A Novel Method for Simultaneous Removal of NO and SO2 from Marine Exhaust Gas via In-Site Combination of Ozone Oxidation and Wet Scrubbing Absorption

 ,
,  ,
,
Abstract
:1. Introduction
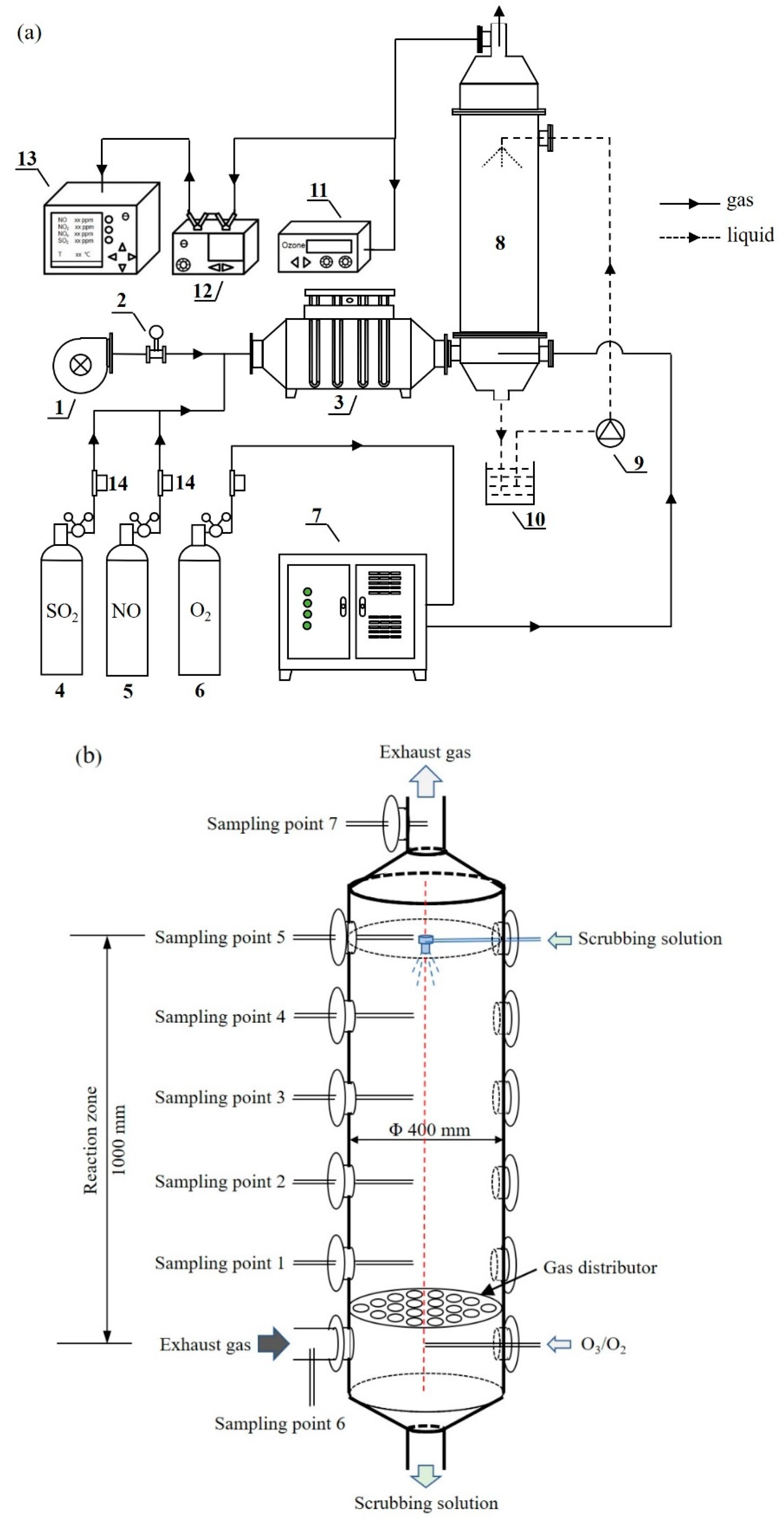
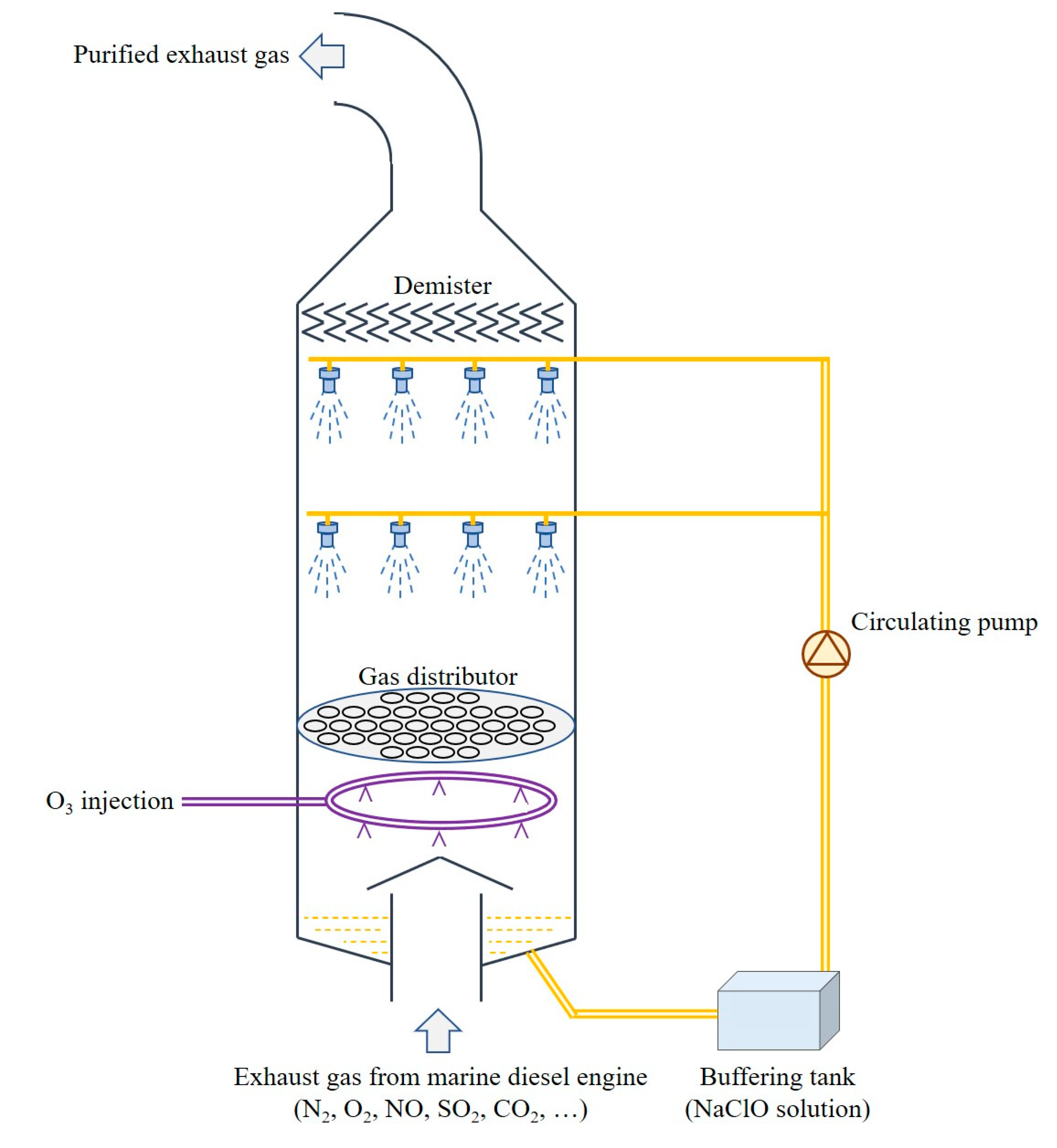
2. Materials and Methods
3. Results
3.1. The Oxidation Performance of NO by Ozone Injection
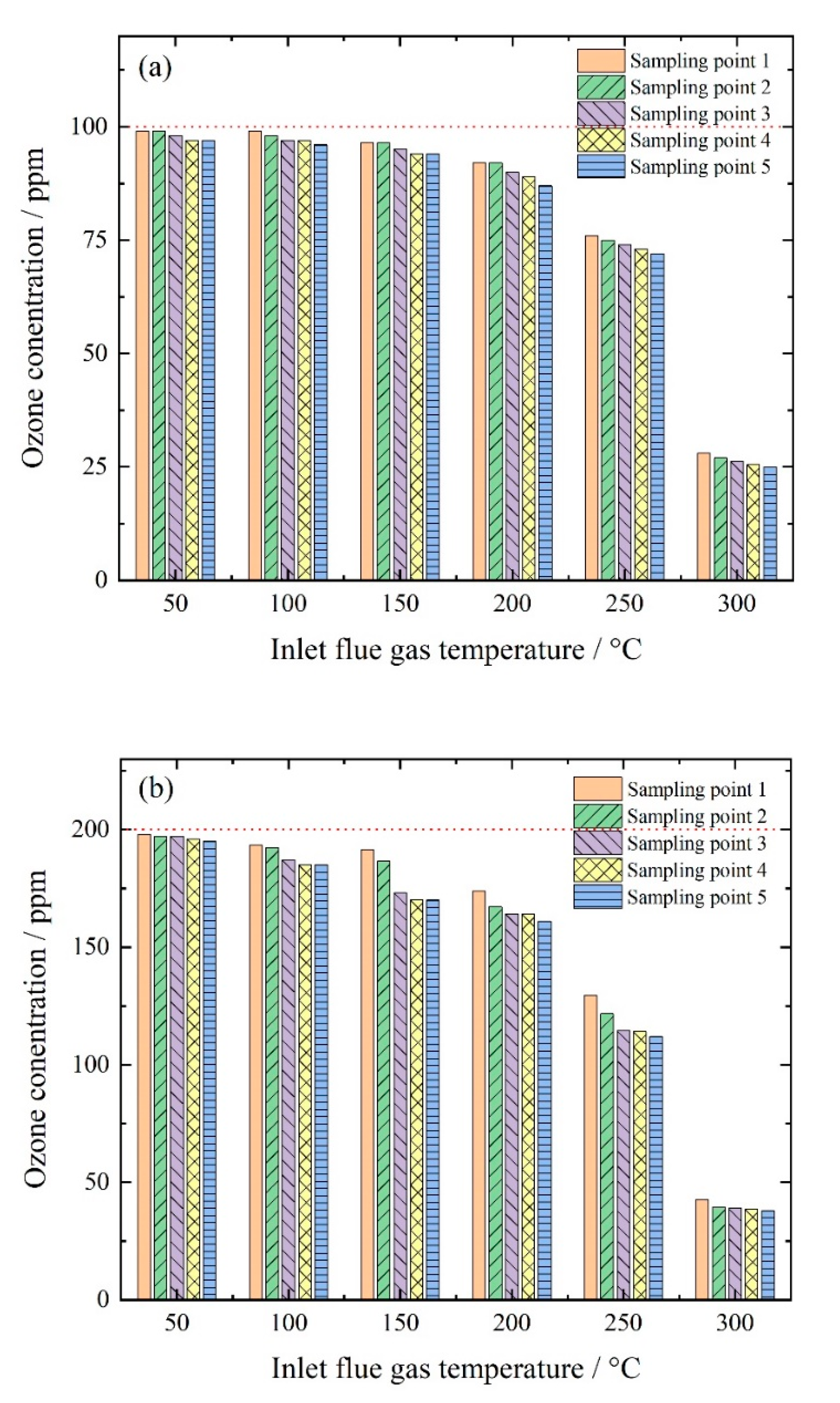
3.1.1. Thermal Decomposition of Ozone
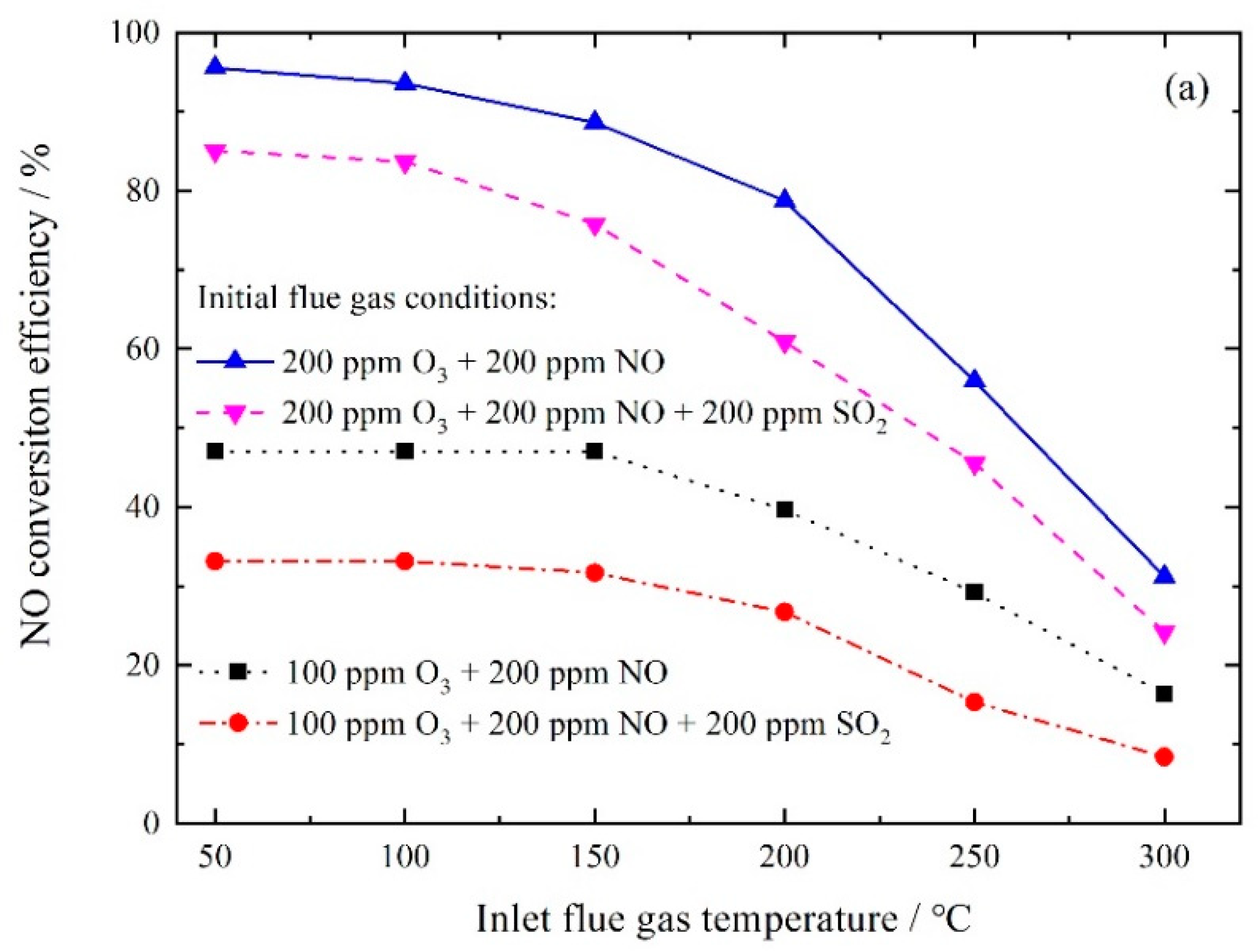
3.1.2. NO Conversion by Ozone Oxidation
3.2. Simultaneous Removal of NO and SO2 by In-Situ Combination of Ozone Injection with Wet Scrubbing
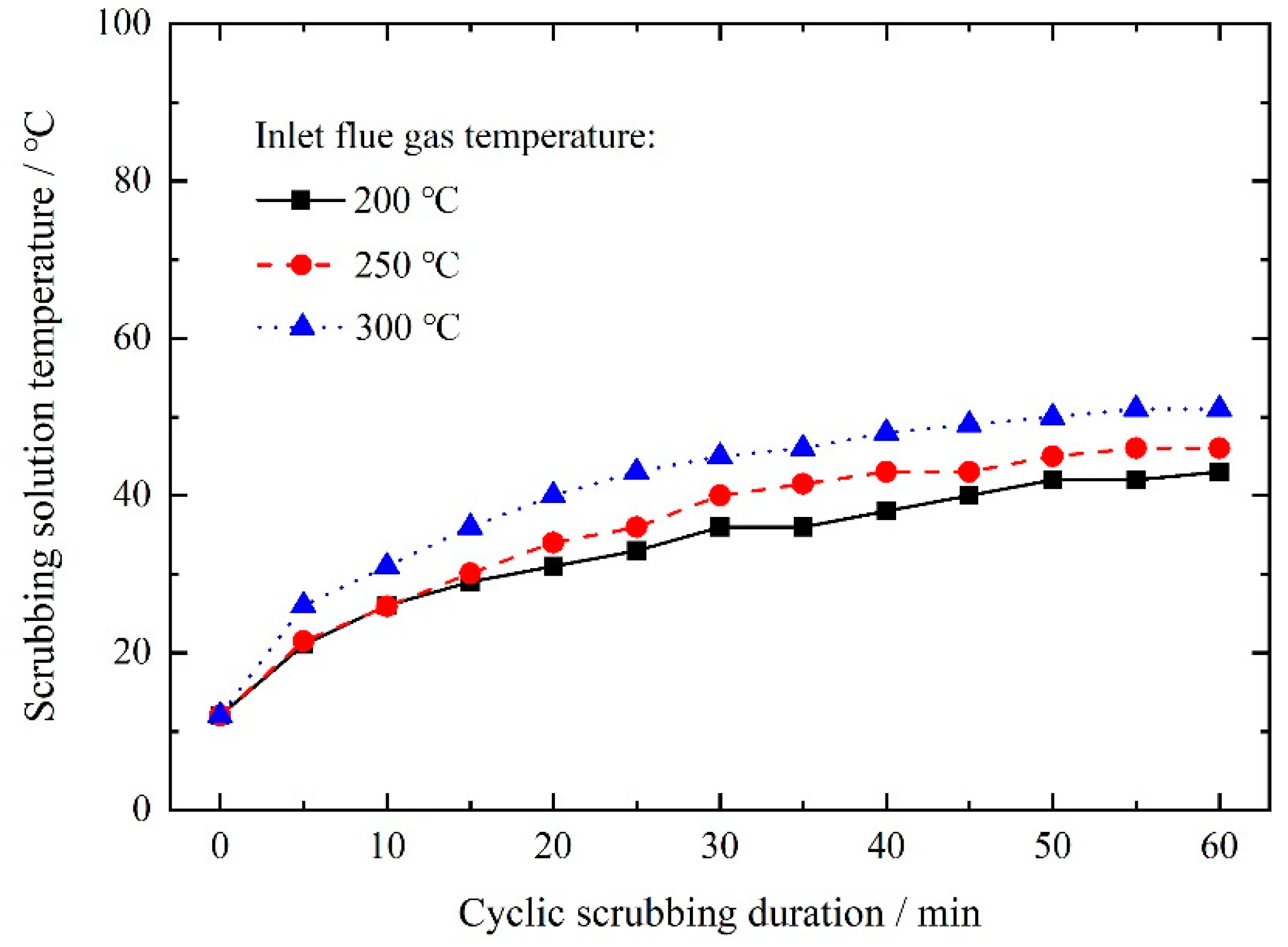
3.2.1. Effect of Wet Scrubbing on Solution Temperature
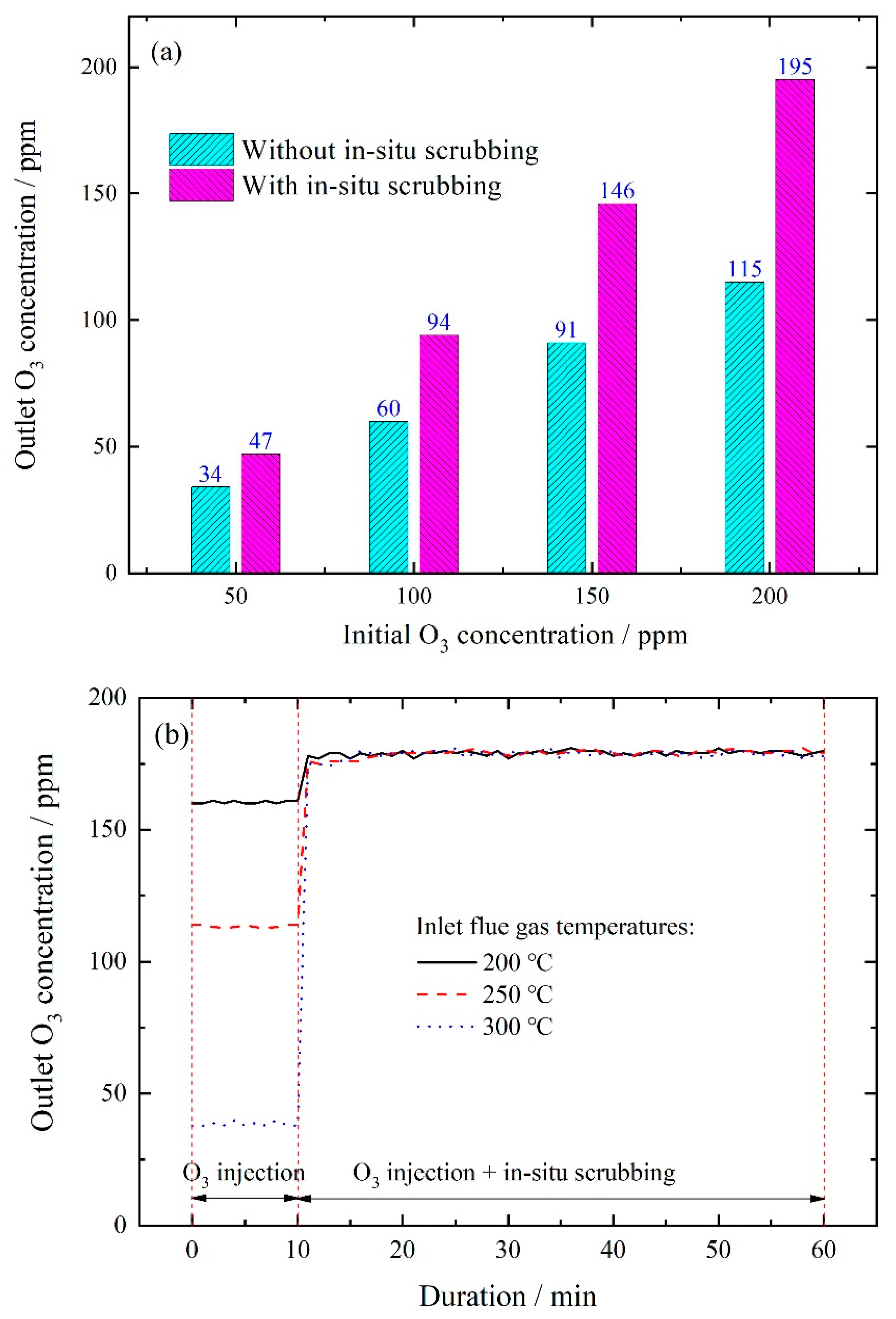
3.2.2. Effect of Wet Scrubbing on Outlet Ozone Concentration
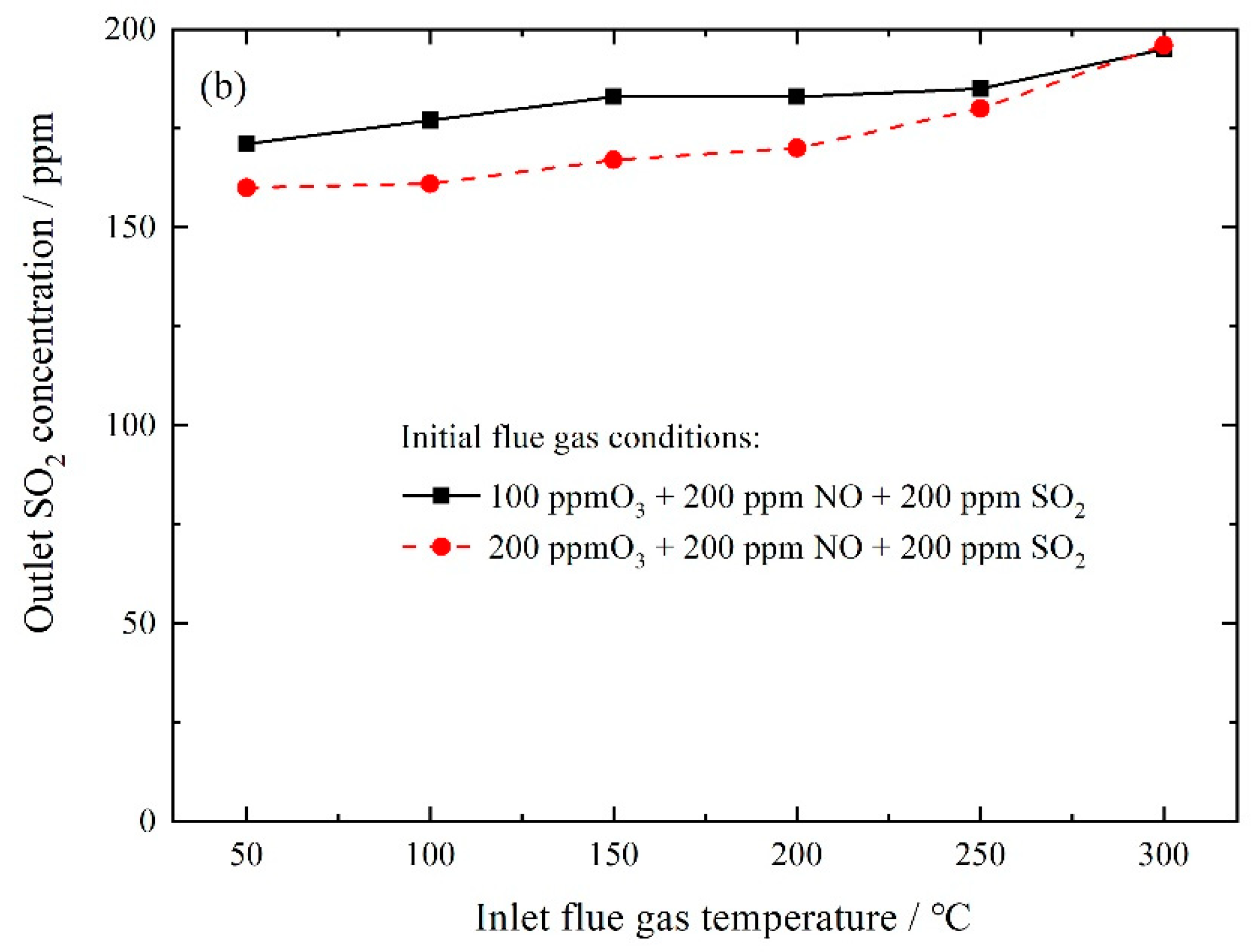
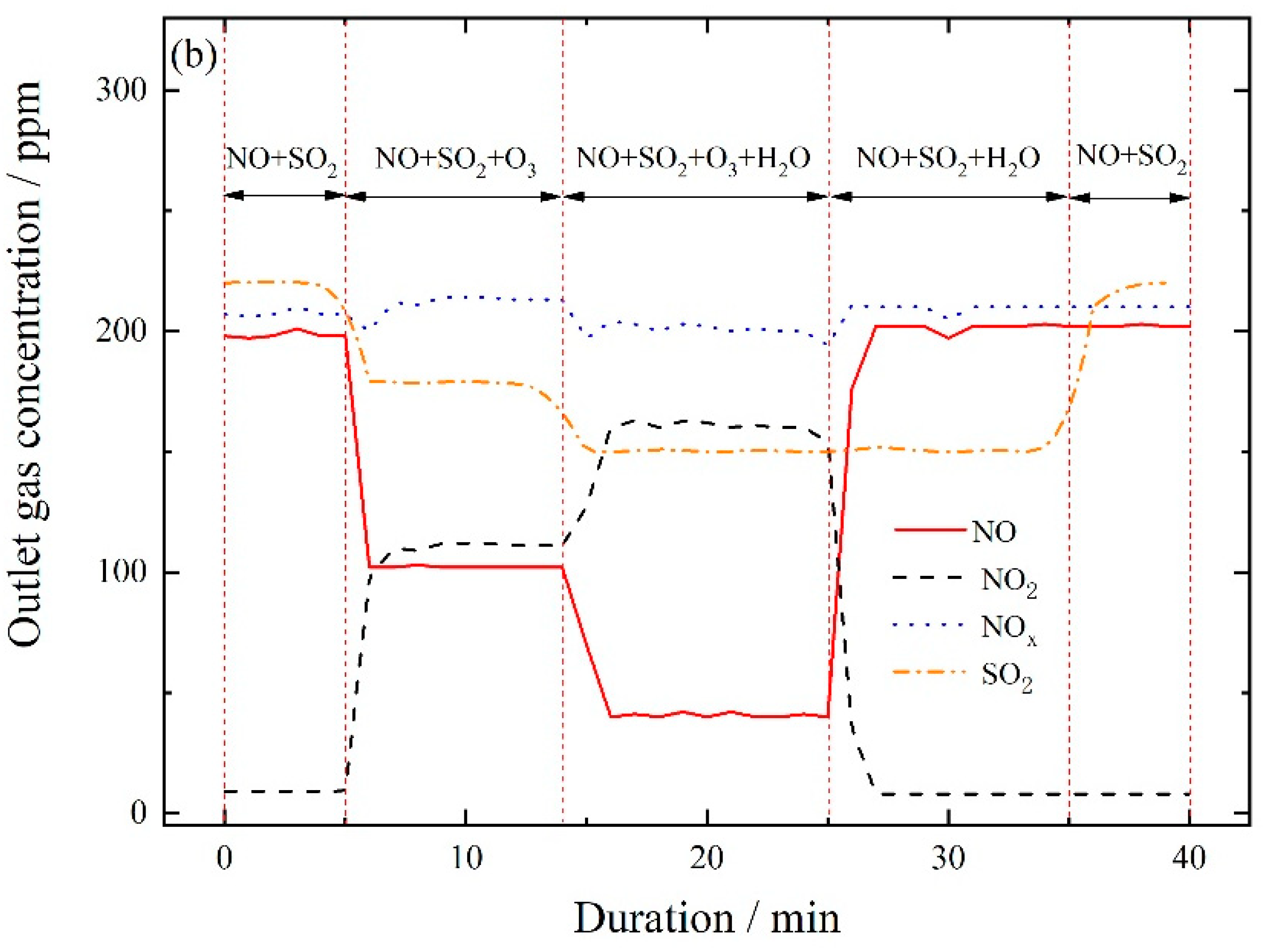
3.2.3. NO Oxidation and SO2 Removal by In-Situ Ozone Oxidation and Wet Scrubbing
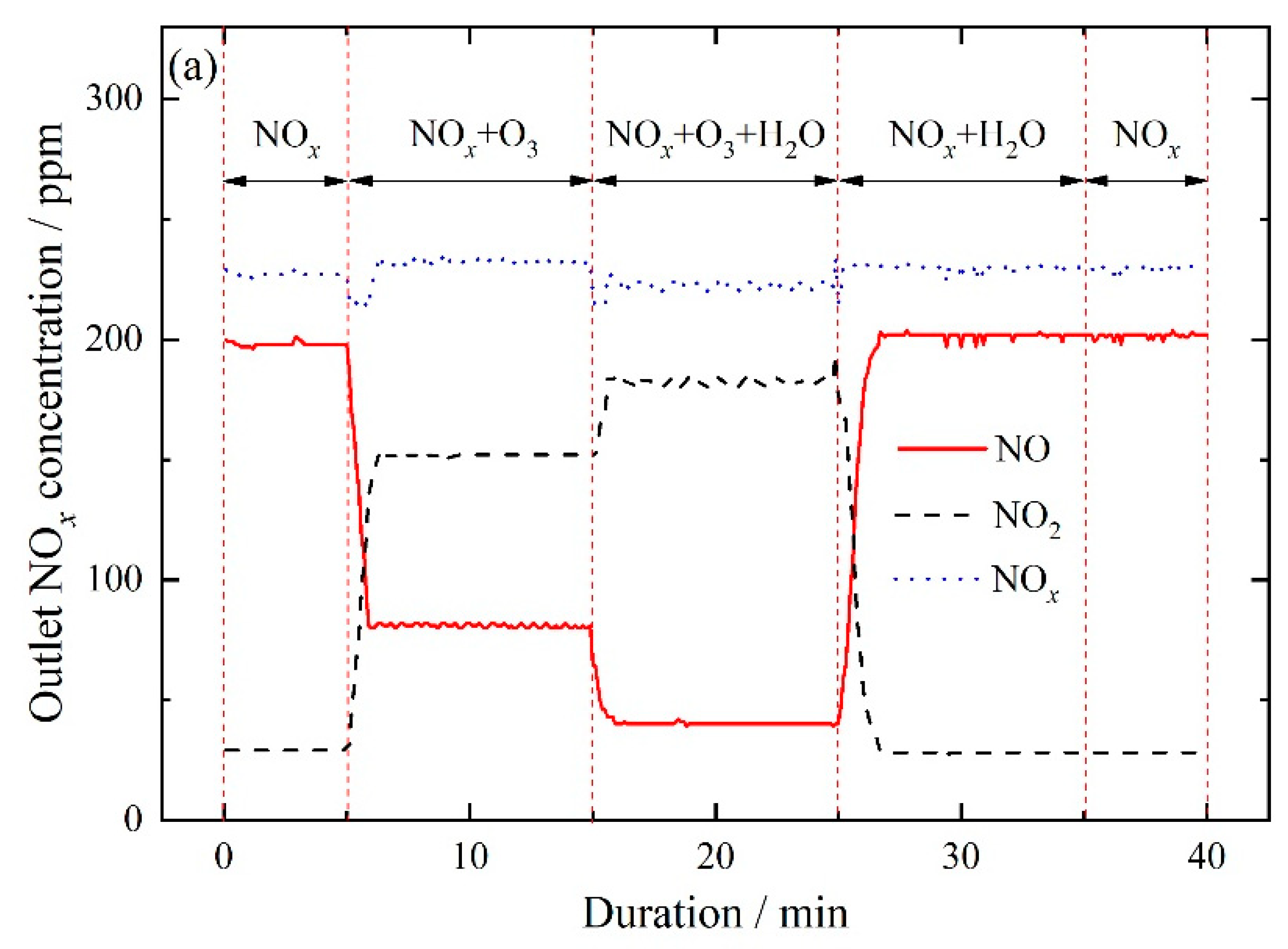
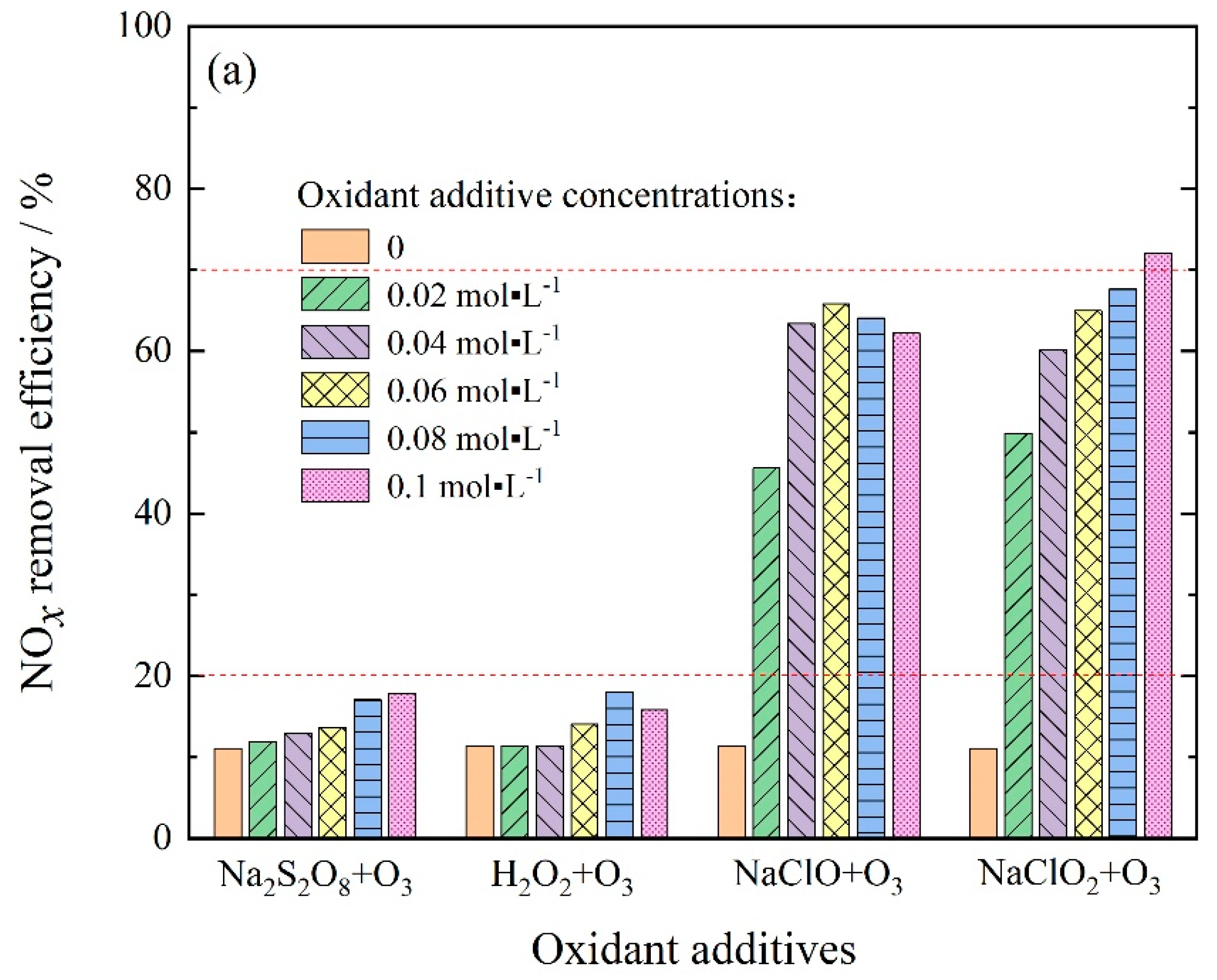
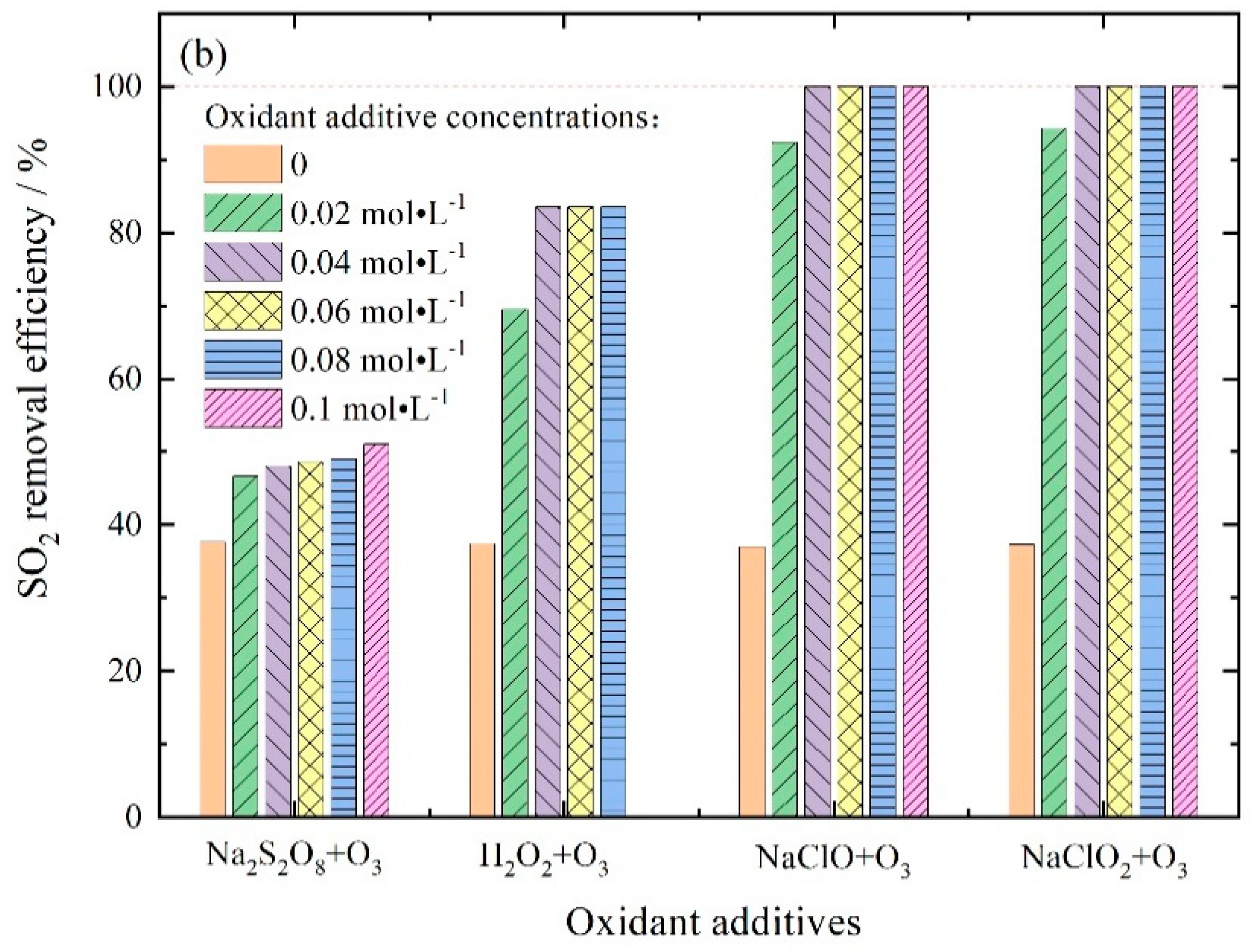
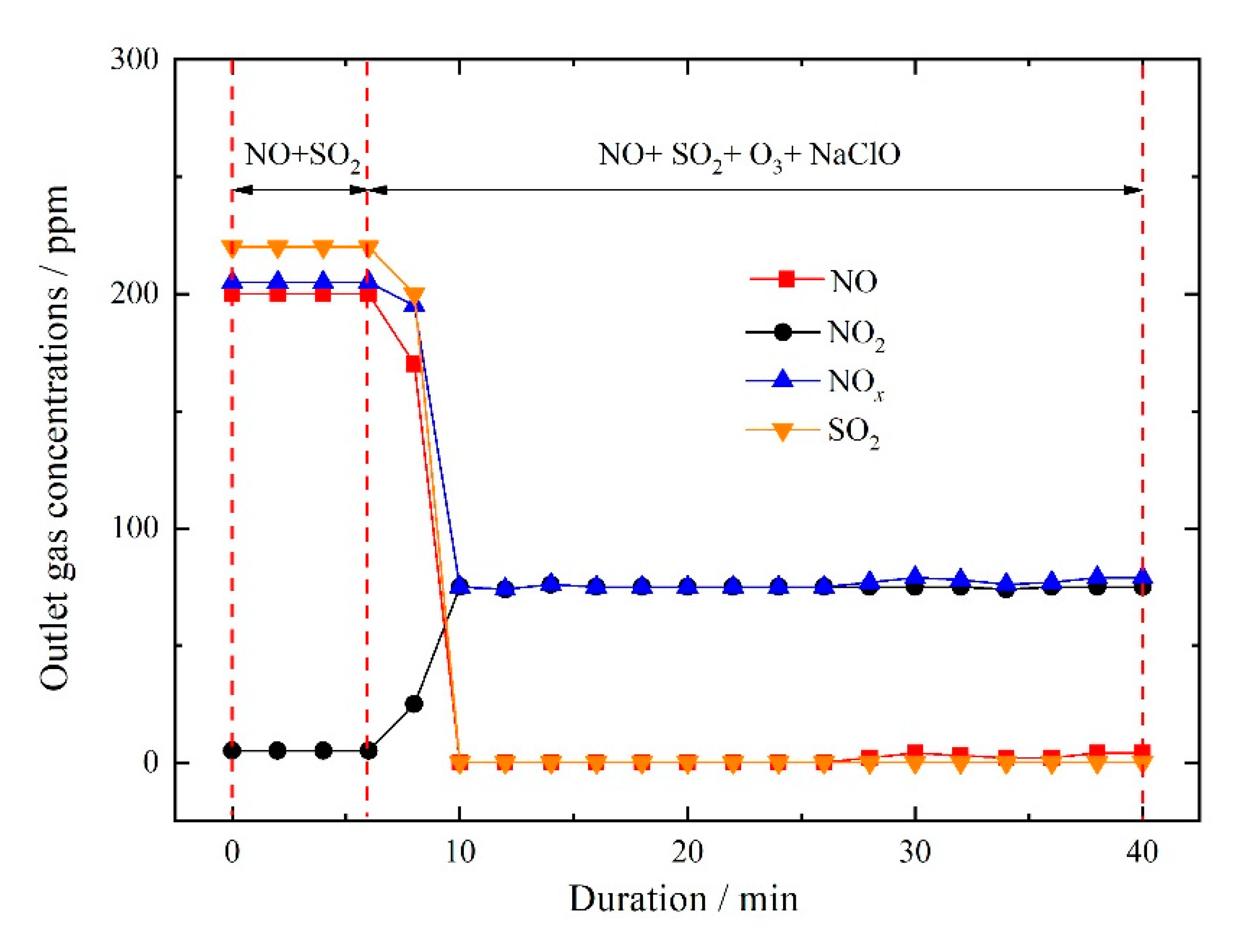
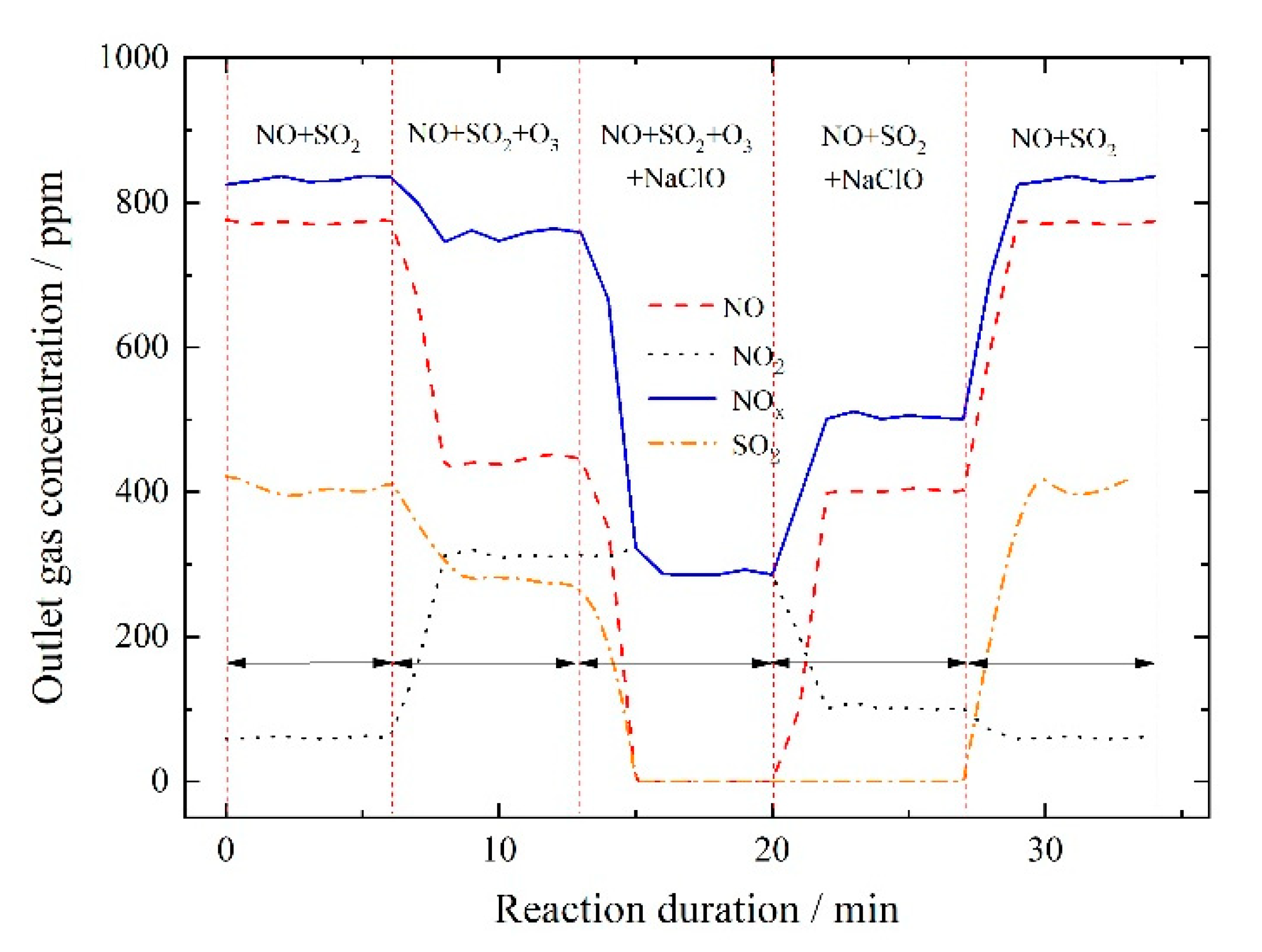
3.2.4. NOx and SO2 Removal by Ozone Injection and In-Situ Scrubbing with Oxidant Additives
4. Discussion
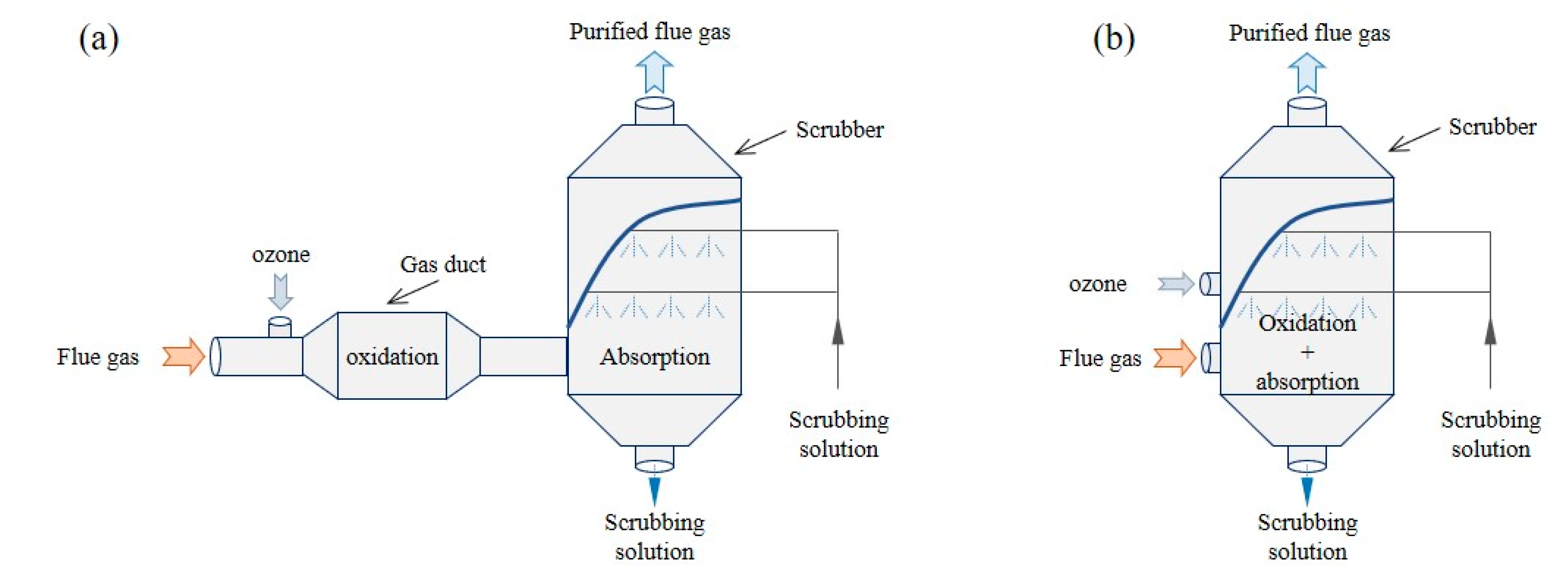
4.1. New Concept of an Integrated System
4.2. Application Prospect of the Integrated System
