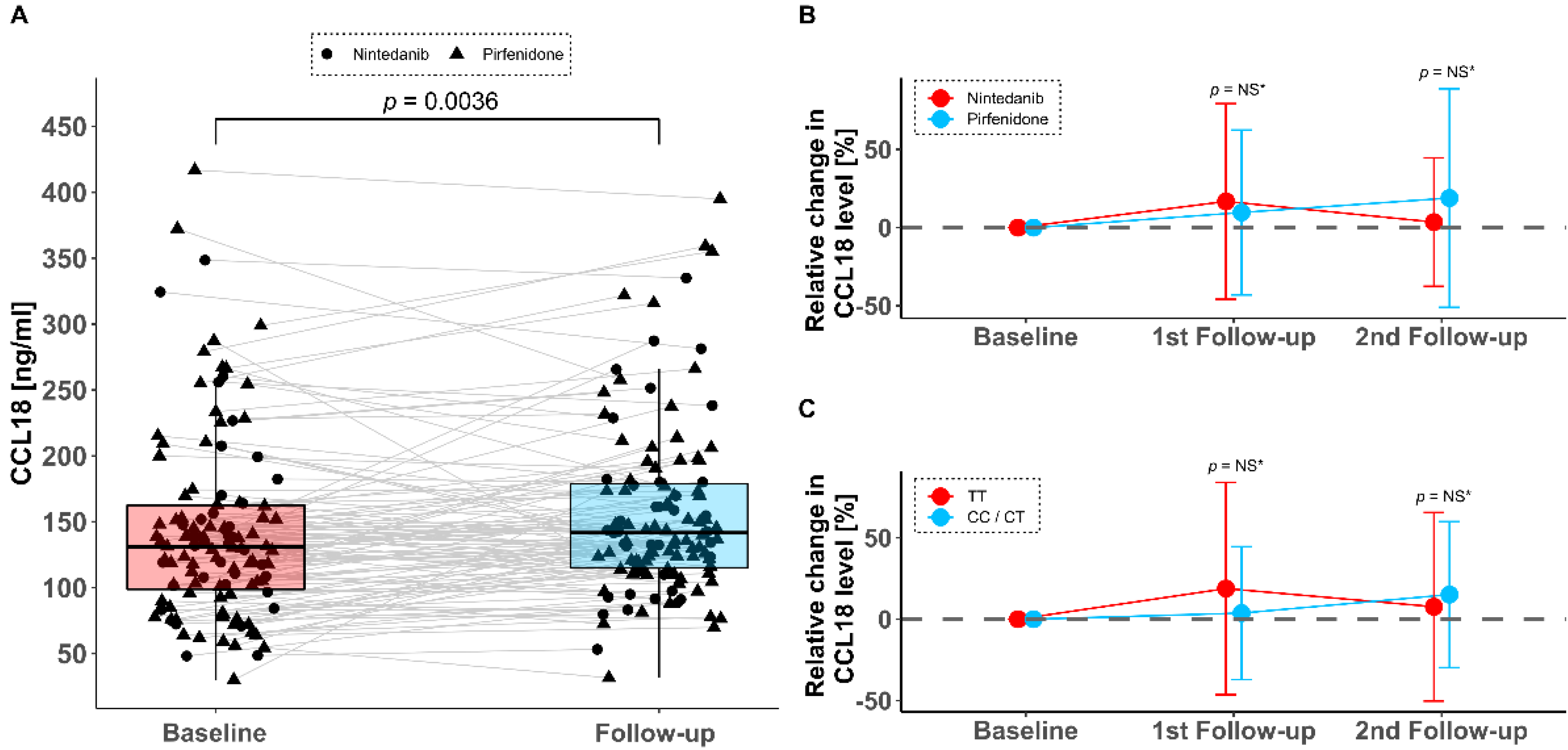
3.3. Serum CCL18 Levels under Antifibrotic Therapy
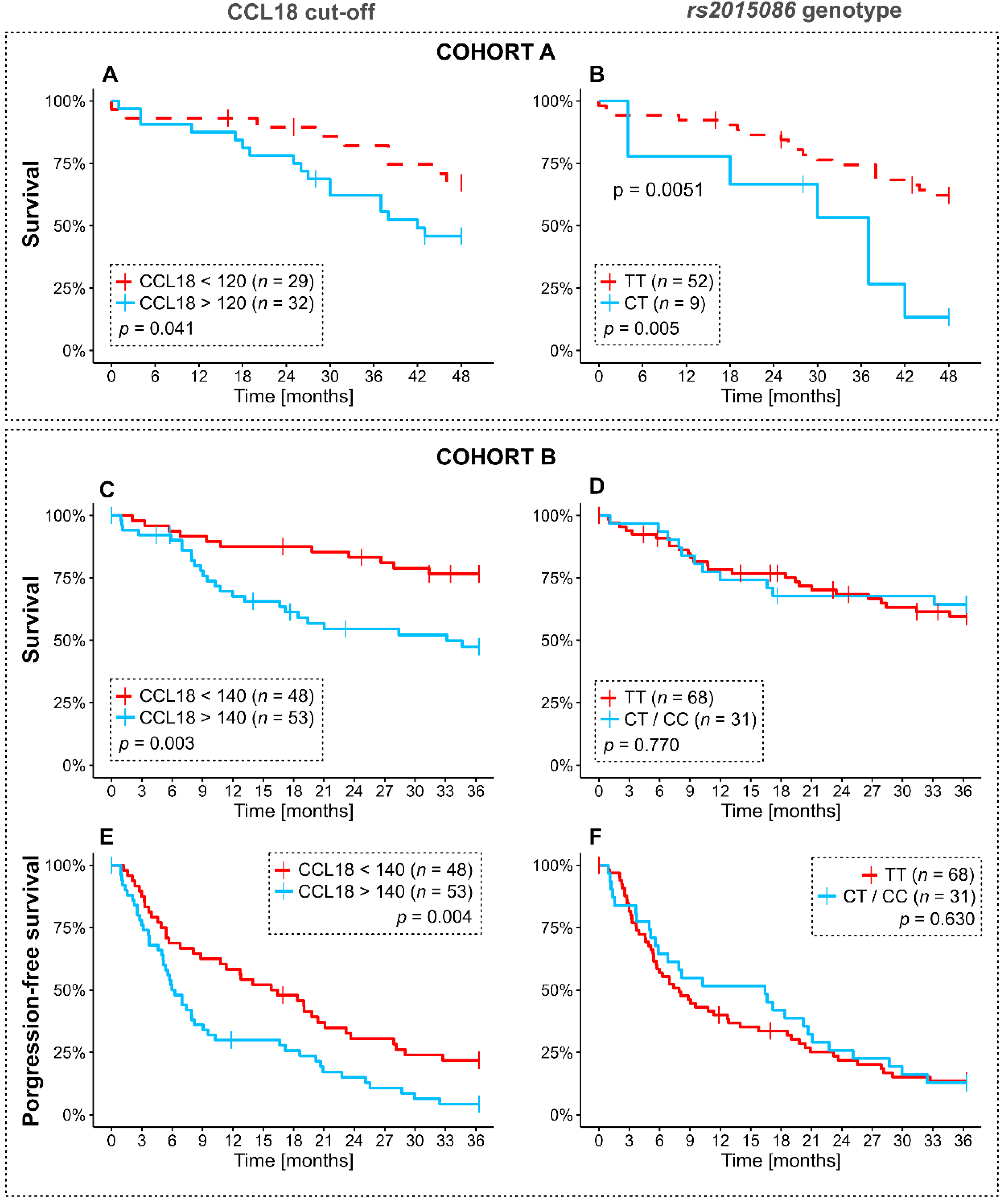
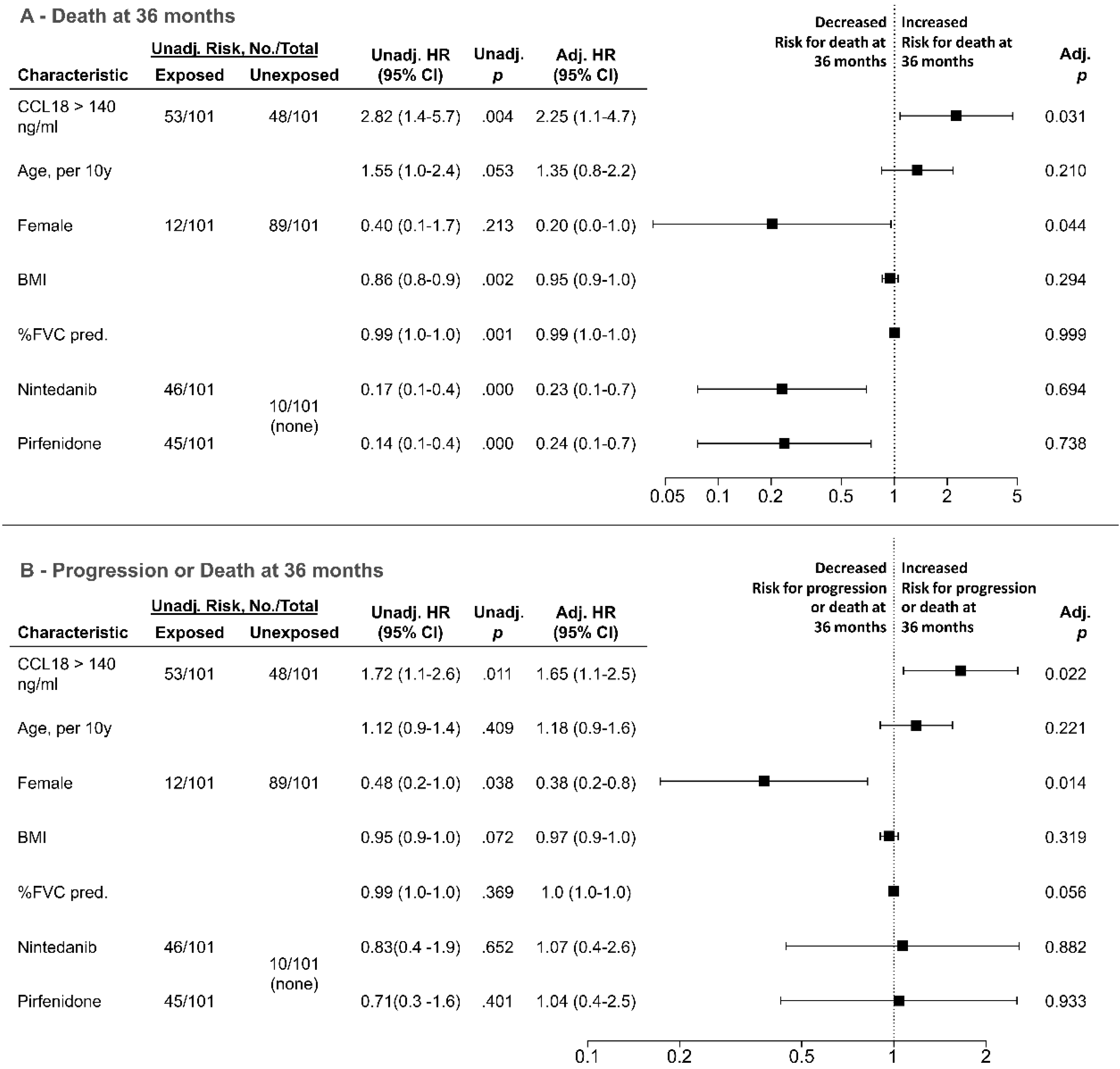
3.4. Analysis of Overall and Progression-Free Survival Depending on CCL18 Cutoffs and rs2015085 Genotype
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Raghu, G.; Remy-Jardin, M.; Myers, J.L.; Richeldi, L.; Ryerson, C.J.; Lederer, D.J.; Behr, J.; Cottin, V.; Danoff, S.K.; Morell, F.; et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am. J. Respir. Crit. Care Med. 2018, 198, e44–e68. [Google Scholar] [CrossRef] [PubMed]
- Lederer, D.J.; Martinez, F.J. Idiopathic pulmonary fibrosis. N. Engl. J. Med. 2018, 378, 1811–1823. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Anstrom, K.J.; King, T.E.; Lasky, J.A.; Martinez, F.J. Prednisone, azathioprine, and N-acetylcysteine for pulmonary fibrosis. N. Engl. J. Med. 2012, 366, 1968–1977. [Google Scholar] [PubMed]
- Richeldi, L.; Du Bois, R.M.; Raghu, G.; Azuma, A.; Brown, K.K.; Costabel, U.; Cottin, V.; Flaherty, K.R.; Hansell, D.M.; Inoue, Y.; et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2071–2082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- King, T.E.; Bradford, W.Z.; Castro-Bernardini, S.; Fagan, E.A.; Glaspole, I.; Glassberg, M.K.; Gorina, E.; Hopkins, P.M.; Kardatzke, D.; Lancaster, L.; et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N. Engl. J. Med. 2014, 370, 2083–2092. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, L.; Crestani, B.; Hernandez, P.; Inoue, Y.; Wachtlin, D.; Loaiza, L.; Quaresma, M.; Stowasser, S.; Richeldi, L. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: Pooled data from six clinical trials. BMJ Open Respir. Res. 2019, 6, e000397. [Google Scholar] [CrossRef] [Green Version]
