

Deep-Learning-Based Human Chromosome Classification: Data Augmentation and Ensemble



Abstract
:1. Introduction
2. Materials and Methods
2.1. A Brief Introduction to CNNs
ResNet-50
2.2. A Brief Introduction to Transformers
Swin
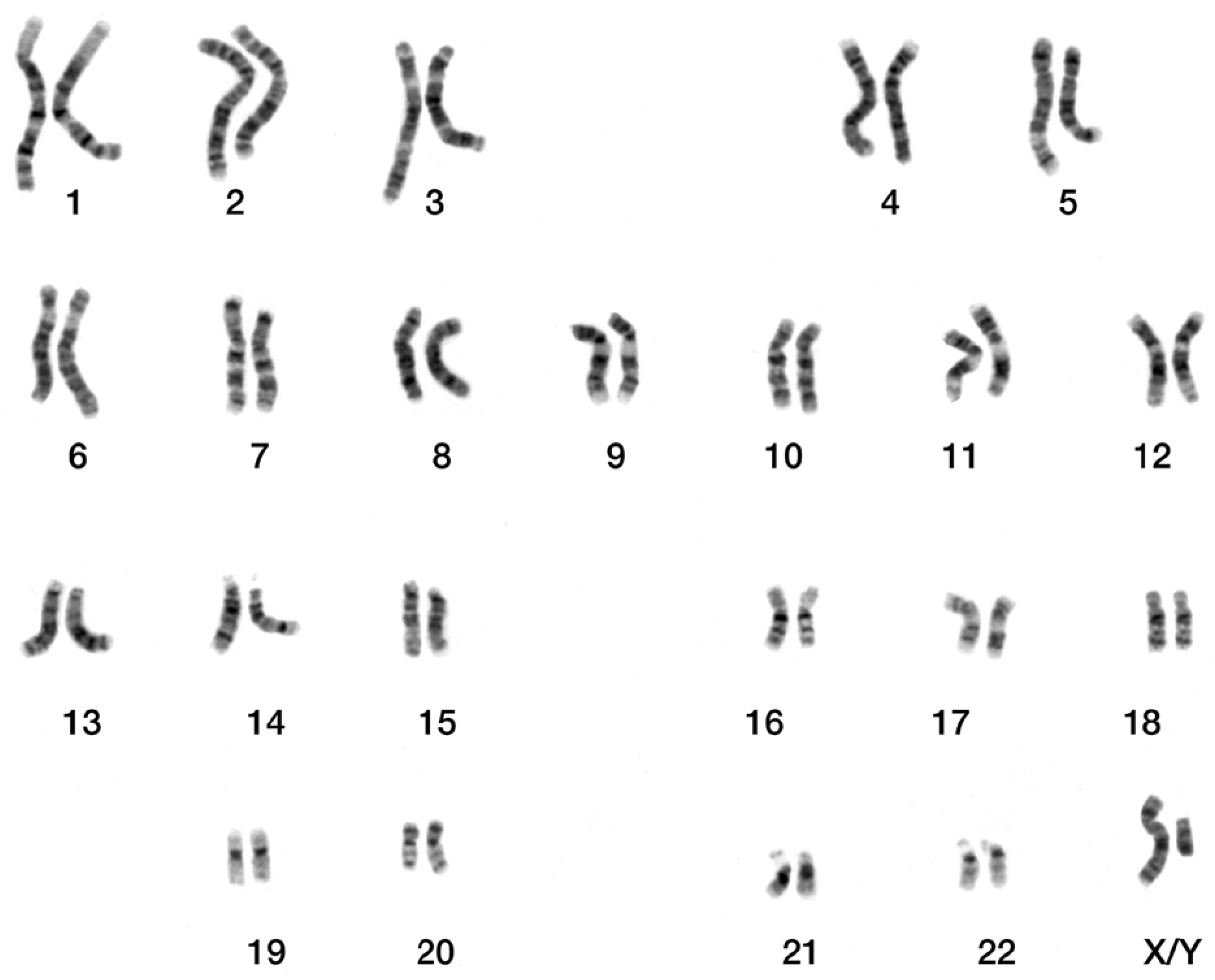
2.3. The Dataset
2.4. Data Augmentation
| Algorithm 1 Chromosome Data Augmentation |
|
2.5. Straightening
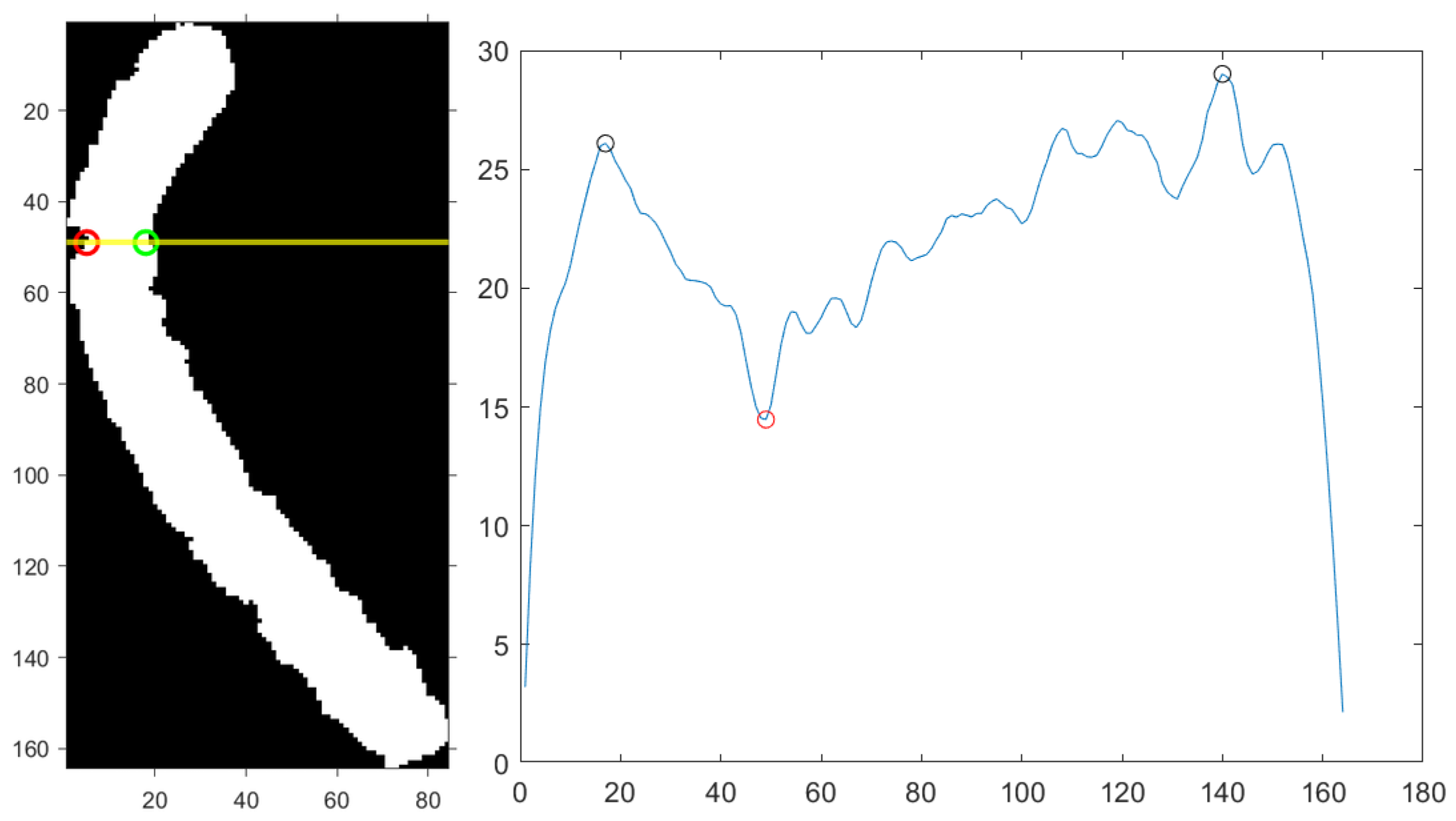
