Kidney Involvement in Hypocomplementemic Urticarial Vasculitis Syndrome—A Case-Based Review

 , ,
, ,
Abstract
:1. Introduction
2. Case Presentations
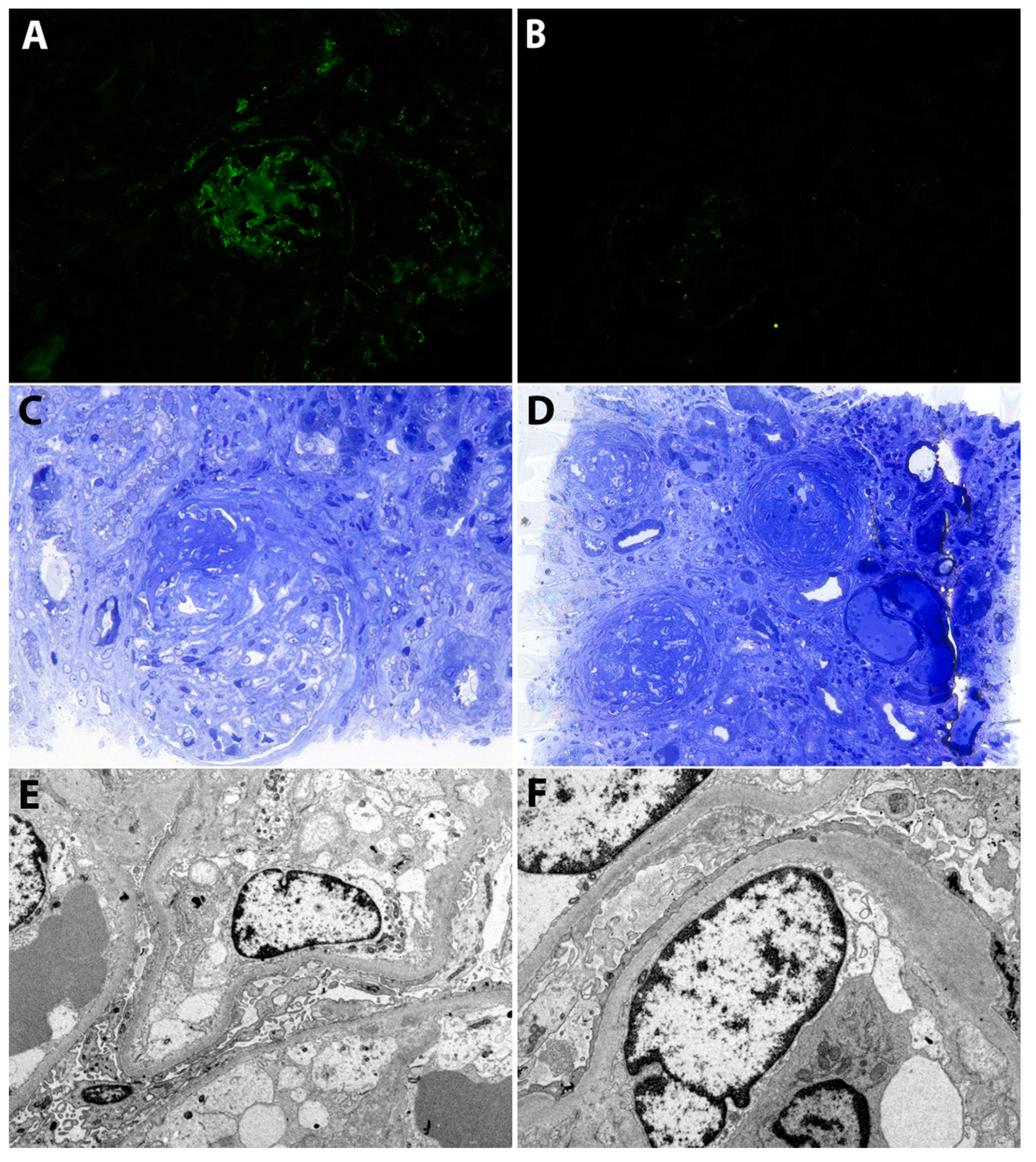
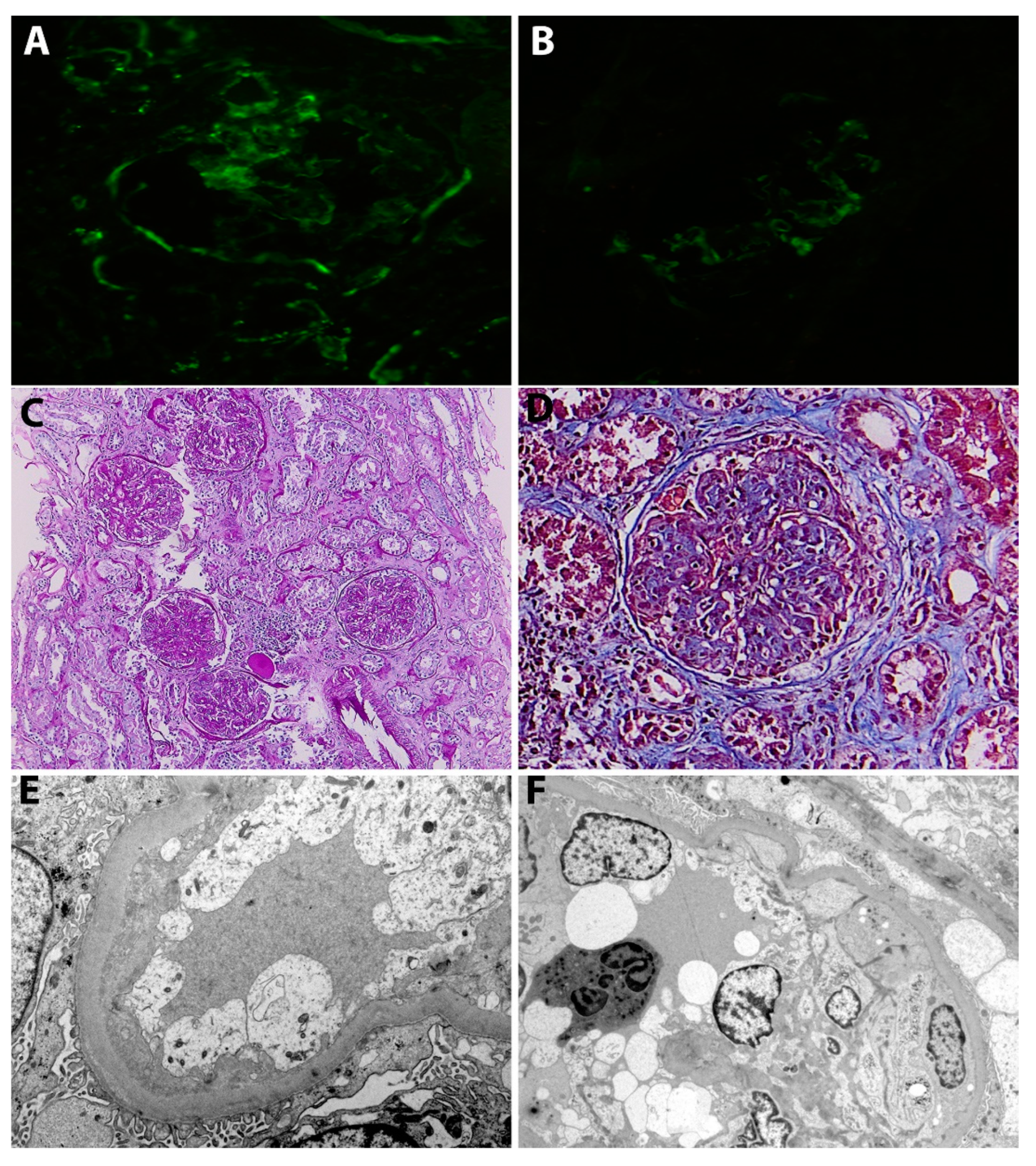
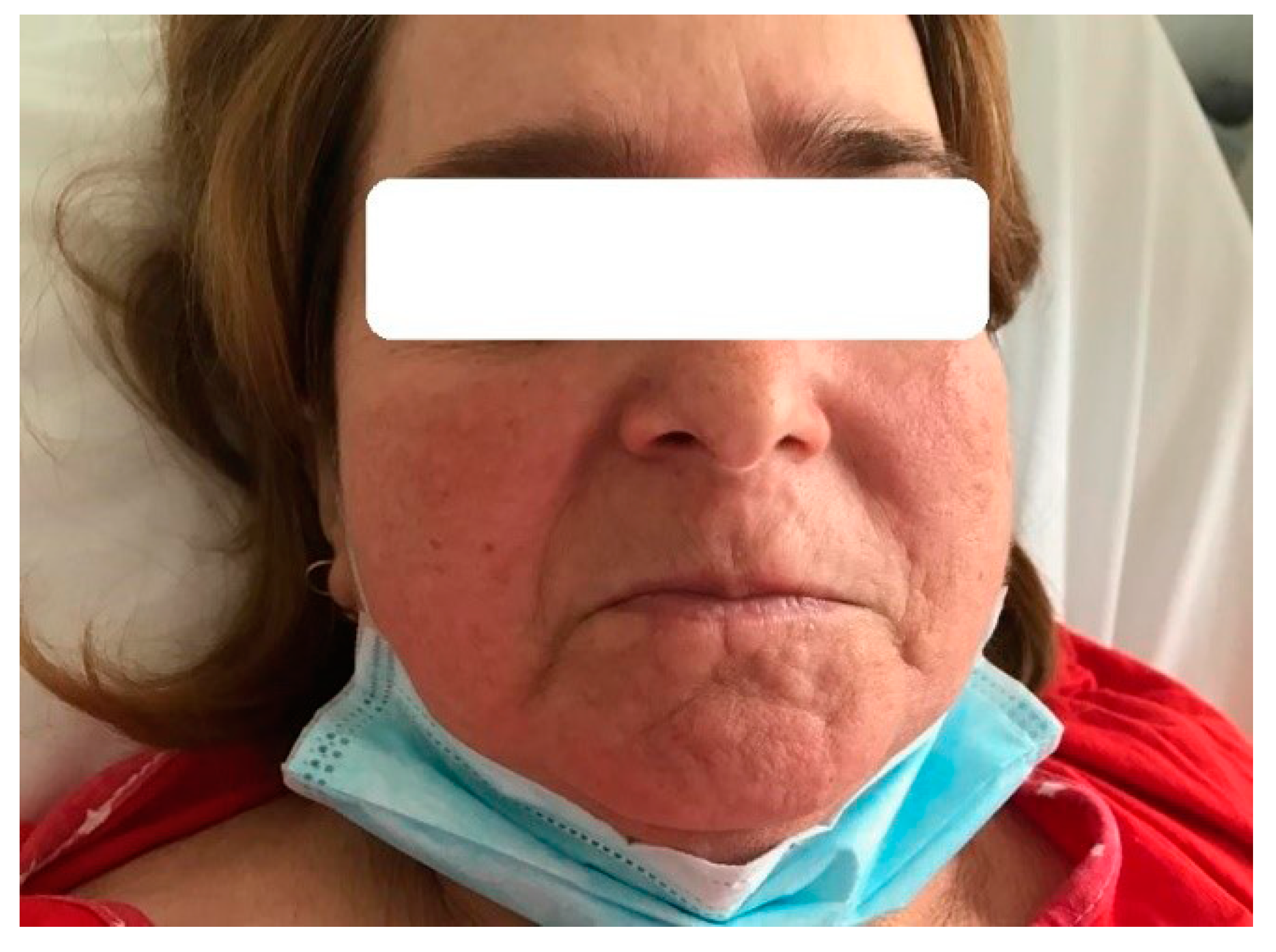
2.1. Case 1
2.2. Case 2
2.3. Case 3
3. Discussion
3.1. HUVS Clinical Presentation
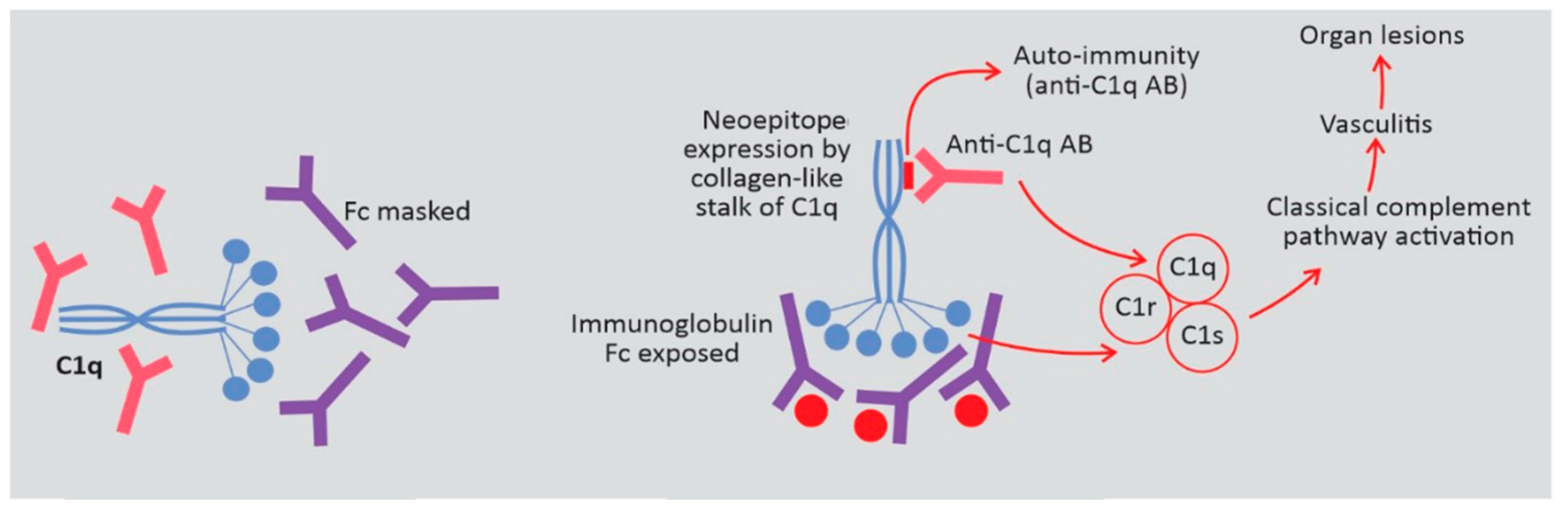
3.2. HUVS Pathogenesis
3.3. Renal Involvement in HUVS
Author Contributions
Funding
Conflicts of Interest
References
- Schwartz, H.R.; McDuffie, F.C.; Black, L.F.; Schroeter, A.L.; Conn, D.L. Hypocomplementemic urticarial vasculitis. Association with chronic obstructive pulmonary disease. Mayo Clin. Proc. 1982, 57, 231–238. [Google Scholar] [PubMed]
- Loricera, J.; Calvo-Río, V.; Mata, C.; Ortiz-Sanjuán, F.; González-López, M.A.; Alvarez, L.; González-Vela, M.C.; Armesto, S.; Fernández-Llaca, H.; Rueda-Gotor, J.; et al. Urticarial vasculitis in Northern Spain: Clinical study of 21 cases. Medicine 2014, 93, 53–60. [Google Scholar] [CrossRef] [PubMed]
- Jachiet, M.; Flageul, B.; Deroux, A.; Le Quellec, A.; Maurier, F.; Cordoliani, F.; Godmer, P.; Abasq, C.; Astudillo, L.; Belenotti, P.; et al. The clinical spectrum and therapeutic management of hypocomplementemic urticarial vasculitis: Data from a french nationwide study of fifty-seven patients. Arthritis Rheumatol. 2015, 67, 527–534. [Google Scholar] [CrossRef]
- McDuffie, F.C.; Sams, W.M.; Maldonado, J.E.; Andreini, P.H.; Conn, D.L.; Samayoa, E.A. Hypocomplementemia with cutaneous vasculitis and arthritis. Possible immune complex syndrome. Mayo Clin. Proc. 1973, 48, 340–348. [Google Scholar]
- Jennette, J.C.; Falk, R.J.; Bacon, P.A.; Basu, N.; Cid, M.C.; Ferrario, F.; Flores-Suárez, L.F.; Gross, W.L.; Guillevin, L.; Hagen, E.C.; et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013, 65, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Jara, L.J.; Navarro-Mateu, F.; Medina, G.; Vera-Lastra, O.; Saavedra, M.A. Hypocomplementemic urticarial vasculitis syndrome. Curr. Rheumatol. Rep. 2009, 11, 410–415. [Google Scholar] [CrossRef]
- Trendelenburg, M. Antibodies against C1q in patients with systemic lupus erythematosus. Springer Semin. Immunopathol. 2005, 27, 276–285. [Google Scholar] [CrossRef] [PubMed]
- Sjöwall, C.; Mandl, T.; Skattum, L.; Olsson, M.; Mohammad, A.J. Epidemiology of hypocomplementaemic urticarial vasculitis (anti-C1q vasculitis). Rheumatology 2018, 57, 1400–1407. [Google Scholar] [CrossRef] [Green Version]
- Jennette, J.C.; Hipp, C.G. Clq Nephropathy: A Distinct Pathologic Entity Usually Causing Nephrotic Syndrome. Am. J. Kidney Dis. 1985, 6, 103–110. [Google Scholar] [CrossRef]
- Strife, C.F.; Leahy, A.E.; West, C.D. Antibody to a cryptic, solid phase C1Q antigen in membranoproliferative nephritis. Kidney Int. 1989, 35, 836–842. [Google Scholar] [CrossRef] [Green Version]
- Enríquez, R.; Sirvent, A.E.; Amorós, F.; Pérez, M.; Matarredona, J.; Reyes, A. Crescentic membranoproliferative glomerulonephritis and hypocomplementemic urticarial vasculitis. J. Nephrol. 2005, 18, 318–322. [Google Scholar] [PubMed]
- Grotz, W.; Baba, H.A.; Becker, J.U.; Baumgärtel, M.W. Hypocomplementemic urticarial vasculitis syndrome: An interdisciplinary challenge. Dtsch. Arztebl. Int. 2009, 106, 756–763. [Google Scholar] [PubMed]