- Margaritopoulos, G.A.; Trachalaki, A.; Wells, A.U.; Vasarmidi, E.; Bibaki, E.; Papastratigakis, G.; Detorakis, S.; Tzanakis, N.; Antoniou, K.M. Pirfenidone improves survival in IPF: Results from a real-life study. BMC Pulm. Med. 2018, 18, 177. [Google Scholar] [CrossRef] [Green Version]
- Somogyi, V.; Chaudhuri, N.; Torrisi, S.E.; Kahn, N.; Müller, V.; Kreuter, M. The therapy of idiopathic pulmonary fibrosis: What is next? Eur. Respir. Rev. 2019, 28. [Google Scholar] [CrossRef] [Green Version]
- Schutyser, E.; Richmond, A.; van Damme, J. Involvement of CC chemokine ligand 18 (CCL18) in normal and pathological processes. J. Leukoc. Biol. 2005, 78, 14–26. [Google Scholar] [CrossRef] [Green Version]
- Prasse, A.; Pechkovsky, D.V.; Toews, G.B.; Jungraithmayr, W.; Kollert, F.; Goldmann, T.; Vollmer, E.; Müller-Quernheim, J.; Zissel, G. A vicious circle of alveolar macrophages and fibroblasts perpetuates pulmonary fibrosis via CCL18. Am. J. Respir. Crit. Care Med. 2006, 173, 781–792. [Google Scholar] [CrossRef] [Green Version]
- Prasse, A.; Probst, C.; Bargagli, E.; Zissel, G.; Toews, G.B.; Flaherty, K.R.; Olschewski, M.; Rottoli, P.; Müller-Quernheim, J. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009, 179, 717–723. [Google Scholar] [CrossRef] [PubMed]
- Prasse, A.; Pechkovsky, D.V.; Toews, G.B.; Schäfer, M.; Eggeling, S.; Ludwig, C.; Germann, M.; Kollert, F.; Zissel, G.; Müller-Quernheim, J. CCL18 as an indicator of pulmonary fibrotic activity in idiopathic interstitial pneumonias and systemic sclerosis. Arthritis Rheum. 2007, 56, 1685–1693. [Google Scholar] [CrossRef] [PubMed]