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Ma, D.; Ma, W.; Jin, S.; Ma, X. Method for Simultaneously Optimizing Ship Route and Speed with Emission Control Areas. Ocean Eng. 2020, 202, 107170. [Google Scholar] [CrossRef]
- Moldanova, J.; Fridell, E.; Popovicheva, O.; Demirdjian, B.; Tishkova, V.; Faccinetto, A.; Focsa, C. Characterisation of Particulate Matter and Gaseous Emissions from a Large Ship Diesel Engine. Atmos. Environ. 2009, 43, 2632–2641. [Google Scholar] [CrossRef]
- Brynolf, S.; Magnusson, M.; Fridell, E.; Andersson, K. Compliance Possibilities for the Future ECA Regulations through the Use of Abatement Technologies or Change of Fuels. Transp. Res. Part D Transp. Environ. 2014, 28, 6–18. [Google Scholar] [CrossRef]
- Lack, D.A.; Corbett, J.J.; Onasch, T.; Lerner, B.; Massoli, P.; Quinn, P.K.; Bates, T.S.; Covert, D.S.; Coffman, D.; Sierau, B.; et al. Particulate Emissions from Commercial Shipping: Chemical, Physical, and Optical Properties. J. Geophys. Res. Atmos. 2009, 114, D00F04. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Tang, X.; Li, T.; Ji, Y.; Liu, Q.; Guo, L.; Zhao, J. Shipboard Trials of Magnesium-Based Exhaust Gas Cleaning System. Ocean Eng. 2016, 128, 124–131. [Google Scholar] [CrossRef]
- Guan, B.; Zhan, R.; Lin, H.; Huang, Z. Review of State of the Art Technologies of Selective Catalytic Reduction of NOx from Diesel Engine Exhaust. Appl. Therm. Eng. 2014, 66, 395–414. [Google Scholar] [CrossRef]