- Davis, M.D.; Brewer, J.D. Urticarial vasculitis and hypocomplementemic urticarial vasculitis syndrome. Immunol. Allergy Clin. N. Am. 2004, 24, 183–213. [Google Scholar] [CrossRef] [PubMed]
- Khasnis, A.; Langford, C.A. Update on vasculitis. J. Allergy Clin. Immunol. 2009, 123, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Wisnieski, J.J.; Baer, A.N.; Christensen, J.; Cupps, T.R.; Flagg, D.N.; Jones, J.V.; Katzenstein, P.L.; McFadden, E.R.; McMillen, J.J.; Pick, M.A.; et al. Hypocomplementemic urticarial vasculitis syndrome: Clinical and serologic findings in 18 patients. Medicine 1995, 74, 24–41. [Google Scholar] [CrossRef] [PubMed]
- Potlukova, E.; Kralikova, P. Complement component C1q and anti-C1q antibodies in theory and in clinical practice. Scand. J. Immunol. 2008, 67, 423–430. [Google Scholar] [CrossRef]
- Pickering, M.C.; Botto, M.; Taylor, P.; Lachmann, P.; Walport, M. Systemic lupus erythematosus, complement deficiency, and apoptosis. Adv. Immunol. 2001, 76, 227–324. [Google Scholar]
- Botto, M.; Walport, M.J. C1q, autoimmunity and apoptosis. Immunobiology 2002, 205, 395–406. [Google Scholar] [CrossRef]
- Mitchell, D.; Taylor, P.R.; Cook, H.T.; Moss, J.; Bygrave, A.E.; Walport, M.J.; Botto, M. Cutting edge: C1q protects against the development of glomerulonephritis independently of C3 activation. J. Immunol. 1999, 162, 5676–5679. [Google Scholar]
- Botto, M.; Agnola, C.D.; Bygrave, A.E.; Thompson, E.M.; Cook, H.T.; Petry, F.; Loos, M.; Pandolfi, P.P.; Walport, M. Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet. 1998, 19, 56–59. [Google Scholar] [CrossRef]
- Taylor, P.R.; Carugati, A.; Fadok, V.A.; Cook, H.T.; Andrews, M.; Carroll, M.C.; Savill, J.S.; Henson, P.M.; Botto, M.; Walport, M. A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J. Exp. Med. 2000, 192, 359–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurskens, F.J.; Van Schaarenburg, R.A.; Trouw, L.A. C1q, antibodies and anti-C1q autoantibodies. Mol. Immunol. 2015, 68, 6–13. [Google Scholar] [CrossRef] [PubMed]
- Siegert, C.E.; Daha, M.R.; Halma, C.; Van Der Voort, E.A.; Breedveld, F.C. IgG and IgA autoantibodies to C1q in systemic and renal diseases. Clin. Exp. Rheumatol. 1992, 10, 19–23. [Google Scholar]
- Verlemyr, A.; Truedsson, L.; Skattum, L. Analysis of anti-C1q autoantibodies by western blot. Methods Mol. Biol. 2019, 1901, 183–189. [Google Scholar]
- Golan, M.D.; Burger, R.; Loos, M. Conformational changes in C1q after binding to immune complexes: Detection of neoantigens with monoclonal antibodies. J. Immunol. 1982, 129, 445–447. [Google Scholar] [PubMed]
- Trouw, L.A.; Groeneveld, T.W.L.; Seelen, M.A.; Duijs, J.M.G.J.; Bajema, I.M.; Prins, F.A.; Kishore, U.; Salant, D.J.; Verbeek, J.S.; van Kooten, C.; et al. Anti-C1q autoantibodies deposit in glomeruli but are only pathogenic in combination with glomerular C1q-containing immune complexes. J. Clin. Investig. 2004, 114, 679–688. [Google Scholar] [CrossRef] [Green Version]
- Holers, V.M. Anti-C1q autoantibodies amplify pathogenic complement activation in systemic lupus erythematosus. J. Clin. Investig. 2004, 114, 616–619. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Nagase, M.; Hidaka, S.; Arai, T.; Ikegaya, N.; Hishida, A.; Honda, N. Membranous nephropathy associated with hypocomplementemic urticarial vasculitis: Report of two cases and a review of the literature. Nephron 1994, 66, 1–7. [Google Scholar] [CrossRef]