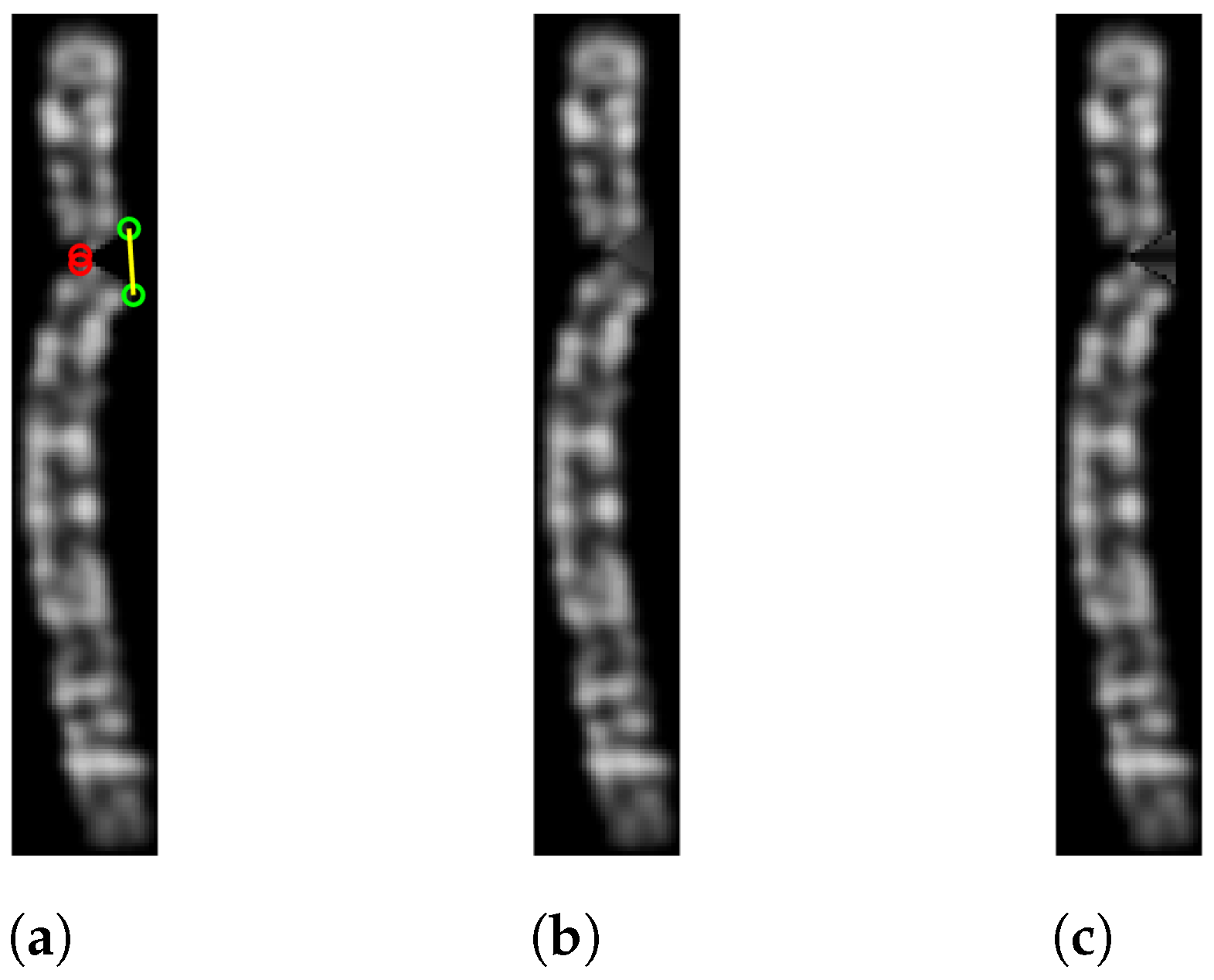
2.5.1. Chromosome Image Binarization and Bending Centre Locating
| Algorithm 2 Rotation Score Calculation |
|
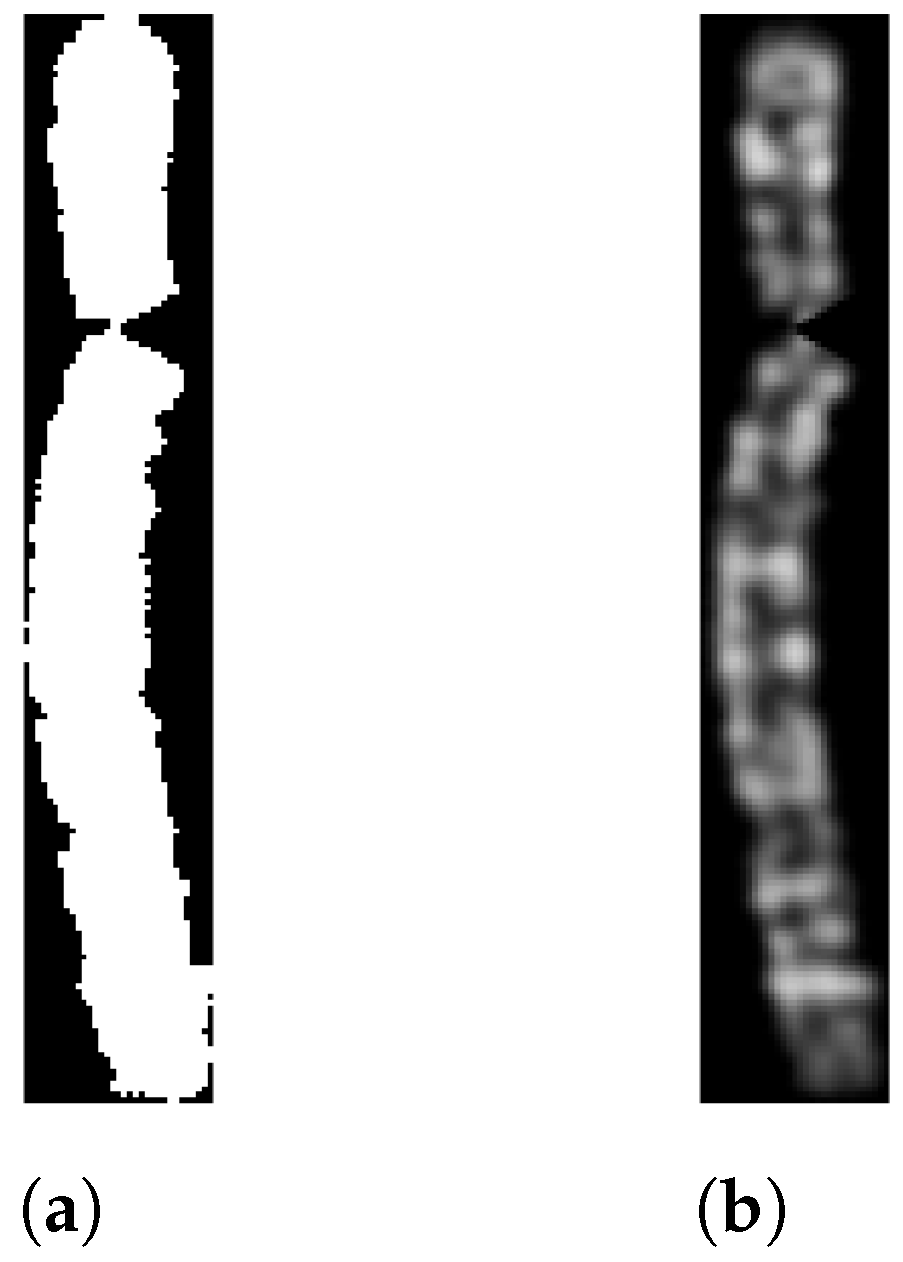
