
Exploring CVD Method for Synthesizing Carbon–Carbon Composites as Materials to Contact with Nerve Tissue

 , , , and
, , , and
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.1.1. Carbon Fibers
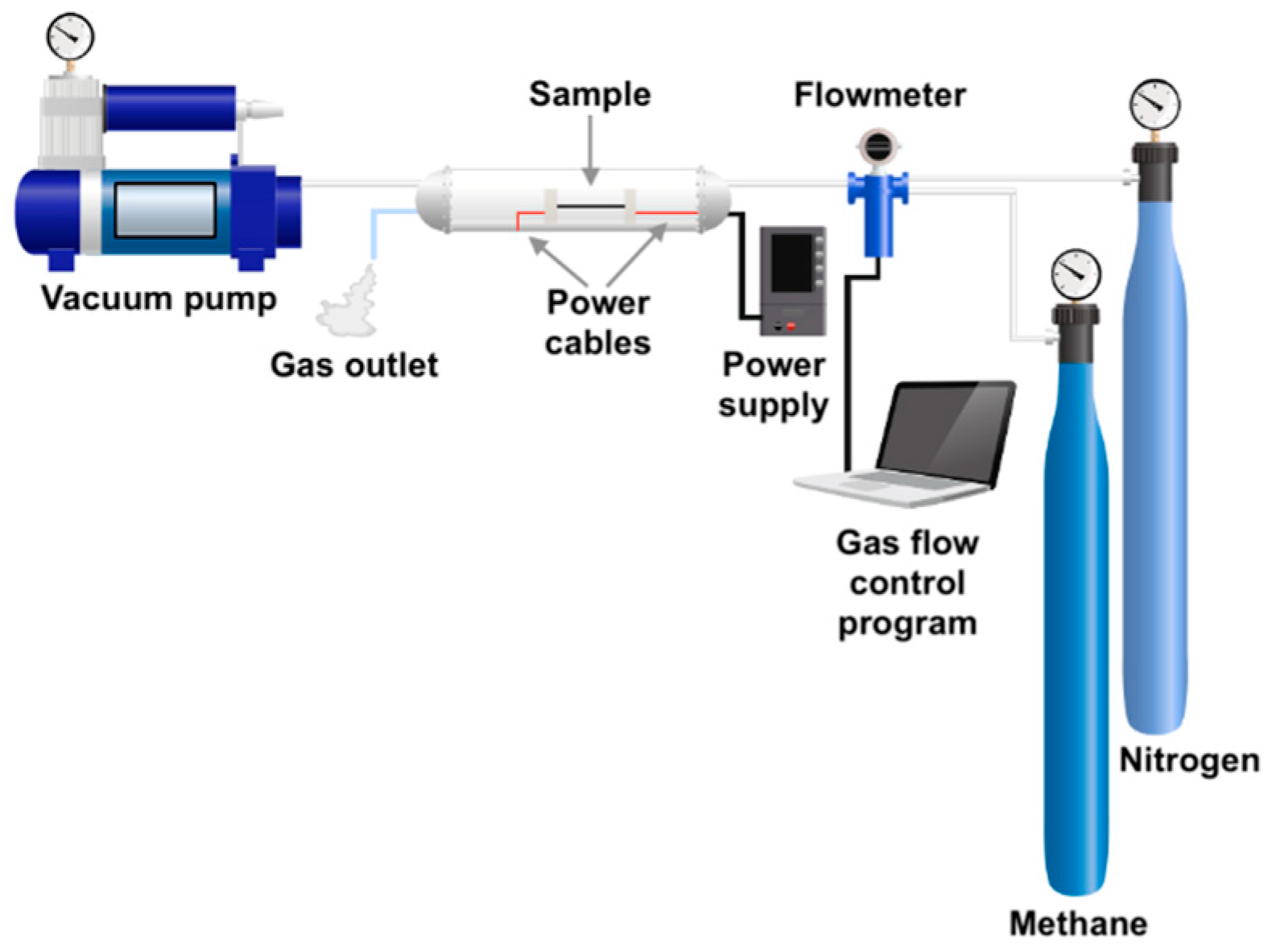
2.1.2. CVD Method with Direct Electrical Heating of Carbon Fibers
- CF_PyC30—rod-shaped C/C composite based on carbon fiber and PyC obtained after 30 s of synthesis.