- Sissons, J.; Peters, D.; Williams, D.G.; Boulton-Jones, J.; Goldsmith, H. Skin lesions, angio-oedema, and hypocomplementaemia. Lancet (Lond. Engl.) 1974, 2, 1350–1352. [Google Scholar] [CrossRef]
- Feig, P.U.; Soter, N.A.; Yager, H.M.; Caplan, L.; Rosen, S. Vasculitis with urticaria, hypocomplementemia, and multiple system involvement. JAMA 1976, 236, 2065–2068. [Google Scholar] [CrossRef]
- Ludivico, C.L.; Myers, A.R.; Maurer, K. Hypocomplementemic urticarial vasculitis with glomerulonephritis and pseudotumor cerebri. Arthritis Rheum. 1979, 22, 1024–1028. [Google Scholar] [CrossRef]
- Schultz, D.R.; Perez, G.O.; Volanakis, J.E.; Pardo, V.; Moss, S.H. Glomerular disease in two patients with urticaria-cutaneous vasculitis and hypocomplementemia. Am. J. Kidney Dis. 1981, 1, 157–165. [Google Scholar] [CrossRef]
- Meyrier, A.; Français, P.; Lesavre, P.; Mougenot, B.; Ronco, P.; Jeanmougin, M.; Français, C. Hypocomplementemic urticarial vasculitis with glomerulopathy and renal venulitis. Nephrologie 1984, 5, 1–7. [Google Scholar] [PubMed]
- Waldo, F.B.; Leist, P.A.; Strife, C.F.; Forristal, J.; West, C.D. Atypical hypocomplementemic vasculitis syndrome in a child. J. Pediatr. 1985, 106, 745–750. [Google Scholar] [CrossRef]
- Fortson, J.S.; Zone, J.J.; Hammond, M.E.; Groggel, G.C. Hypocomplementemic urticarial vasculitis syndrome responsive to dapsone. J. Am. Acad. Derm. 1986, 15, 1137–1142. [Google Scholar] [CrossRef]
- Ramirez, G.; Saba, S.R.; Espinoza, L. Hypocomplementemic vasculitis and renal involvement. Nephron 1987, 45, 147–150. [Google Scholar] [CrossRef]
- Wisnieski, J.J.; Emancipator, S.N.; Korman, N.J.; Lass, J.H.; Zaim, T.M.; McFadden, E.R. Hypocomplementemic urticarial vasculitis syndrome in identical twins. Arthritis Rheum. 1994, 37, 1105–1111. [Google Scholar] [CrossRef]
- Martini, A.; Ravelli, A.; Albani, S.; De Benedetti, F.; Massa, M.; Wisnieski, J.J. Hypocomplementemic urticarial vasculitis syndrome with severe systemic manifestations. J. Pediatr. 1994, 124, 742–744. [Google Scholar] [CrossRef]
- Mituiki, K.; Hirakata, H.; Oochi, N.; Nagashima, A.; Onoyama, K.; Abe, M.; Okuda, S.; Fujishima, M. Nephrotic syndrome due to membranous glomerulopathy in hypocomplementemic urticarial vasculitis syndrome;—A case report. Nihon Jinzo Gakkai Shi 1994, 36, 863–870. [Google Scholar]
- Eiser, A.R.; Singh, P.; Shanies, H.M. Sustained dapsone-induced remission of hypocomplementemic urticarial vasculitis—A case report. Angiology 1997, 48, 1019–1022. [Google Scholar] [CrossRef]
- Renard, M.; Wouters, C.; Proesmans, W. Rapidly progressive glomerulonephritis in a boy with hypocomplementaemic urticarial vasculitis. Eur. J. Pediatr. 1998, 157, 243–245. [Google Scholar] [CrossRef]
- Jovanović, D.; Kovacević, Z.; Zecević, R.; Dimitrijević, J.; Romanović, R.; Marić, M. Urticarial vasculitis—a syndrome with low complement levels and secondary glomerulopathy. Vojnosanit. Pregl. 1999, 56, 551–554. [Google Scholar]
- Soma, J.; Sato, H.; Ito, S.; Saito, T. Nephrotic syndrome associated with hypocomplementaemic urticarial vasculitis syndrome: Successful treatment with cyclosporin A. Nephrol. Dial. Transpl. 1999, 14, 1753–1757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadnapaphornchai, M.A.; Saulsbury, F.T.; Norwood, V.F. Hypocomplementemic urticarial vasculitis: Report of a pediatric case. Pediatr. Nephrol. 2000, 14, 328–331. [Google Scholar] [CrossRef] [PubMed]