- Neighbors, M.; Cabanski, C.R.; Ramalingam, T.R.; Sheng, X.R.; Tew, G.W.; Gu, C.; Jia, G.; Peng, K.; Ray, J.M.; Ley, B.; et al. Prognostic and predictive biomarkers for patients with idiopathic pulmonary fibrosis treated with pirfenidone: Post-hoc assessment of the CAPACITY and ASCEND trials. Lancet Respir. Med. 2018, 6, 615–626. [Google Scholar] [CrossRef]
- Kodera, M.; Hasegawa, M.; Komura, K.; Yanaba, K.; Takehara, K.; Sato, S. Serum pulmonary and activation-regulated chemokine/CCL18 levels in patients with systemic sclerosis: A sensitive indicator of active pulmonary fibrosis. Arthritis Rheum. 2005, 52, 2889–2896. [Google Scholar] [CrossRef]
- Tiev, K.P.; Hua-Huy, T.; Kettaneh, A.; Gain, M.; Duong-Quy, S.; Tolédano, C.; Cabane, J.; Dinh-Xuan, A.T. Serum CC chemokine ligand-18 predicts lung disease worsening in systemic sclerosis. Eur. Respir. J. 2011, 38, 1355–1360. [Google Scholar] [CrossRef]
- Schupp, J.C.; Binder, H.; Jäger, B.; Cillis, G.; Zissel, G.; Müller-Quernheim, J.; Prasse, A. Macrophage activation in acute exacerbation of idiopathic pulmonary fibrosis. PLoS ONE 2015, 10, e0116775. [Google Scholar] [CrossRef] [Green Version]
- Modi, W.S.; Lautenberger, J.; An, P.; Scott, K.; Goedert, J.J.; Kirk, G.D.; Buchbinder, S.; Phair, J.; Donfield, S.; O’Brien, S.J.; et al. Genetic variation in the CCL18-CCL3-CCL4 chemokine gene cluster influences HIV Type 1 transmission and AIDS disease progression. Am. J. Hum. Genet. 2006, 79, 120–128. [Google Scholar] [CrossRef] [Green Version]
- Wiertz, I.A.; Moll, S.A.; Seeliger, B.; Barlo, N.P.; van der Vis, J.J.; Korthagen, N.M.; Rijkers, G.T.; Ruven, H.J.; Grutters, J.C.; Prasse, A.; et al. Genetic variation in CCL18 gene influences CCL18 expression and correlates with survival in idiopathic pulmonary fibrosis: Part A. J. Clin. Med. 2020, 9, 1940. [Google Scholar] [CrossRef]