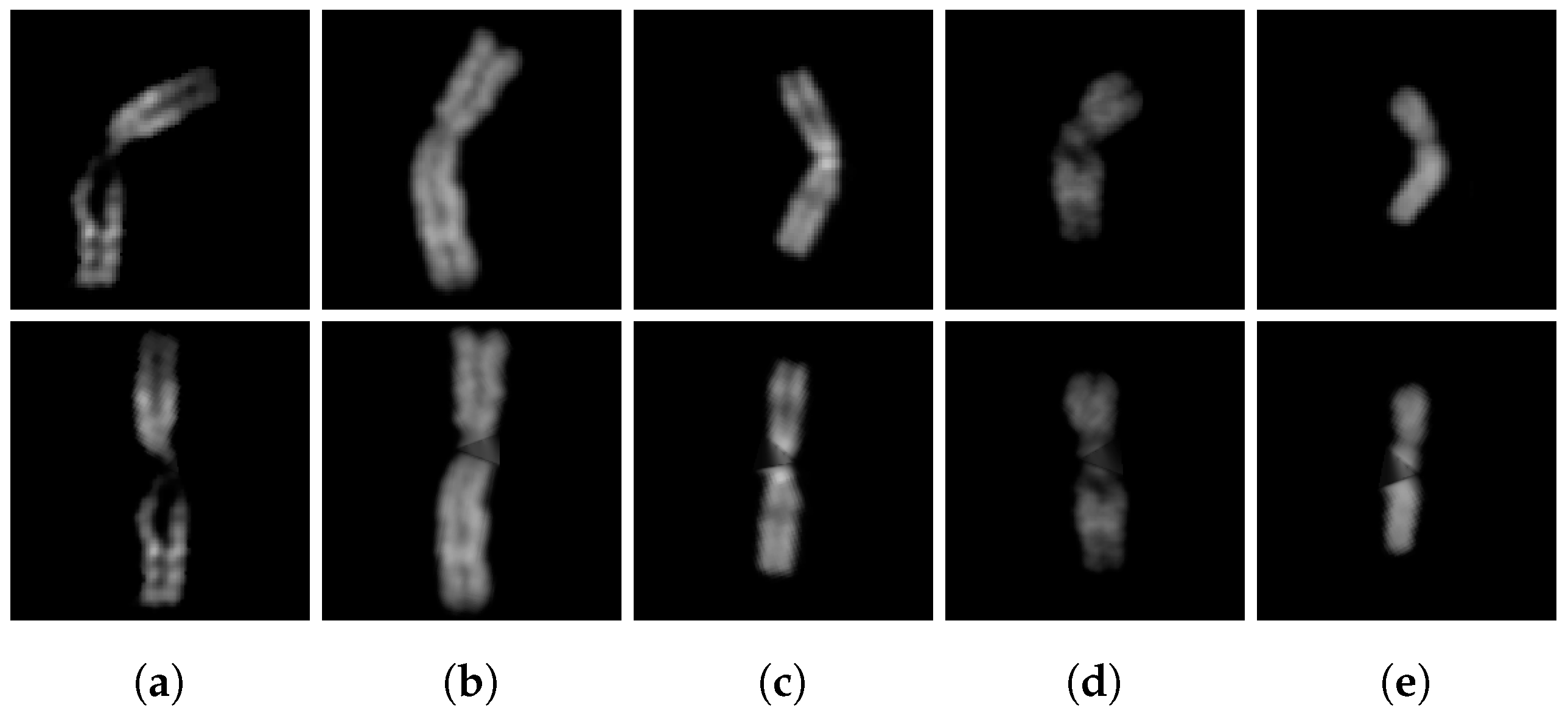
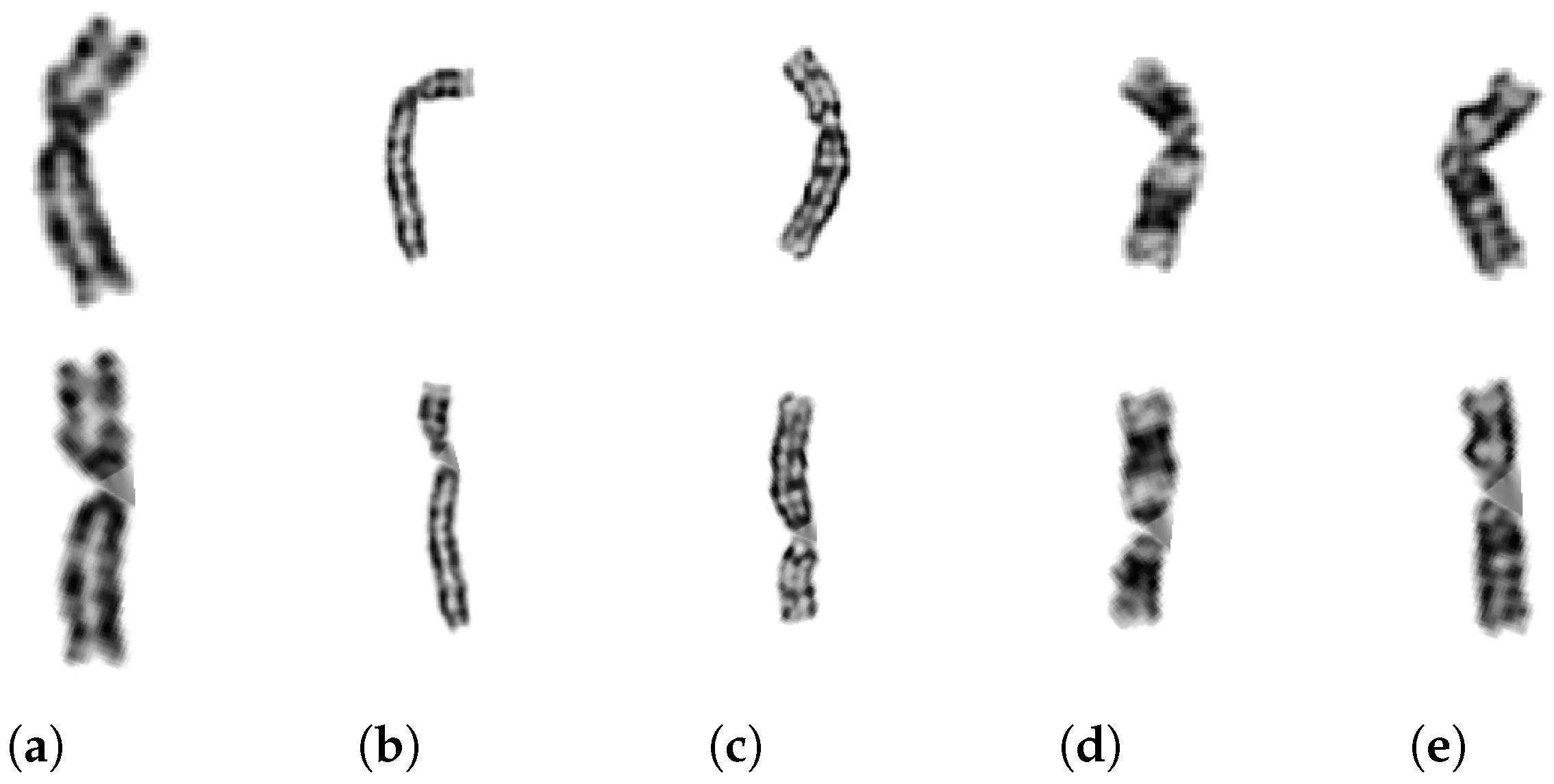
2.5.2. Chromosome Arms Straightening and Final Image Creation
| Algorithm 3 Chromosome Arm Straightening |
|