- Wang, Z.; Zhou, S.; Feng, Y.; Zhu, Y. Research of NOx Reduction on a Low-Speed Two-Stroke Marine Diesel Engine by Using EGR (Exhaust Gas Recirculation)–CB (Cylinder Bypass) and EGB (Exhaust Gas Bypass). Int. J. Hydrogen Energy 2017, 42, 19337–19345. [Google Scholar] [CrossRef]
- Ni, P.; Wang, X.; Li, H. A Review on Regulations, Current Status, Effects and Reduction Strategies of Emissions for Marine Diesel Engines. Fuel 2020, 279, 118477. [Google Scholar] [CrossRef]
- Han, Z.; Liu, B.; Yang, S.; Pan, X.; Yan, Z. NOx Removal from Simulated Marine Exhaust Gas by Wet Scrubbing Using NaClO Solution. J. Chem. 2017, 2017, 9340856. [Google Scholar] [CrossRef] [Green Version]
- Han, Z.; Lan, T.; Han, Z.; Yang, S.; Dong, J.; Sun, D.; Yan, Z.; Pan, X.; Song, L. Simultaneous Removal of NO and SO2 from Exhaust Gas by Cyclic Scrubbing and Online Supplementing pH-Buffered NaClO2 Solution. Energy Fuels 2019, 33, 6591–6599. [Google Scholar] [CrossRef]
- Xie, W.X.; Xu, C.W.; Zhang, J.; Liu, Y.X.; Xi, J.F.; Lv, J.H.; Gu, Z.Z. Simultaneous Removal of SO2 and NO from Flue Gas Using H2O2/Urea Activated by Vacuum Ultraviolet Light in a Pilot-Scale Spraying Tower. Energy Fuels 2019, 33, 1325–1333. [Google Scholar] [CrossRef]
- Xi, H.Y.; Zhou, S.; Zhang, Z. A Novel Method Using Na2S2O8 as an Oxidant to Simultaneously Absorb SO2 and NO from Marine Diesel Engine Exhaust Gases. Energy Fuels 2020, 34, 1984–1991. [Google Scholar] [CrossRef]
- Sun, S.; Ma, S.; Yang, B.; Cui, R.; Wang, J. NO Removal from Flue Gas by Using Chlorine Dioxide Solution. Energy Fuels 2019, 33, 10004–10010. [Google Scholar] [CrossRef]