- CF_PyC60—rod-shaped C/C composite based on carbon fiber and PyC obtained after 60 s of synthesis.
- CF_PyC120—rod-shaped C/C composite based on carbon fiber and PyC obtained after 120 s of synthesis.
- CF_PyC180—rod-shaped C/C composite based on carbon fiber and PyC obtained after 180 s of synthesis.
- CF—bundle of carbon fibers.
2.2. Methods
2.2.1. SEM and Digital Microscope
2.2.2. TEM and HRTEM
2.2.3. Selected Area Electron Diffraction (SAED)
- -
- Determining the position of the center of the diffraction pattern and the radius of the diffraction ring with indices (002);
- -
- Determining (using a self-developed script in Python) the profile of intensity changes along the perimeter of a circle with a predetermined center and radius (values read in 0.2 degree steps);
- -
- -
- Determination of their average value, equal to orientation angle OA.
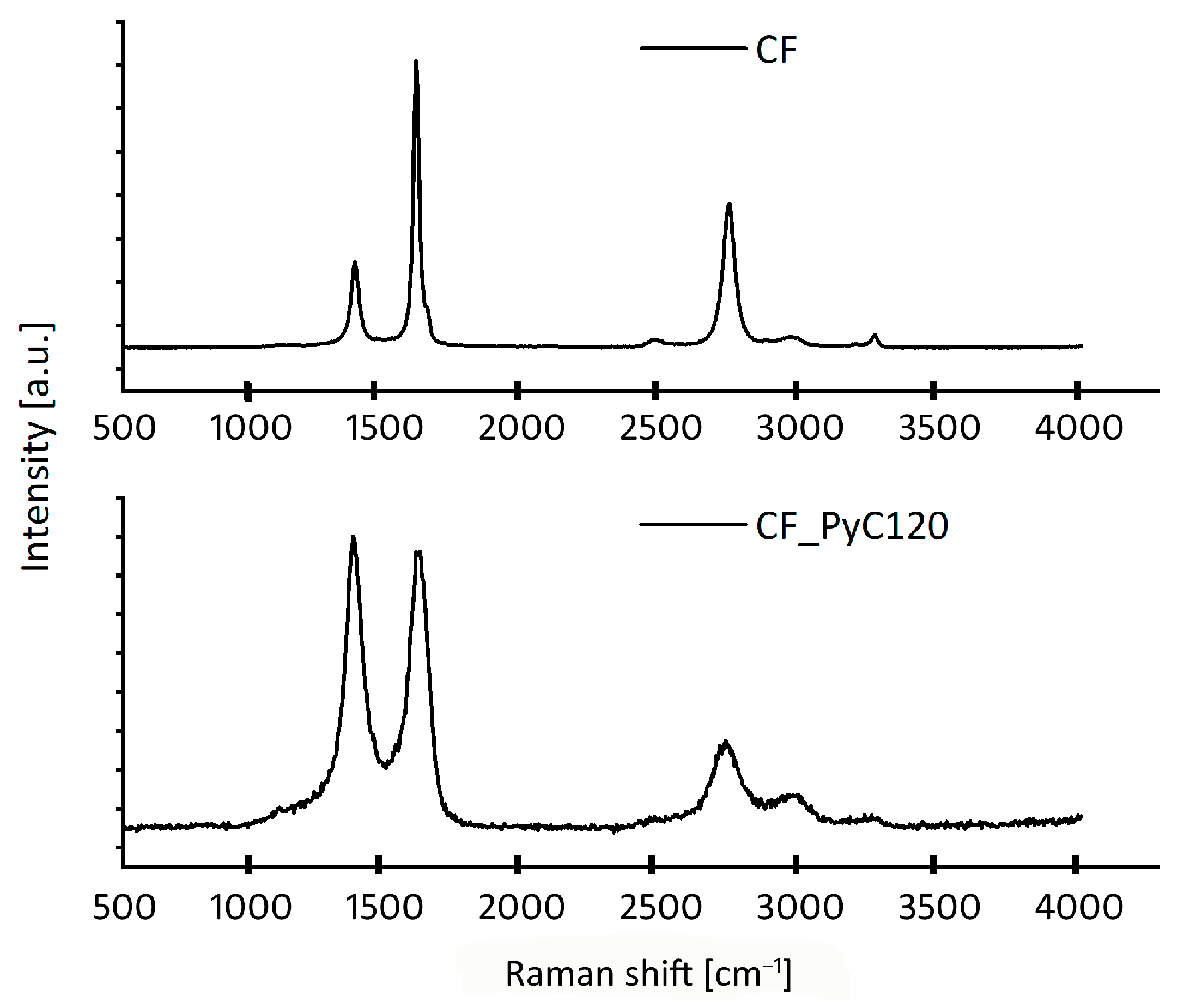