- Sessler, R.; Hasche, G.; Olbricht, C.J. Hypocomplementemic urticarial vasculitis syndrome. Dtsch. Med. Wochenschr. 2000, 125, 1003–1006. [Google Scholar] [CrossRef] [PubMed]
- Messiaen, T.; Van Damme, B.; Kuypers, D.; Maes, B.; Vanrenterghem, Y. Crescentic glomerulonephritis complicating the course of a hypocomplementemic urticarial vasculitis. Clin. Nephrol. 2000, 54, 409–412. [Google Scholar]
- Chew, G.Y.J.; Gatenby, P.A. Inflammatory myositis complicating hypocomplementemic urticarial vasculitis despite on-going immunosuppression. Clin. Rheumatol. 2007, 26, 1370–1372. [Google Scholar] [CrossRef]
- Boulay, V.; Lauque, D.; Reynaud, F.; Carles, P.; Pourrat, J. Hypocomplementemic urticarial vasculitis. Presse Med. 2000, 29, 1507–1509. [Google Scholar]
- El Maghraoui, A.; Abouzahir, A.; Mahassine, F.; Tabache, F.; Bezza, A.; Ghafir, D.; Ohayon, V.; Archane, M. Vascularite urticarienne hypocomplémentémique de McDuffie. Deux observations et revue de la littérature. Rev. Med. Interne 2001, 22, 70–74. [Google Scholar] [CrossRef]
- Brass, H.; Uppenkamp, M.; Voigtländer, V. Nierenbeteiligung bei hypokomplementärem urtikaria-vaskulitis-syndrom—Einer spielart des systemischen lupus erythematodes. Med. Klin. 2001, 96, 238–241. [Google Scholar] [CrossRef]
- Saeki, T.; Ueno, M.; Shimada, H.; Nishi, S.; Imai, N.; Miyamura, S.; Gejou, F.; Arakawa, M. Membranoproliferative glomerulonephritis associated with hypocomplementemic urticarial vasculitis after complete remission of membranous nephropathy. Nephron 2001, 88, 174–177. [Google Scholar] [CrossRef]
- Grimbert, P.; Schulte, K.-M.; Buisson, C.; Desvaux, D.; Baron, C.; Pastural, M.; Dhamane, D.; Rémy, P.; Weil, B.; Lang, P. Renal transplantation in a patient with hypocomplementemic urticarial vasculitis syndrome. Am. J. Kidney Dis. 2001, 37, 144–148. [Google Scholar] [CrossRef]
- Toprak, O.; Cirit, M.; Uzunel, H.; Ersoy, R.; Ermete, M.; Unsal, B.; Guven, F. Hypocomplementaemic urticarial vasculitis syndrome and acute renal failure with cryoglobulin (–) hepatitis C infection. Nephrol. Dial. Transplant. 2004, 19, 2680–2682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiederkehr, M.; Nicosia, R.F.; Munschauer, C.; Moe, O.W. An Unusual Case of Urticaria and Nephrotic Syndrome. Am. J. Kidney Dis. 2006, 48, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Balsam, L.; Karim, M.; Miller, F.W.; Rubinstein, S. Crescentic Glomerulonephritis Associated With Hypocomplementemic Urticarial Vasculitis Syndrome. Am. J. Kidney Dis. 2008, 52, 1168–1173. [Google Scholar] [CrossRef]
- Özçakar, Z.B.; Yalçınkaya, F.; Altugan, F.Ş.; Kavaz, A.; Ensari, A.; Ekim, M.; Yalcinkaya, F.; Kavaz, A. Hypocomplementemic urticarial vasculitis syndrome in three siblings. Rheumatol. Int. 2013, 33, 763–766. [Google Scholar]
- Al Mosawi, Z.S.A.; Al Hermi, B.E.A. Hypocomplementemic Urticarial Vasculitis Syndrome in an 8-year-old Boy: A Case Report and Review of Literature. Oman Med. J. 2013, 28, 275–277. [Google Scholar] [CrossRef]
- Park, C.; Choi, S.W.; Kim, M.; Park, J.; Lee, J.S.; Chung, H.C. Membranoproliferative glomerulonephritis presenting as arthropathy and cardiac valvulopathy in hypocomplementemic urticarial vasculitis: A case report. J. Med. Case Rep. 2014, 8, 352. [Google Scholar] [CrossRef] [Green Version]