- Zhou, S.; Zhou, J.; Feng, Y.; Zhu, Y. Marine Emission Pollution Abatement Using Ozone Oxidation by a Wet Scrubbing Method. Ind. Eng. Chem. Res. 2016, 55, 5825–5831. [Google Scholar] [CrossRef]
- Wu, Q.; Sun, C.; Wang, H.; Wang, T.; Wang, Y.; Wu, Z. The Role and Mechanism of Triethanolamine in Simultaneous Absorption of NOx and SO2 by Magnesia Slurry Combined with Ozone Gas-Phase Oxidation. Chem. Eng. J. 2018, 341, 157–163. [Google Scholar] [CrossRef]
- Meng, Z.H.; Wang, C.Y.; Wang, X.R.; Chen, Y.; Wu, W.F.; Li, H.Q. Simultaneous Removal of SO2 and NOx from Flue Gas Using (NH4)2S2O3/Steel Slag Slurry Combined with Ozone Oxidation. Fuel 2019, 255, 115760. [Google Scholar] [CrossRef]
- Lin, F.; Wang, Z.; Zhang, Z.; He, Y.; Zhu, Y.; Shao, J.; Yuan, D.; Chen, G.; Cen, K. Flue Gas Treatment with Ozone Oxidation: An Overview on NO, Organic Pollutants, and Mercury. Chem. Eng. J. 2020, 382, 123030. [Google Scholar] [CrossRef]
- Yuan, B.; Mao, X.; Wang, Z.; Hao, R.; Zhao, Y. Radical-Induced Oxidation Removal of Multi-Air-Pollutant: A Critical Review. J. Hazard. Mater. 2020, 383, 121162. [Google Scholar] [CrossRef]
- Ji, R.; Wang, J.; Xu, W.; Liu, X.; Zhu, T.; Yan, C.; Song, J. Study on the Key Factors of NO Oxidation Using O3: The Oxidation Product Composition and Oxidation Selectivity. Ind. Eng. Chem. Res. 2018, 57, 14440–14447. [Google Scholar] [CrossRef]
- Sun, C.; Zhao, N.; Zhuang, Z.; Wang, H.; Liu, Y.; Weng, X.L.; Wu, Z. Mechanisms and Reaction Pathways for Simultaneous Oxidation of NOx and SO2 by Ozone Determined by in Situ IR Measurements. J. Hazard. Mater. 2014, 274, 376–383. [Google Scholar] [CrossRef]