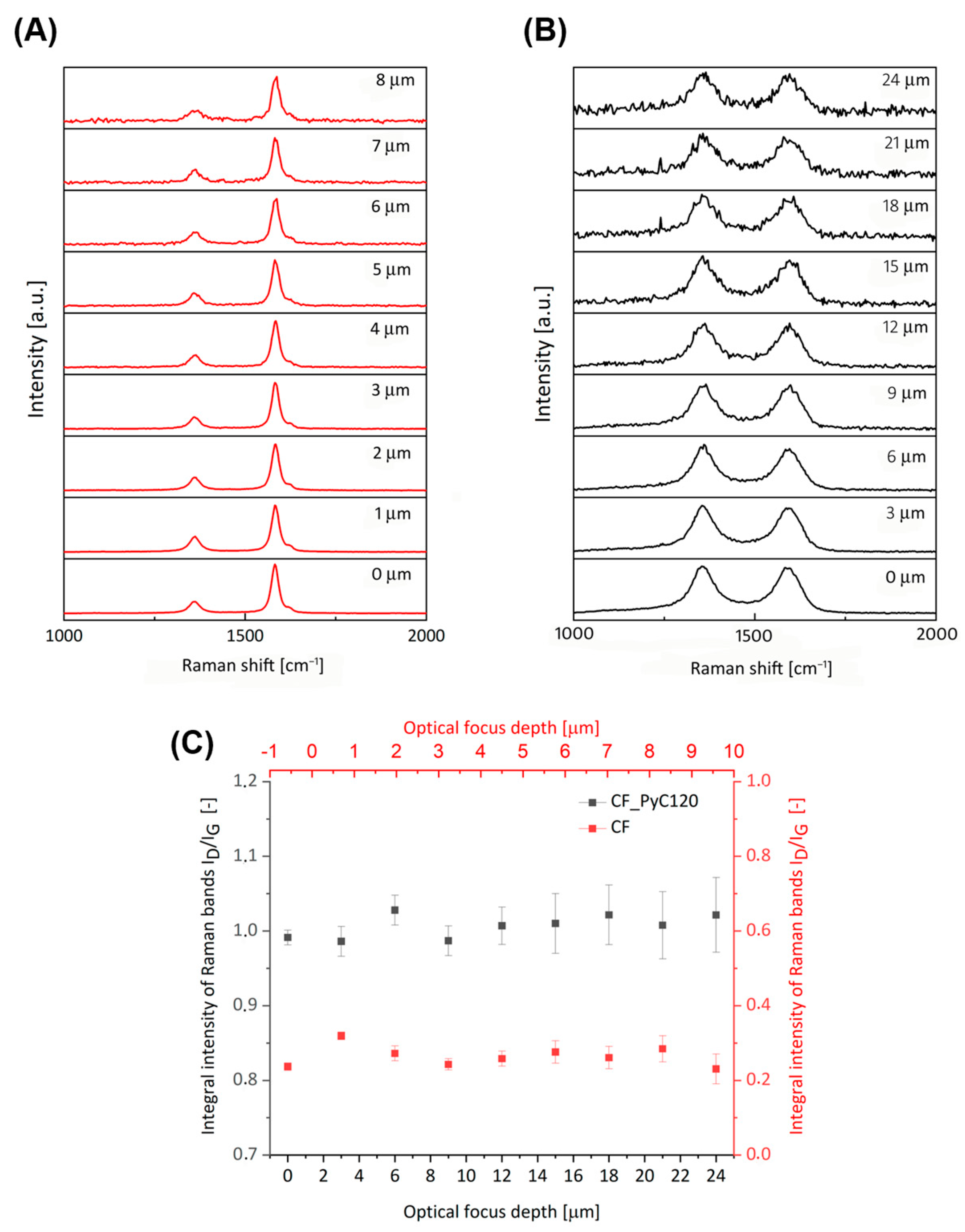
2.2.4. Raman Spectroscopy
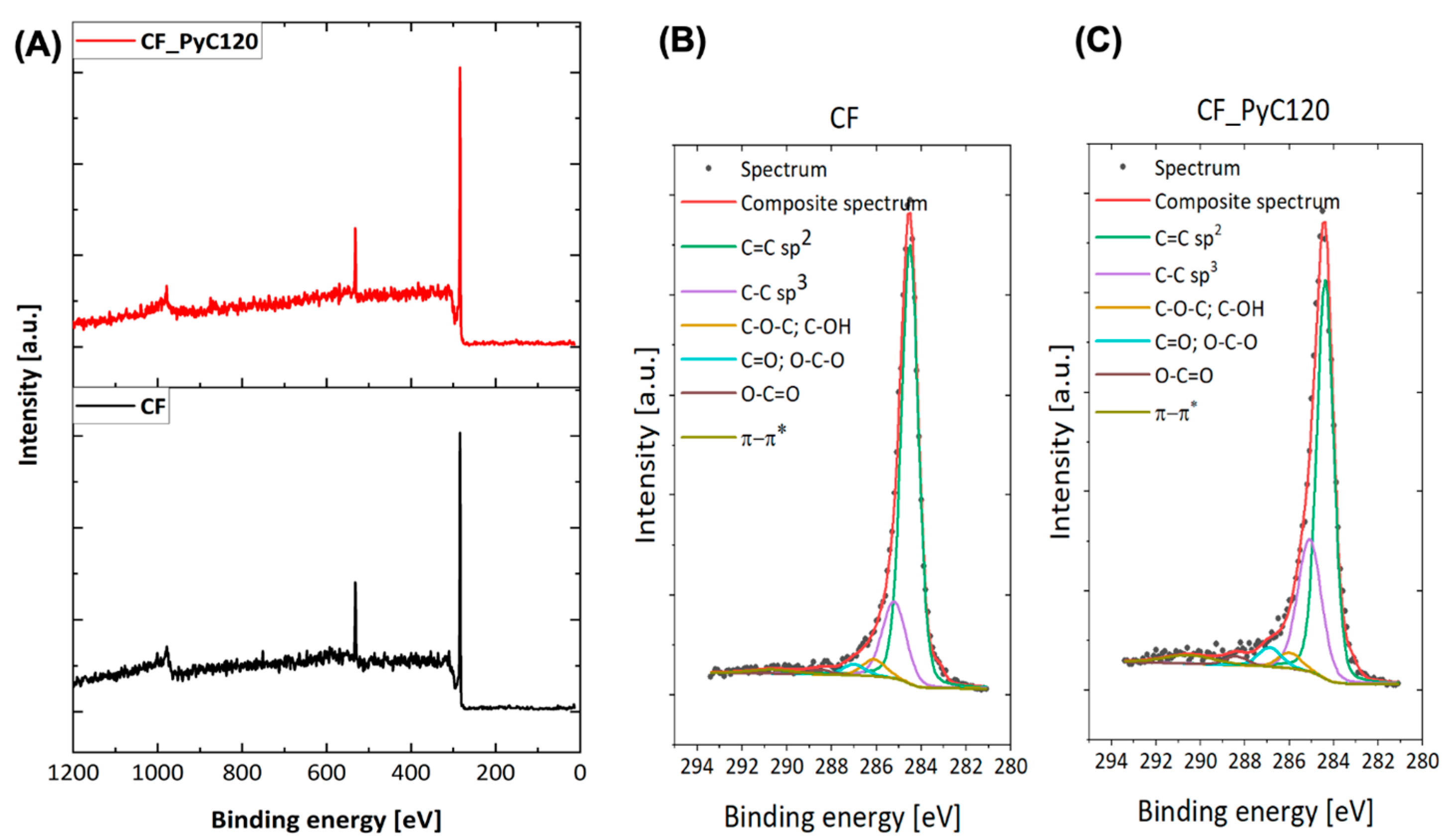
2.2.5. XPS
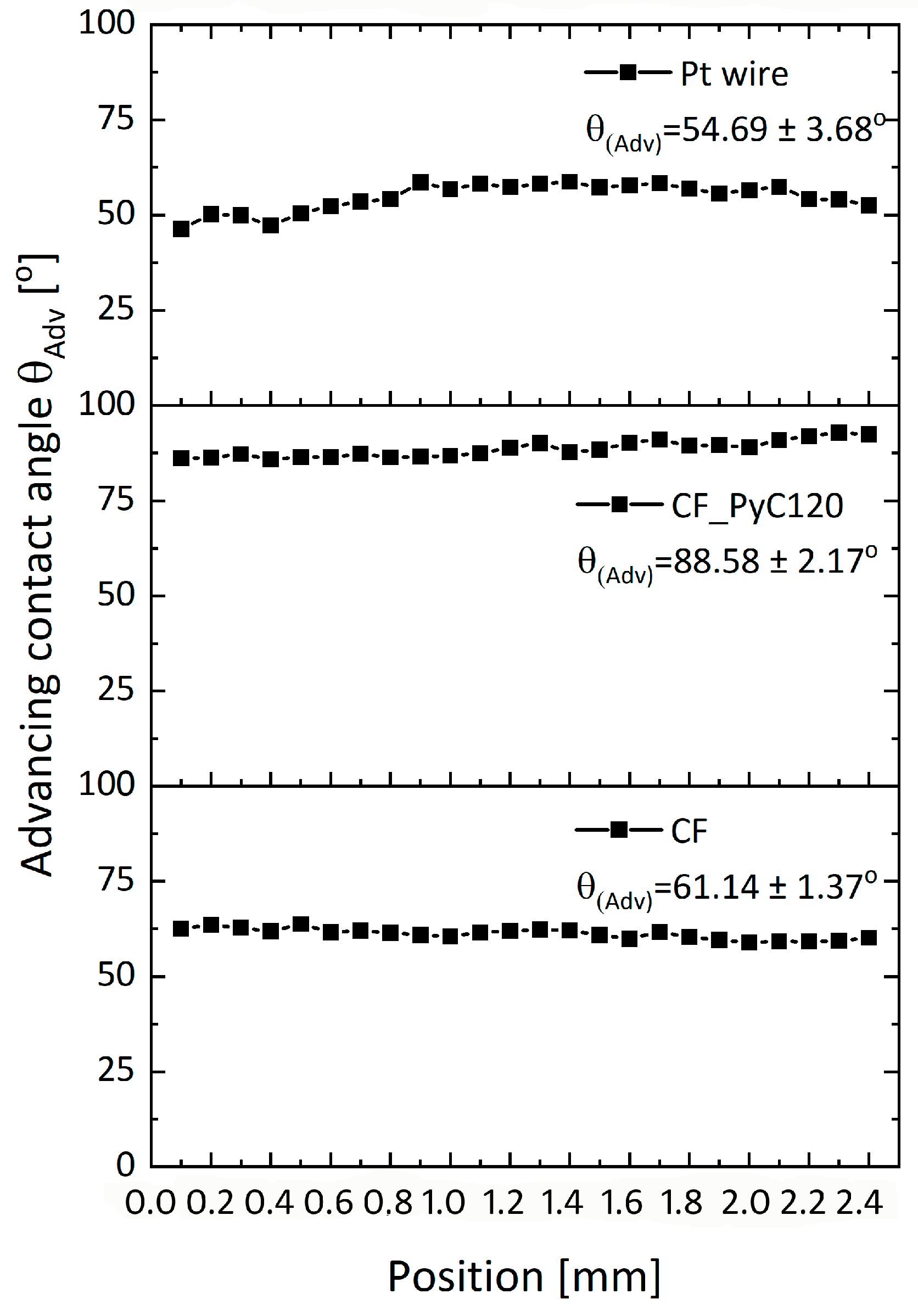
2.2.6. Contact Angle Measurement
2.2.7. In Vitro Study
Cell Culture and Experimental Groups