2.5.3. Algorithm Improvements
2.5.4. Additional Tests
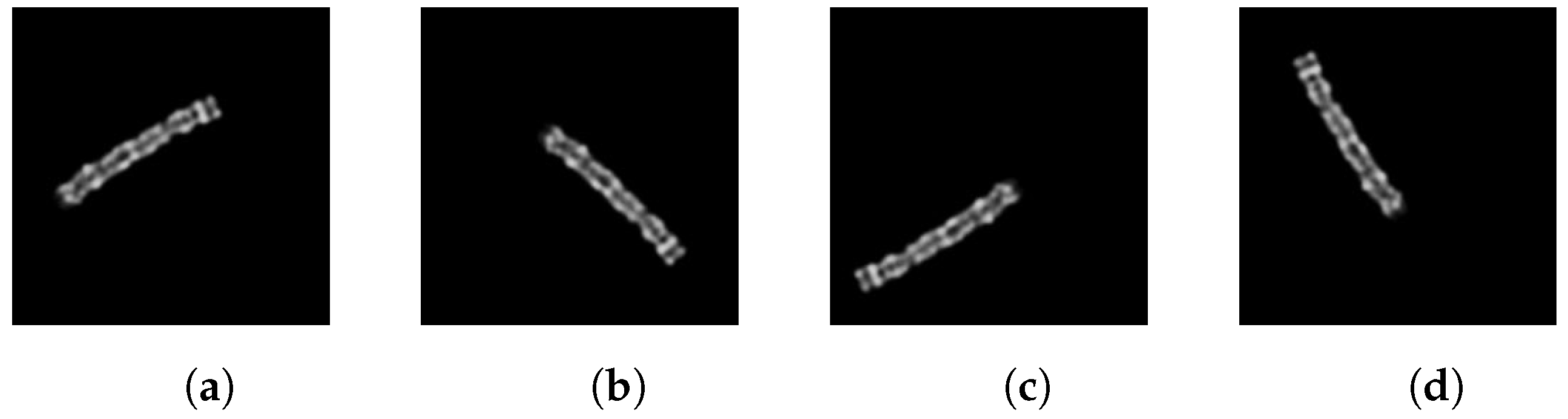
2.6. Feature-Transform-Based Techniques
- First technique
- Apply FFT to the grayscale chromosome image.