- Shao, J.; Yang, Y.; Whiddon, R.; Wang, Z.; Lin, F.; He, Y.; Kumar, S.; Cen, K. Investigation of NO Removal with Ozone Deep Oxidation in Na2CO3 Solution. Energy Fuels 2019, 33, 4454–4461. [Google Scholar] [CrossRef]
- Fujishima, H.; Takekoshi, K.; Kuroki, T.; Tanaka, A.; Otsuka, K.; Okubo, M. Towards Ideal NOx Control Technology for Bio-Oils and a Gas Multi-Fuel Boiler System Using a Plasma-Chemical Hybrid Process. Appl. Energy 2013, 111, 394–400. [Google Scholar] [CrossRef]
- Sun, B.-C.; Dong, K.; Zhao, W.; Wang, J.; Chu, G.-W.; Zhang, L.; Zou, H.; Chen, J.-F. Simultaneous Absorption of NOx and SO2 into Na2SO3 Solution in a Rotating Packed Bed with Preoxidation by Ozone. Ind. Eng. Chem. Res. 2019, 58, 8332–8341. [Google Scholar] [CrossRef]
- Han, Z.; Wang, J.; Zou, T.; Zhao, D.; Gao, C.; Dong, J.; Pan, X. NOx Removal from Flue Gas Using an Ozone Advanced Oxidation Process with Injection of Low Concentration of Ethanol: Performance and Mechanism. Energy Fuels 2020, 34, 2080–2088. [Google Scholar] [CrossRef]
- Huang, X.; Ding, J.; Zhong, Q. Catalytic Decomposition of H2O2 over Fe-Based Catalysts for Simultaneous Removal of NOx and SO2. Appl. Surf. Sci. 2015, 326, 66–72. [Google Scholar] [CrossRef]
- Ding, J.; Cai, H.; Zhong, Q.; Lin, J.; Xiao, J.; Zhang, S.; Fan, M. Selective Denitrification of Flue Gas by O3 and Ethanol Mixtures in a Duct: Investigation of Processes and Mechanisms. J. Hazard. Mater. 2016, 311, 218–229. [Google Scholar] [CrossRef]