- American Thoracic Society. Idiopathic pulmonary fibrosis: Diagnosis and treatment. International consensus statement. American Thoracic Society (ATS), and the European Respiratory Society (ERS). Am. J. Respir. Crit. Care Med. 2000, 161, 646–664. [Google Scholar] [CrossRef] [Green Version]
- Graham, B.L.; Steenbruggen, I.; Miller, M.R.; Barjaktarevic, I.Z.; Cooper, B.G.; Hall, G.L.; Hallstrand, T.S.; Kaminsky, D.A.; McCarthy, K.; McCormack, M.C.; et al. Standardization of spirometry 2019 update. An official American thoracic society and European respiratory society technical statement. Am. J. Respir. Crit. Care Med. 2019, 200, e70–e88. [Google Scholar] [CrossRef]
- Smits, K.M.; Schouten, J.S.; Smits, L.J.; Stelma, F.F.; Nelemans, P.; Prins, M.H. A review on the design and reporting of studies on drug-gene interaction. J. Clin. Epidemiol. 2005, 58, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Oldham, J.M.; Ma, S.-F.; Martinez, F.J.; Anstrom, K.J.; Raghu, G.; Schwartz, D.A.; Valenzi, E.; Witt, L.; Lee, C.; Vij, R.; et al. TOLLIP, MUC5B, and the response to N-Acetylcysteine among individuals with idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2015, 192, 1475–1482. [Google Scholar] [CrossRef] [Green Version]
- Mantovani, A.; Bonecchi, R. One clever macrophage checkpoint. Clin. Cancer Res. 2019, 25, 3202–3204. [Google Scholar] [CrossRef] [Green Version]
- Schupp, J.; Becker, M.; Günther, J.; Müller-Quernheim, J.; Riemekasten, G.; Prasse, A. Serum CCL18 is predictive for lung disease progression and mortality in systemic sclerosis. Eur. Respir. J. 2014, 43, 