- Shift the zero frequency component to the centre of the frequency domain.
- Create a mask made of 1 s with the same dimensions as the transformed and frequency-shifted image; this will preserve only selected frequencies in step 7.
- Create two 2-D grids to represent the x and y coordinates.
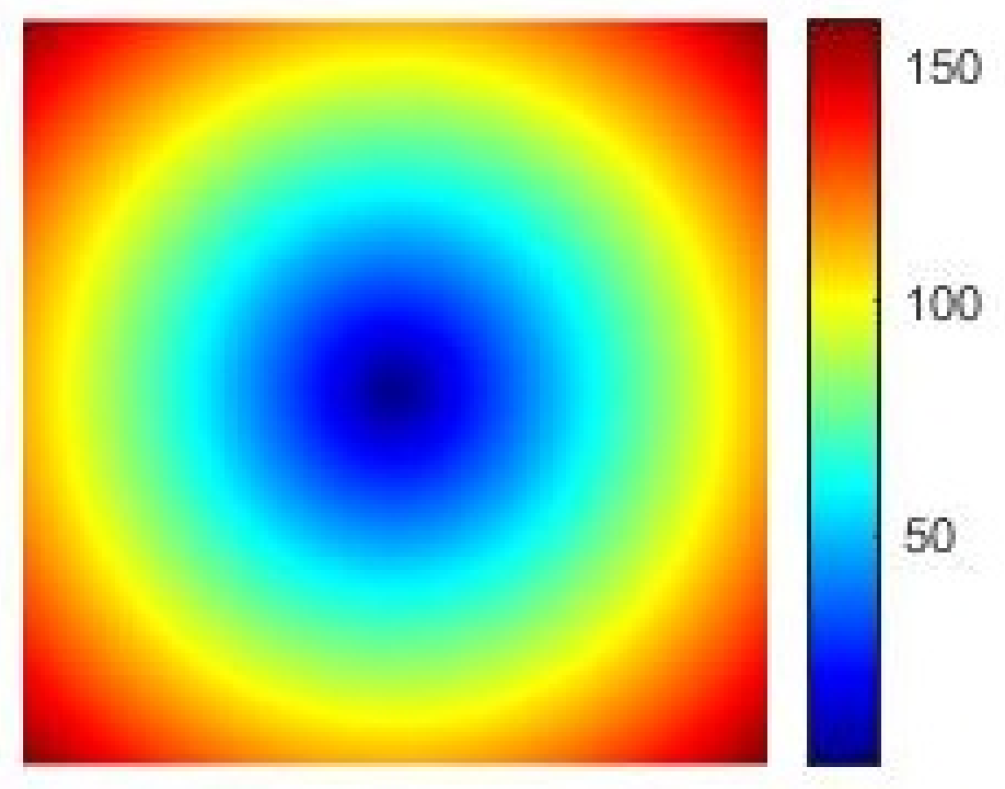
- Use the grids to compute the Euclidean distance and obtain a colored distance matrix R (see Figure 12).
- Select a radius (threshold) and set to zero the values of the mask inside this radius using the values of distance matrix R as coordinates.
- Apply the mask to the transformed image.
- Apply the inverse Fourier transform (iFFT) to obtain the blurred grayscale image, visible in Figure 13b.
- Second techniqueThis technique is similar to the previous one, except that the zero frequency is not shifted:
- Apply FFT to the grayscale image.
- Sort and store in an array the elements (IDs) of the transformed image by their intensity values.
- Select a value that represents the percentage of points in the transformed image that will be set to zero.
- Randomize the IDs array and set a portion of the elements corresponding to the selected percentage p to zero in the transformed image.
- Apply the inverse Fourier transform (iFFT) to recover the filtered grayscale image. The result is shown in Figure 13c.
- Third techniqueThis method uses the discrete cosine transform (DCT) for image processing instead of the Fourier transform:
- Convert the input RGB image to grayscale.
- Apply the DCT to the grayscale image to obtain its frequency components.
- Set to zero a low frequency range, which is a 10 × 10 square in this case.
- Apply the inverse DCT (iDCT) to obtain the processed grayscale image (Figure 13d).
3. Results
3.1. Metrics
3.2. Experiment Settings
3.3. Experimental Results
- None(1) achieves significantly lower performance than the network trained using an expanded training set. None(10) outperforms None(1), but its performance is not comparable to that achieved by CDA(10). CDA(1) outperforms both CDAst8(1) and CDAst3(1); it is clear that, in this application, data augmentation is a very important step.
- CDA(10) clearly outperforms CDA(1).
- The best performance, considering a single data augmentation approach, is obtained by STR(10).
- The CNN ensemble trained with different augmentation methods can outperform each of its components; e.g., CDA(3) + STR(3) + FT(3) outperforms STR(10).
- The best result among CNN methods is obtained by CDA(3) + STR(3) + FT(3), which achieved an accuracy of 98.56%.
- Swin performs significantly better than ResNet-50, confirming the same conclusions: the ensemble performs significantly better than the stand-alone network; furthermore, we calculated the area under the ROC curve of the Swin ensemble, obtaining an excellent 0.9999.
4. Conclusions
- A data augmentation algorithm called CDA was used to generate additional samples from the original dataset. This algorithm introduces different spatial orientations to the chromosomes, effectively diversifying the training data.