- Pasini, A.; Bracaglia, C.; Aceti, A.; Vivarelli, M.; Lavacchini, A.; Miniaci, A.; De Benedetti, F.; Montini, G. Renal involvement in hypocomplementaemic urticarial vasculitis syndrome: A report of three paediatric cases. Rheumatology 2014, 53, 1409–1413. [Google Scholar] [CrossRef] [Green Version]
- Zakharova, E.; Es, S.; Ol, V.; Ev, Z. Hypocomplementemic Urticarial Vasculitis with Crescentic Glomerulonephritis, Interstitial Nephritis and Small Vessel Vasculopathy: Case Report and Mini-Review. J. Nephrol. Ther. 2016, 6. [Google Scholar] [CrossRef]
- Jung, S.W.; Choi, Y.Y.; Choi, I.S.; Kim, S.; Jeong, K.H.; Song, R.; Lee, S.H.; Yang, H.I.; Hong, S.-J.; Lee, Y.-A. Hypocomplementemic Urticarial Vasculitis Syndrome with Membranous Nephropathy: Case Report. J. Korean Med. Sci. 2017, 32, 2064–2068. [Google Scholar] [CrossRef] [Green Version]
- Gheerbrant, H.; Giovannini, D.; Falque, L.; Andry, F.; Lugosi, M.; Deroux, A. Vascularite hypocomplémentémique urticarienne associée à une glomérulonéphrite membrano-proliférative sévère et polyadénopathies. Presse Med. 2017, 46, 547–550. [Google Scholar] [CrossRef]
- Tanaka, M.; Moniwa, N.; Mita, T.; Tobisawa, T.; Matsumoto, T.; Mochizuki, A.; Yamashita, T.; Yano, T.; Furuhashi, M.; Miura, T. A Case of Crescentic Glomerulonephritis Complicated with Hypocomplementemic Urticarial Vasculitis Syndrome and ANCA-Associated Vasculitis. Case Rep. Nephrol. Dial. 2017, 7, 144–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AlHermi, B.; Al Mosawi, Z.; Mohammed, D. Renal manifestations in hypocomplementic urticarial vasculitis syndrome: Is it a distinct pathology? Saudi J. Kidney Dis. Transpl. 2017, 28, 929–933. [Google Scholar] [PubMed]
- Salim, S.; Yousuf, T.; Patel, A.; Fülöp, T.; Agarwal, M. Hypocomplementemic Urticarial Vasculitis Syndrome with Crescentic Glomerulonephritis. Am. J. Med. Sci. 2018, 355, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Khoury, F.G.; Hopkins, A.M.; Gibbs, A.M.; Griffiths, R.S.; Avasare, R.S. A case report of Hypocomplementemic urticarial vasculitic syndrome presenting with Renal failure. J. Clin. Nephrol. 2018, 2, 39–43. [Google Scholar] [CrossRef] [Green Version]
- López-Romero, L.C.; Poma-Saavedra, F.H.; Panizo-González, N.; Pérez-Rojasb, J.; Moll-Guillema, J.L.; Bea-Granella, S.; Peris-Fernándeza, M.; Aledon-Viñesa, P.; Hernández-Jarasa, J. Hypocomplementemic urticarial vasculitis with polyadenopathies and renal involvement. NefroPlus 2019, 11, 88–93. [Google Scholar]
- Ueki, K.; Tsuchimoto, A.; Matsukuma, Y.; Torisu, K.; Fujisaki, K.; Torisu, T.; Yamada, Y.; Oda, Y.; Masutani, K.; Nakano, T.; et al. Hypocomplementemic urticarial vasculitis syndrome with gastrointestinal vasculitis and crescentic membranoproliferative glomerulonephritis without immune complex deposits. CEN Case Rep. 2020, 9, 30–35. [Google Scholar] [CrossRef]
- Boyer, A.; Gautier, N.; Comoz, F.; de Ligny, B.H.; Aouba, A.; Lanot, A. Nephropathy associated with hypocomplementemic urticarial vasculitis: A case report and literature review. Nephrol. Ther. 2020, 16, 124–135. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| At Presentation | Follow-Up | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 6 mo | 6 mo | 6 mo | 27 mo | 16 mo | 29 mo | ||||