1530–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tandon, K.; Herrmann, F.E.; Ayaub, E.; Parthasarathy, P.; Ackermann, M.; Inman, M.D.; Kolb, M.R.J.; Wollin, L.; Ask, K. Nintedanib attenuates the polarization of profibrotic macrophages through the inhibition of tyrosine phosphorylation on CSF1 receptor. Am. J. Respir. Crit. Care Med. 2017, 201, A2397. [Google Scholar]
- Saito, Y.; Azuma, A.; Matsuda, K.; Kamio, K.; Abe, S.; Gemma, A. Pirfenidone exerts a suppressive effect on CCL18 expression in U937-derived macrophages partly by inhibiting STAT6 phosphorylation. Immunopharmacol. Immunotoxicol. 2016, 38, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Denton, C.P.; Jahreis, A.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): A phase 2, randomised, controlled trial. Lancet 2016, 387, 2630–2640. [Google Scholar] [CrossRef]
- Khanna, D.; Allanore, Y.; Denton, C.P.; Kuwana, M.; Matucci-Cerinic, M.; Pope, J.E.; Atsumi, T.; Bečvář, R.; Czirják, L.; Hachulla, E.; et al. Riociguat in patients with early diffuse cutaneous systemic sclerosis (RISE-SSc): Randomised, double-blind, placebo-controlled multicentre trial. Ann. Rheum. Dis. 2020, 79, 618–625. [Google Scholar] [CrossRef] [Green Version]
- Khanna, D.; Denton, C.P.; Lin, C.J.F.; van Laar, J.M.; Frech, T.M.; Anderson, M.E.; Baron, M.; Chung, L.; Fierlbeck, G.; Lakshminarayanan, S.; et al. Safety and efficacy of subcutaneous tocilizumab in systemic sclerosis: Results from the open-label period of a phase II randomised controlled trial (faSScinate). Ann. Rheum. Dis. 2018, 77, 212–220. [Google Scholar] [CrossRef]
- Khanna, D.; Jahreis, A.; Furst, D.E. Tocilizumab treatment of patients with systemic sclerosis: Clinical data. J. Scleroderma Relat. Disord. 2017, 2, S29–S35. [Google Scholar] [CrossRef] [Green Version]