- We implemented a straightening procedure that utilizes projection vectors to straighten the chromosomes. This step removes curves from the subjects, allowing the neural network to learn other important features.
- In the end, we employed three feature-transform-based techniques to create more images and alter their appearance through manipulations, such as blur and contrast adjustments. These techniques contributed to further enhancing the diversity and variability of the training data.
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Zheng, B.; Li, S.; Mulvihill, J.J.; Liu, H. A rule-based computer scheme for centromere identification and polarity assignment of metaphase chromosomes. Comput. Methods Programs Biomed. 2008, 89, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Tjio, J.H.; Levan, A. The Chromosome Number in Man. Hereditas 2010, 42, 1–6. [Google Scholar] [CrossRef]
- Remani Sathyan, R.; Chandrasekhara Menon, G.; Hariharan, S.; Thampi, R.; Duraisamy, J.H. Traditional and deep-based techniques for end-to-end automated karyotyping: A review. Expert Syst. 2022, 39, e12799. [Google Scholar] [CrossRef]
- Agam, G.; Dinstein, I. Geometric separation of partially overlapping nonrigid objects applied to automatic chromosome classification. IEEE Trans. Pattern Anal. Mach. Intell. 1997, 19, 1212–1222. [Google Scholar] [CrossRef]
- Errington, P.A.; Graham, J. Application of artificial neural networks to chromosome classification. Cytometry 1993, 14, 627–639. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Song, S.; Bai, T.; Zhao, Y.; Ma, F.; Su, J.; Yu, L. Chromosome Classification with Convolutional Neural Network Based Deep Learning. In Proceedings of the 2018 11th International Congress on Image and Signal Processing, BioMedical Engineering and Informatics (CISP-BMEI), Beijing, China, 13–15 October 2018; pp. 1–5. [Google Scholar] [CrossRef]
- Swati.; Gupta, G.; Yadav, M.; Sharma, M.; Vig, L. Siamese Networks for Chromosome Classification. In Proceedings of the 2017 IEEE International Conference on Computer Vision Workshops (ICCVW), Venice, Italy, 22–29 October 2017; pp. 72–81. [Google Scholar] [CrossRef]
- Huang, K.; Lin, C.; Huang, R.; Zhao, G.; Yin, A.; Chen, H.; Guo, L.; Shan, C.; Nie, R.; Li, S. A novel chromosome instance segmentation method based on geometry and deep learning. In Proceedings of the 2021 International Joint Conference on Neural Networks (IJCNN), Shenzhen, China, 18–22 July 2021; pp. 1–8. [Google Scholar] [CrossRef]
- Abid, F.; Hamami, L. A survey of neural network based automated systems for human chromosome classification. Artif. Intell. Rev. 2018, 49, 41–56. [Google Scholar] [CrossRef]
- Anh, L.Q.; Thanh, V.D.; Son, N.H.H.; Phuong, D.T.K.; Anh, L.T.L.; Ram, D.T.; Minh, N.T.B.; Tung, T.H.; Thinh, N.H.; Ha, L.V.; et al. Efficient Type and Polarity Classification of Chromosome Images using CNNs: A Primary Evaluation on Multiple Datasets. In Proceedings of the 2022 IEEE Ninth International Conference on Communications and Electronics (ICCE), Nha Trang, Vietnam, 27–29 July 2022; pp. 400–405. [Google Scholar] [CrossRef]
- Lin, C.; Zhao, G.; Yang, Z.; Yin, A.; Wang, X.; Guo, L.; Chen, H.; Ma, Z.; Zhao, L.; Luo, H.; et al. CIR-Net: Automatic Classification of Human Chromosome Based on Inception-ResNet Architecture. IEEE/ACM Trans. Comput. Biol. Bioinform. 2022, 19, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Javan Roshtkhari, M.; Setarehdan, K. A novel algorithm for straightening highly curved images of human chromosome. Pattern Recognit. Lett. 2008, 29, 1208–1217. [Google Scholar] [CrossRef]
- Sharma, M.; Saha, O.; Sriraman, A.; Hebbalaguppe, R.; Vig, L.; Karande, S. Crowdsourcing for Chromosome Segmentation and Deep Classification. In Proceedings of the 2017 IEEE Conference on Computer Vision and Pattern Recognition Workshops (CVPRW), Honolulu, HI, USA, 21–26 July 2017; pp. 786–793. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Deep Residual Learning for Image Recognition. In Proceedings of the 2016 IEEE Conference on Computer Vision and Pattern Recognition (CVPR), Las Vegas, NV, USA, 27–30 June 2016; pp. 770–778. [Google Scholar] [CrossRef] [Green Version]
- MathWorks. ResNet-50 Convolutional Neural Network—MATLAB Resnet50—MathWorks. Available online: https://it.mathworks.com/help/deeplearning/ref/resnet50.html?lang=en (accessed on 27 April 2023).