| Case | 1 | 2 | 3 | 1 | 2 | 3 | 1 | 2 | 3 |
| Serum creatinine [mg/dL] | 4.29 | 1.27 | 5.96 | 1.16 | 0.86 | 3.9 | 0.9 | 0.85 | 3.9 |
| eGFR (ml/min/1.73 m2) | 12 | 50 | 8 | 56 | 81 | 13 | 76 | 82 | 13 |
| Serum albumin [g/dL] | 2.8 | 1.3 | 2.8 | 3.5 | 2.41 | 4 | 4.61 | 3.3 | 4 |
| Proteinuria [g/24 h] | 13.4 | 6.2 | 4 | 7.4 | 2 | 1.2 | 1.8 | 2.6 | 1.2 |
| Hematuria [RBC/μL] | 1937 | 105 | 773 | 49 | 39 | 25 | 10 | 14 | 25 |
| Anti-C1q antibodies [U/mL] | 75.8 | 356 | 112.8 | 3.5 | 72 | 75 | 9.1 | 157 | 86 |
| C3 [mg/dL] | 58.4 | 41 | 18.7 | 103 | 65.1 | 81.3 | 108 | 65.7 | 81.3 |
| C4 [mg/dL] | 9.24 | 8.94 | 3.61 | 31.9 | 19.6 | 23.3 | 30.2 | 17.1 | 23.3 |
| CRP [mg/L] | 37.5 | 5.06 | 12.9 | 1.74 | 0.43 | 8 | 2.15 | 0.37 | 8 |
| Major Criteria |
| ● Chronic urticarial exanthema |
| ● Hypocomplementemia |
| Minor criteria |
| ● Leuko-cytoclastic vasculitis |
| ● Arthralgia/arthritis |
| ● Uveitis/episcleritis/conjunctivitis |
| ● Glomerulonephritis |
| ● Abdominal pain |
| ● Positive C1q antibodies |
| For a positive diagnosis are necessary: |
| Two major criteria, two minor criteria and exclusion of an autoimmune disease (SLE, Sjögren syndrome, cryoglobulinemia) |
| Author (Year) | Age | Sex | Organ Damage * | Anti-C1q AB | GFR | Renal Manifestations | Kidney Biopsy | Immunosuppressive Therapy | ESRD | Death | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Sissons (1974) [29] | 48 | F | AE | NR | 8 | NN proteinuria | GN | CS (PO), CYC (PO) | No | No |
| 2 | Feig (1976) [30] | 31 | F | J, E, L | NR | NR | Hematuria, NN proteinuria | GN | NR | NR | NR |
| 3 | Ludivico (1979) [31] | 24 | F | D | NR | 90 | Hematuria, N Syndrome | MPGN | CS (PO, IV), AZA | No | No |
| 4 | Schultz (1981) [32] | 36 | M | J, E | NR | 96 | Hematuria, NN proteinuria | MesPGN | CS (PO) | No | No |
| 5 | Schultz (1981) [32] | 54 | F | NR | 64 | Hematuria, NN proteinuria | MesPGN | CS (PO) | No | No | |
| 6 | Meyrier (1984) [33] | 58 | F | J, D, AE | NR | NR | NS proteinuria | GN | CS | No | No |
| 7 | Waldo (1985) [34] | 16 | M | D | NR | NR | Hematuria, NS proteinuria | MPGN | CS (PO) | Yes | No |
| 8 | Kobayashi (1985) [28] | 55 | M | E | NR | 96 | Hematuria, N Syndrome | MN | CS (PO) | No | No |
| 9 | Kobayashi (1985) [28] | 28 | F | NR | 102 | Hematuria, NS proteinuria | MN | CS (PO) | NR | NR | |
| 10 | Fortson (1986) [35] | 45 | F | J, L | NR | 68 | Hematuria, NS proteinuria | MPGN | CS (PO) | No | No |
| 11 | Ramirez (1987) [36] | 59 | M | E | + | 20 | Hematuria, NN proteinuria | MPGN | CS (PO), CYC, AZA | No | Yes |
| 12 | Wisnieski (1994) [37] | 32 | F | J, E, L | + | 85 | NN proteinuria | NR | CS (PO), HCQ, IG | No | No |
| 13 | Wisnieski (1994) [37] | 31 | F | J, E, AE | + | 25 | N Syndrome | GN | CS (PO), HCQ, IG | No | No |
| 14 | Martini (1994) [38] | 14 | M | J, E, D | + | NR | Hematuria, NS proteinuria | MesPGN, (FC) | CS (PO, IV), CYC (PO, IV) | Yes | No |
| 15 | Martini (1994) [38] | 15 | M | J, E, L, D, AE | + | NR | Hematuria | NR | CS (PO, IV), CYC (IV), AZA | No | No |
| 16 | Mituiki (1994) [39] | 66 | M | L | + | 44 | N Syndrome | MN | CS | No | Yes |
| 17 | Wisnieski (1995) [15] | 53 | M | J, E, L, AE | + | 107 | Hematuria, NN proteinuria | MPGN | CS (PO, IV) | No | Yes |
| 18 | Wisnieski (1995) [15] | 41 | F | J, L, AE | + | 92 | NN proteinuria | MesPGN | NR | No | Yes |