- Pechkovsky, D.V.; Prasse, A.; Kollert, F.; Engel, K.M.Y.; Dentler, J.; Luttmann, W.; Friedrich, K.; Müller-Quernheim, J.; Zissel, G. Alternatively activated alveolar macrophages in pulmonary fibrosis-mediator production and intracellular signal transduction. Clin. Immunol. 2010, 137, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Epstein Shochet, G.; Brook, E.; Bardenstein-Wald, B.; Shitrit, D. TGF-β pathway activation by idiopathic pulmonary fibrosis (IPF) fibroblast derived soluble factors is mediated by IL-6 trans-signaling. Respir. Res. 2020, 21, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flaherty, K.R.; Wells, A.U.; Cottin, V.; Devaraj, A.; Walsh, S.L.F.; Inoue, Y.; Richeldi, L.; Kolb, M.; Tetzlaff, K.; Stowasser, S.; et al. Nintedanib in progressive fibrosing interstitial lung diseases. N. Engl. J. Med. 2019, 381, 1718–1727. [Google Scholar] [CrossRef] [Green Version]
- Khan, K.; Xu, S.; Nihtyanova, S.; Derrett-Smith, E.; Abraham, D.; Denton, C.P.; Ong, V.H. Clinical and pathological significance of interleukin 6 overexpression in systemic sclerosis. Ann. Rheum. Dis. 2012, 71, 1235–1242. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | Cohort A (n = 61) | Cohort B (n = 101) | ||||
|---|---|---|---|---|---|---|
| rs2015086 Genotype | TT (n = 52) | CT (n = 9) | p | TT (n = 68) | CT/CC (n = 31) | p |
| Male, n (%) | 44 (85) | 8 (89) | 0.739 | 58 (85) | 29 (94) | 0.243 |
| Age, years (IQR) | 68 (63–76) | 67 (58–72) | 0.600 | 69 (61–74) | 68 (59–76) | 0.963 |
| Former or active smoker, n (%) | 32 (63) | 5 (56) | 0.683 | 47 (69) | 21 (68) | 0.633 |
| Baseline %FVC predicted, (IQR) | 79 (62–88) | 55 (36–68) | 0.014 | 65 (53–77) | 69 (58–82) | 0.341 |