- Liu, B.; Xu, X.C.; Xue, Y.; Liu, L.F.; Yang, F.L. Simultaneous Desulfurization and Denitrification from Flue Gas by Catalytic Ozonation Combined with NH3/(NH4)2S2O8 Absorption: Mechanisms and Recovery of Compound Fertilizer. Sci. Total Environ. 2020, 706, 136072. [Google Scholar] [CrossRef]
- Shao, J.; Ma, Q.; Wang, Z.; Tang, H.; He, Y.; Zhu, Y.; Cen, K. A Superior Liquid Phase Catalyst for Enhanced Absorption of NO2 Together with SO2 after Low Temperature Ozone Oxidation for Flue Gas Treatment. Fuel 2019, 247, 1–9. [Google Scholar] [CrossRef]
- Eyring, V.; Isaksen, I.S.A.; Berntsen, T.; Collins, W.J.; Corbett, J.J.; Endresen, O.; Grainger, R.G.; Moldanova, J.; Schlager, H.; Stevenson, D.S. Transport Impacts on Atmosphere and Climate: Shipping. Atmos. Environ. 2010, 44, 4735–4771. [Google Scholar] [CrossRef]
- Itoh, H.; Taguchi, M.; Suzuki, S. Thermal Decomposition of Ozone at High Temperature Leading to Ozone Zero Phenomena. J. Phys. D Appl. Phys. 2020, 53, 185206. [Google Scholar] [CrossRef]
- Yamamoto, H.; Kuroki, T.; Fujishima, H.; Yamamoto, Y.; Yoshida, K.; Okubo, M. Pilot-Scale Exhaust Gas Treatment for a Glass Manufacturing System Using a Plasma Combined Semi-Dry Chemical Process. IEEE Trans. Ind. Appl. 2016, 53, 1416–1423. [Google Scholar] [CrossRef]