| 19 | Wisnieski (1995) [15] | 40 | F | J, L, AE | + | 37 | Hematuria, NS proteinuria | MPGN | AZA | No | Yes |
| 20 | Wisnieski (1995) [15] | 37 | F | J, L | + | NR | Hematuria, NN proteinuria | NR | CS (PO), CYC | No | Yes |
| 21 | Wisnieski (1995) [15] | 26 | F | J, E, D, AE | + | 69 | Hematuria | MesPGN | CS (PO), HCQ | No | No |
| 22 | Wisnieski (1995) [15] | 35 | F | J, L, AE | + | 95 | Hematuria, N proteinuria | MN | MET | No | Yes |
| 23 | Eiser (1997) [40] | 35 | F | J, E, L | NR | NR | Hematuria, NN proteinuria | MN | CS (PO), CYC (IV) | No | No |
| 24 | Renard (1998) [41] | 13 | M | J, E | NR | NR | Hematuria, N proteinuria | MesPGN (FC, CC) | CS (PO, IV), CSA, AZA | No | No |
| 25 | Jovanovic (1999) [42] | 41 | F | J | NR | NR | Hematuria, NS proteinuria | MesPGN | CS (PO) | No | No |
| 26 | Trendelenburg (1999) [7] | 37 | F | J, E, D, L | + | NR | Hematuria, NS proteinuria | MesPGN | CS (PO), HCQ, AZA, PLEX, IVIG | No | No |
| 27 | Soma (1999) [43] | 43 | M | NR | NR | 109 | Hematuria, N Syndrome | MN | CS (PO, IV), CSA | No | No |
| 28 | Cadnapaphornchai (2000) [44] | 11 | F | J | NR | NR | Hematuria, NN proteinuria | MPGN | CS (PO) | No | No |
| 29 | Sessler (2000) [45] | 40 | F | J, AE | + | NR | NR | MPGN | CS, CYC | No | No |
| 30 | Messiaen (2000) [46] | 27 | F | J, E, L | NR | 9 | Hematuria, NS proteinuria | MPGN (FC) | CS, CYC | Yes | No |
| 31 | Chew (2000) [47] | 55 | F | L, AE | + | 90 | NN proteinuria | MPGN | CS, CYC (IV), MET | No | No |
| 32 | Boulay (2000) [48] | 34 | F | L | NR | 22 | NR | MPGN | CS | No | Yes |
| 33 | El Maghraoui (2001) [49] | 41 | F | J | NR | NR | NN proteinuria | MN | CS (PO), HCQ, CYC (PO) | No | No |
| 34 | Brass (2001) [50] | 44 | F | J, E, AE | + | NR | Hematuria, NN proteinuria | MN | CS (PO), HCQ, CYC, CSA, MET, IVIG | No | No |
| 35 | Saeki (2001) [51] | 49 | M | NR | 59 | Hematuria, N Syndrome | MPGN | CS (PO) | No | No | |
| 36 | Grimbert (2001) [52] | 36 | M | J, AE | + | 15 | Hematuria, N proteinuria | MPGN | CS (PO, IV), CSA, AZA, PLEX | Yes | Yes |
| 37 | Toprak (2004) [53] | 53 | F | D | NR | 9 | Hematuria, NN proteinuria | GN | CS (PO, IV), CYC (IV) | Yes | Yes |
| 38 | Enriquez (2005) [11] | 39 | F | J, E | + | 71 | Hematuria, N Syndrome | MPGN (C) | CS, CYC, MMF | No | No |
| 39 | Wiederkehr (2006) [54] | 63 | M | J, E, L, AE | NR | 42 | Hematuria, N Syndrome | MN | CS (PO), MMF | No | No |
| 40 | Balsam (2008) [55] | 23 | F | J, D | + | 29 | Hematuria, N Syndrome | GN (C) | CS (PO, IV), CYC (IV), PLEX | Yes | No |
| 41 | Özçakar (2010) [56] | 6 | F | D | NR | NR | Hematuria, NN proteinuria | GN (C) | CS, CYC, AZA | No | Yes |
| 42 | Al Mosawi (2013) [57] | 8 | M | J, E, AE | NR | NR | Hematuria, NN proteinuria | GN (C) | CS (PO, IV), MMF, AZA | No | No |
| 43 | Loricera (2014) [2] | 35 | F | J | NR | NR | Hematuria | NR | CS (PO), HCQ | No | No |
| 44 | Park (2014) [58] | 30 | M | J, AE | NR | 44 | Hematuria, NN proteinuria | MPGN | CS (PO, IV), HCQ, CYC (PO), MMF | No | No |
| 45 | Pasini (2014) [59] | 9 | F | J, E, D | + | NR | Hematuria | MesPGN | CS (PO), MMF | No | No |
| 46 | Pasini (2014) [59] | 9 | F | J, E, L, D | + | NR | Hematuria, NN proteinuria | GN | CS (PO), CYC (PO), AZA | No | No |
| 47 | Pasini (2014) [59] | 9 | M | J, E, D, L, AE | + | NR | Hematuria, N Syndrome | GN (C) | CS (PO, IV), HCQ, CYC (IV), CSA, AZA | No | No |