| Baseline %DLCO predicted (IQR) | 48 (35–60) | 48 (41–58) | 0.710 | 48 (36–58) | 48 (36–60) | 0.925 |
| Antifibrotic therapy | - | 0.669 | ||||
| None | 52 (100) | 9 (100) | 8 (12) | 2 (6) | ||
| Nintedanib | - | 32 (47) | 14 (45) | |||
| Pirfenidone | - | 28 (41) | 15 (49) | |||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Caliskan, C.; Seeliger, B.; Jäger, B.; Fuge, J.; Welte, T.; Terwolbeck, O.; Freise, J.; van Moorsel, C.H.M.; Zhang, Y.; Prasse, A. Genetic Variation in CCL18 Gene Influences CCL18 Expression and Correlates with Survival in Idiopathic Pulmonary Fibrosis—Part B. J. Clin. Med. 2020, 9, 1993. https://doi.org/10.3390/jcm9061993
Caliskan C, Seeliger B, Jäger B, Fuge J, Welte T, Terwolbeck O, Freise J, van Moorsel CHM, Zhang Y, Prasse A. Genetic Variation in CCL18 Gene Influences CCL18 Expression and Correlates with Survival in Idiopathic Pulmonary Fibrosis—Part B. Journal of Clinical Medicine. 2020; 9(6):1993. https://doi.org/10.3390/jcm9061993
Chicago/Turabian StyleCaliskan, Canay, Benjamin Seeliger, Benedikt Jäger, Jan Fuge, Tobias Welte, Oliver Terwolbeck, Julia Freise, Coline H. M. van Moorsel, Yingze Zhang, and Antje Prasse. 2020. "Genetic Variation in CCL18 Gene Influences CCL18 Expression and Correlates with Survival in Idiopathic Pulmonary Fibrosis—Part B" Journal of Clinical Medicine 9, no. 6: 1993. https://doi.org/10.3390/jcm9061993
APA StyleCaliskan, C., Seeliger, B., Jäger, B., Fuge, J., Welte, T., Terwolbeck, O., Freise, J., van Moorsel, C. H. M., Zhang, Y., & Prasse, A. (2020). Genetic Variation in CCL18 Gene Influences CCL18 Expression and Correlates with Survival in Idiopathic Pulmonary Fibrosis—Part B. Journal of Clinical Medicine, 9(6), 1993. https://doi.org/10.3390/jcm9061993