- Han, Z.; Yang, S.; Pan, X.; Zhao, N.; Yu, J.; Zhou, Y.; Xia, P.; Zheng, D.; Song, Y.; Yan, Z. New Experimental Results of NO Removal from Simulated Flue Gas by Wet Scrubbing Using NaClO Solution. Energy Fuels 2017, 31, 3047–3054. [Google Scholar] [CrossRef]
- Liu, Y.X.; Wang, Q.; Yin, Y.S.; Pan, J.F.; Zhang, J. Advanced Oxidation Removal of NO and SO2 from Flue Gas by Using Ultraviolet/H2O2/NaOH Process. Chem. Eng. Res. Des. 2014, 92, 1907–1914. [Google Scholar] [CrossRef]
- Wu, B.; Xiong, Y.; Ge, Y. Simultaneous Removal of SO2 and NO from Flue Gas with OH from the Catalytic Decomposition of Gas-Phase H2O2 over Solid-Phase Fe2(SO4)3. Chem. Eng. J. 2018, 331, 343–354. [Google Scholar] [CrossRef]
- Han, Z.; Yang, S.; Zheng, D.; Pan, X.; Yan, Z. An Investigation on NO Removal by Wet Scrubbing Using NaClO2 Seawater Solution. SpringerPlus 2016, 5, 751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Han, Z.; Pan, X.; Yan, Z.; Yu, J. Nitrogen Oxide Removal Using Seawater Electrolysis in an Undivided Cell for Ocean-Going Vessels. RSC Adv. 2016, 6, 114623–114631. [Google Scholar] [CrossRef]
- Mondal, M.K.; Chelluboyana, V.R. New Experimental Results of Combined SO2 and NO Removal from Simulated Gas Stream by NaClO as Low-Cost Absorbent. Chem. Eng. J. 2013, 217, 48–53. [Google Scholar] [CrossRef]
- Yang, S.; Han, Z.; Dong, J.-M.; Zheng, Z.-S.; Pan, X. UV-Enhanced NaClO Oxidation of Nitric Oxide from Simulated Flue Gas. J. Chem. 2016, 2016, 6065019. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gas Temperatures (°C) | Gas Flow Rates (m3·h−1) | Gas Velocities (m·s−1) | Residence Time (s) |
|---|---|---|---|