| 48 | Zakharova (2016) [60] | 32 | F | E, D | + | 24 | Hematuria, NN proteinuria | MesPGN (FC) | CS (PO, IV), AZA | No | No |
| 49 | Jung (2017) [61] | 15 | M | E | NR | NR | NN proteinuria | MN | CS (PO, IV), HCQ, AZA, TAC | No | No |
| 50 | Gheerbrant (2017) [62] | 41 | F | E, D | + | 69 | Hematuria, N Syndrome | GN | CS (PO, IV), MMF | No | No |
| 51 | Tanaka (2017) [63] | 64 | F | D | NR | 22 | Hematuria, NN proteinuria | GN (CC) | CS (PO, IV) | No | No |
| 52 | AlHermi (2017) [64] | 6 | M | J, E, D | + | NR | Hematuria, NS proteinuria | MesPGN | CS (PO, IV), AZA | No | No |
| 53 | Salim (2018) [65] | 31 | F | J, D | + | 12 | Hematuria, N Syndrome | MPGN (C) | CS (PO, IV), CYC (IV), MMF, R | No | No |
| 54 | Hopkins (2018) [66] | 66 | M | J, D | NR | 5 | Hematuria, NS proteinuria | NR | CS (PO) | No | No |
| 55 | Sjowall (2018) [8] | 53 | F | L | + | NR | NR | NR | CS (PO), HCQ, MMF | Yes | Yes |
| 56 | Sjowall (2018) [8] | 64 | F | J | + | NR | NR | NR | CS (PO), CYC, PLEX | Yes | Yes |
| 57 | Sjowall (2018) [8] | 49 | F | J | + | NR | NR | NR | CS (PO), HCQ, MMF | No | No |
| 58 | Lopez-Romero (2019) [67] | 52 | M | J, E | NR | 31 | Hematuria, N proteinuria | MPGN | CS (PO, IV) | No | No |
| 59 | Ueki (2020) [68] | 36 | M | D, L | + | 22 | Hematuria, N proteinuria | MPGN (CC) | CS (PO, IV), PLEX | Yes | No |
| 60 | Boyer (2020) [69] | 49 | F | J | + | 35 | Hematuria, N proteinuria | FSGS | CS (PO), HCQ | No | No |
| Hematuria | 76.7% (46) | Isolated 7% (4) | ||
| Associated 70% (42) | Proteinuria (Non-Nephrotic) | 27% (16) | ||
| Proteinuria (Nephrotic) | 10% (6) | |||
| Nephrotic Syndrome | 17% (10) | |||
| Non-Specified Proteinuria | 17%(10) | |||
| Proteinuria (isolated) | 12% (7) | |||
| Nephrotic syndrome | 3% (2) | |||
| Not reported | 8% (5) | |||
| GFR | Mean; 95% CI | 44; 25–69 | ||
| <60 | 33% (20) | |||
| Not reported | 41% (25) | |||
| Data in brackets are numbers of patients; GFR—Glomerular Filtration Rate [mL/min/1.73 m2]; | ||||
| Membranoproliferative GN | 35% (18) |
| GN not specified | 23% (12) |
| Mesangioproliferative GN | 21% (11) |
| Membranous nephropathy | 19% (10) |
| Focal segmental glomerulosclerosis | 2% (1) |
| Crescents | 23% (12) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ion, O.; Obrișcă, B.; Ismail, G.; Sorohan, B.; Bălănică, S.; Mircescu, G.; Sinescu, I. Kidney Involvement in Hypocomplementemic Urticarial Vasculitis Syndrome—A Case-Based Review. J. Clin. Med. 2020, 9, 2131. https://doi.org/10.3390/jcm9072131
Ion O, Obrișcă B, Ismail G, Sorohan B, Bălănică S, Mircescu G, Sinescu I. Kidney Involvement in Hypocomplementemic Urticarial Vasculitis Syndrome—A Case-Based Review. Journal of Clinical Medicine. 2020; 9(7):2131. https://doi.org/10.3390/jcm9072131
Chicago/Turabian StyleIon, Oana, Bogdan Obrișcă, Gener Ismail, Bogdan Sorohan, Sonia Bălănică, Gabriel Mircescu, and Ioanel Sinescu. 2020. "Kidney Involvement in Hypocomplementemic Urticarial Vasculitis Syndrome—A Case-Based Review" Journal of Clinical Medicine 9, no. 7: 2131. https://doi.org/10.3390/jcm9072131
APA StyleIon, O., Obrișcă, B., Ismail, G., Sorohan, B., Bălănică, S., Mircescu, G., & Sinescu, I. (2020). Kidney Involvement in Hypocomplementemic Urticarial Vasculitis Syndrome—A Case-Based Review. Journal of Clinical Medicine, 9(7), 2131. https://doi.org/10.3390/jcm9072131