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. arXiv 2017, arXiv:1706.03762. [Google Scholar]
- Wolf, T.; Debut, L.; Sanh, V.; Chaumond, J.; Delangue, C.; Moi, A.; Cistac, P.; Rault, T.; Louf, R.; Funtowicz, M.; et al. Transformers: State-of-the-Art Natural Language Processing. In Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing: System Demonstrations, Association for Computational Linguistics, Online, 16–18 November 2020; pp. 38–45. [Google Scholar] [CrossRef]
- Dosovitskiy, A.; Beyer, L.; Kolesnikov, A.; Weissenborn, D.; Zhai, X.; Unterthiner, T.; Dehghani, M.; Minderer, M.; Heigold, G.; Gelly, S.; et al. An Image is Worth 16 × 16 Words: Transformers for Image Recognition at Scale. arXiv 2021, arXiv:2010.11929. [Google Scholar]
- Liu, Z.; Lin, Y.; Cao, Y.; Hu, H.; Wei, Y.; Zhang, Z.; Lin, S.; Guo, B. Swin Transformer: Hierarchical Vision Transformer using Shifted Windows. arXiv 2021, arXiv:2103.14030. [Google Scholar]
- Grisan, E.; Poletti, E.; Ruggeri, A. Automatic Segmentation and Disentangling of Chromosomes in Q-Band Prometaphase Images. IEEE Trans. Inf. Technol. Biomed. 2009, 13, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Poletti, E.; Grisan, E.; Ruggeri, A. Automatic classification of chromosomes in Q-band images. In Proceedings of the 2008 30th Annual International Conference of the IEEE Engineering in Medicine and Biology Society, Vancouver, BC, Canada, 20–25 August 2008; pp. 1911–1914. [Google Scholar] [CrossRef]
- Ritter, G.; Gao, L. Automatic segmentation of metaphase cells based on global context and variant analysis. Pattern Recognit. 2008, 41, 38–55. [Google Scholar] [CrossRef]
- Lin, C.; Yin, A.; Wu, Q.; Chen, H.; Guo, L.; Zhao, G.; Fan, X.; Luo, H.; Tang, H. Chromosome Cluster Identification Framework Based on Geometric Features and Machine Learning Algorithms. In Proceedings of the 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Seoul, Republic of Korea, 16–19 December 2020; pp. 2357–2363. [Google Scholar] [CrossRef]
- Shorten, C.; Khoshgoftaar, T.M. A survey on Image Data Augmentation for Deep Learning. J. Big Data 2019, 6. [Google Scholar] [CrossRef] [Green Version]
- Moradi, M.; Setarehdan, S.; Ghaffari, S. Automatic locating the centromere on human chromosome pictures. In Proceedings of the 16th IEEE Symposium Computer-Based Medical Systems, New York, NY, USA, 26–27 June 2003; pp. 56–61. [Google Scholar] [CrossRef] [Green Version]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Precision | Recall | Accuracy | F1 |
|---|---|---|---|---|
| Vanilla-CNN [6] | 0.8800 | 0.8600 | 0.8644 | 0.8700 |
| SiameseNet [7] | 0.8800 | 0.8700 | 0.8763 | 0.8700 |
| CIR-Net [11] | 0.9600 | 0.9600 | 0.9598 | 0.9600 |
| ResNet-50 | ||||
| None(1) | 0.8834 | 0.8767 | 0.8808 | 0.8775 |
| None(10) | 0.9246 | 0.9157 | 0.9216 | 0.9157 |
| CDAst8(1) | 0.9609 | 0.9524 | 0.9588 | 0.9547 |
| CDAst3(1) | 0.9706 | 0.9663 | 0.9715 | 0.9676 |
| CDA(1) | 0.9765 | 0.9743 | 0.9759 | 0.9749 |
| CDA(10) | 0.9822 | 0.9748 | 0.9812 | 0.9772 |
| STR(10) | 0.9834 | 0.9787 | 0.9822 | 0.9803 |
| FT(10) | 0.9846 | 0.9810 | 0.9836 | 0.9822 |
| CDA(5) + STR(5) | 0.9858 | 0.9814 | 0.9849 | 0.9831 |
| CDA(3) + STR(3) + FT(3) | 0.9864 | 0.9831 | 0.9856 | 0.9843 |
| Swin | ||||
| None(1) | 0.9579 | 0.9502 | 0.9578 | 0.9523 |
| CDA(1) | 0.9886 | 0.9854 | 0.9886 | 0.9857 |
| CDA(3) + STR(3) + FT(3) | 0.9966 | 0.9963 | 0.9963 | 0.9964 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
D’Angelo, M.; Nanni, L. Deep-Learning-Based Human Chromosome Classification: Data Augmentation and Ensemble. Information 2023, 14, 389. https://doi.org/10.3390/info14070389
D’Angelo M, Nanni L. Deep-Learning-Based Human Chromosome Classification: Data Augmentation and Ensemble. Information. 2023; 14(7):389. https://doi.org/10.3390/info14070389
Chicago/Turabian StyleD’Angelo, Mattia, and Loris Nanni. 2023. "Deep-Learning-Based Human Chromosome Classification: Data Augmentation and Ensemble" Information 14, no. 7: 389. https://doi.org/10.3390/info14070389
APA StyleD’Angelo, M., & Nanni, L. (2023). Deep-Learning-Based Human Chromosome Classification: Data Augmentation and Ensemble. Information, 14(7), 389. https://doi.org/10.3390/info14070389