| 25 | 50 | 0.111 | 9.01 |
| 50 | 55.12 | 0.122 | 8.20 |
| 100 | 63.66 | 0.141 | 7.09 |
| 150 | 72.18 | 0.160 | 6.25 |
| 200 | 80.72 | 0.178 | 5.62 |
| 250 | 89.25 | 0.197 | 5.07 |
| 300 | 97.78 | 0.216 | 4.63 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Z.; Zou, T.; Wang, J.; Dong, J.; Deng, Y.; Pan, X. A Novel Method for Simultaneous Removal of NO and SO2 from Marine Exhaust Gas via In-Site Combination of Ozone Oxidation and Wet Scrubbing Absorption. J. Mar. Sci. Eng. 2020, 8, 943. https://doi.org/10.3390/jmse8110943
Han Z, Zou T, Wang J, Dong J, Deng Y, Pan X. A Novel Method for Simultaneous Removal of NO and SO2 from Marine Exhaust Gas via In-Site Combination of Ozone Oxidation and Wet Scrubbing Absorption. Journal of Marine Science and Engineering. 2020; 8(11):943. https://doi.org/10.3390/jmse8110943
Chicago/Turabian StyleHan, Zhitao, Tianyu Zou, Junming Wang, Jingming Dong, Yangbo Deng, and Xinxiang Pan. 2020. "A Novel Method for Simultaneous Removal of NO and SO2 from Marine Exhaust Gas via In-Site Combination of Ozone Oxidation and Wet Scrubbing Absorption" Journal of Marine Science and Engineering 8, no. 11: 943. https://doi.org/10.3390/jmse8110943
APA StyleHan, Z., Zou, T., Wang, J., Dong, J., Deng, Y., & Pan, X. (2020). A Novel Method for Simultaneous Removal of NO and SO2 from Marine Exhaust Gas via In-Site Combination of Ozone Oxidation and Wet Scrubbing Absorption. Journal of Marine Science and Engineering, 8(11), 943. https://doi.org/10.3390/jmse8110943