Cytotoxicity Assay
Live Cell Imaging
Cell Viability Assay
Scanning Electron Microscopy
Statistical Analysis
3. Results and Discussion
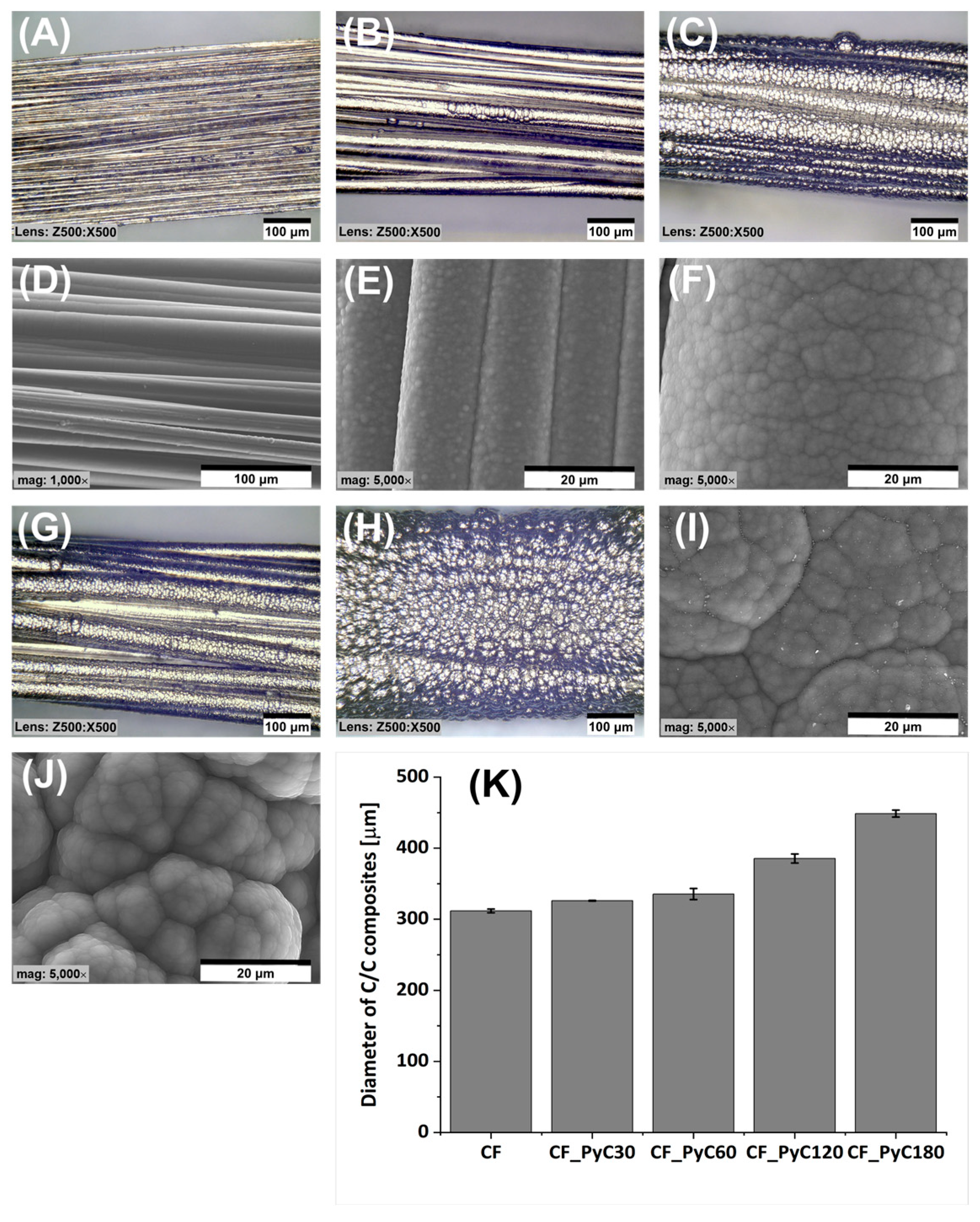
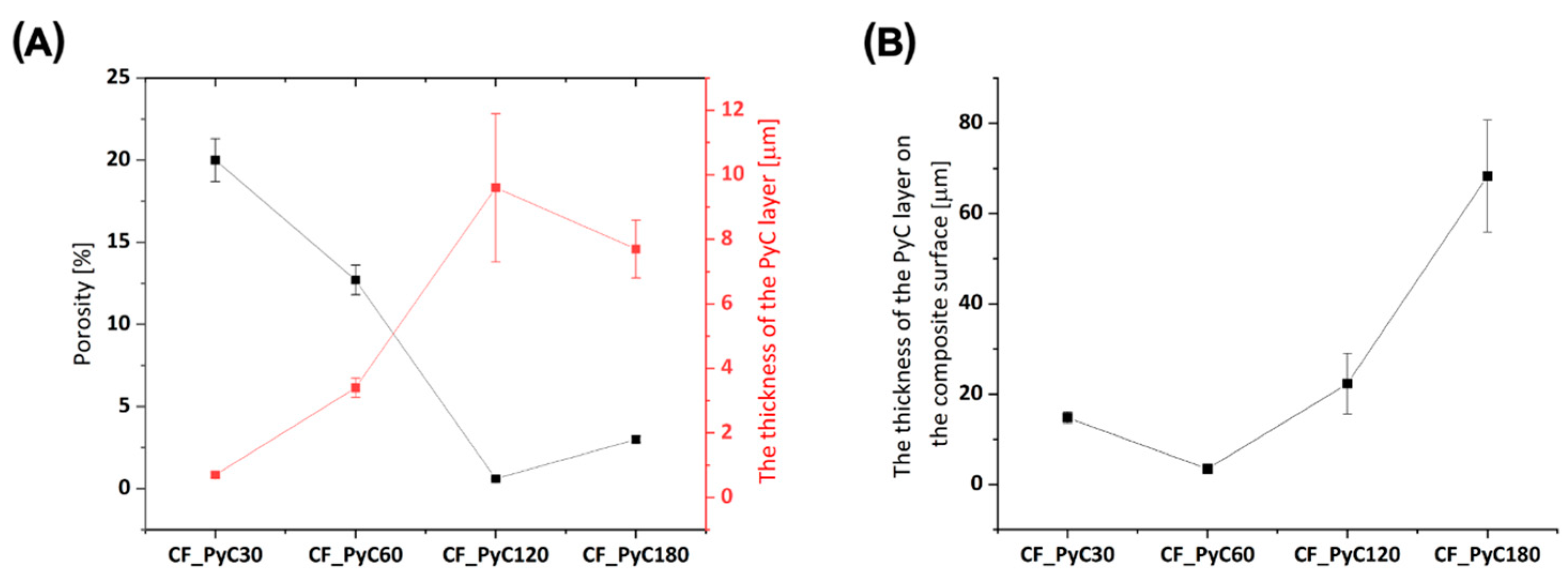
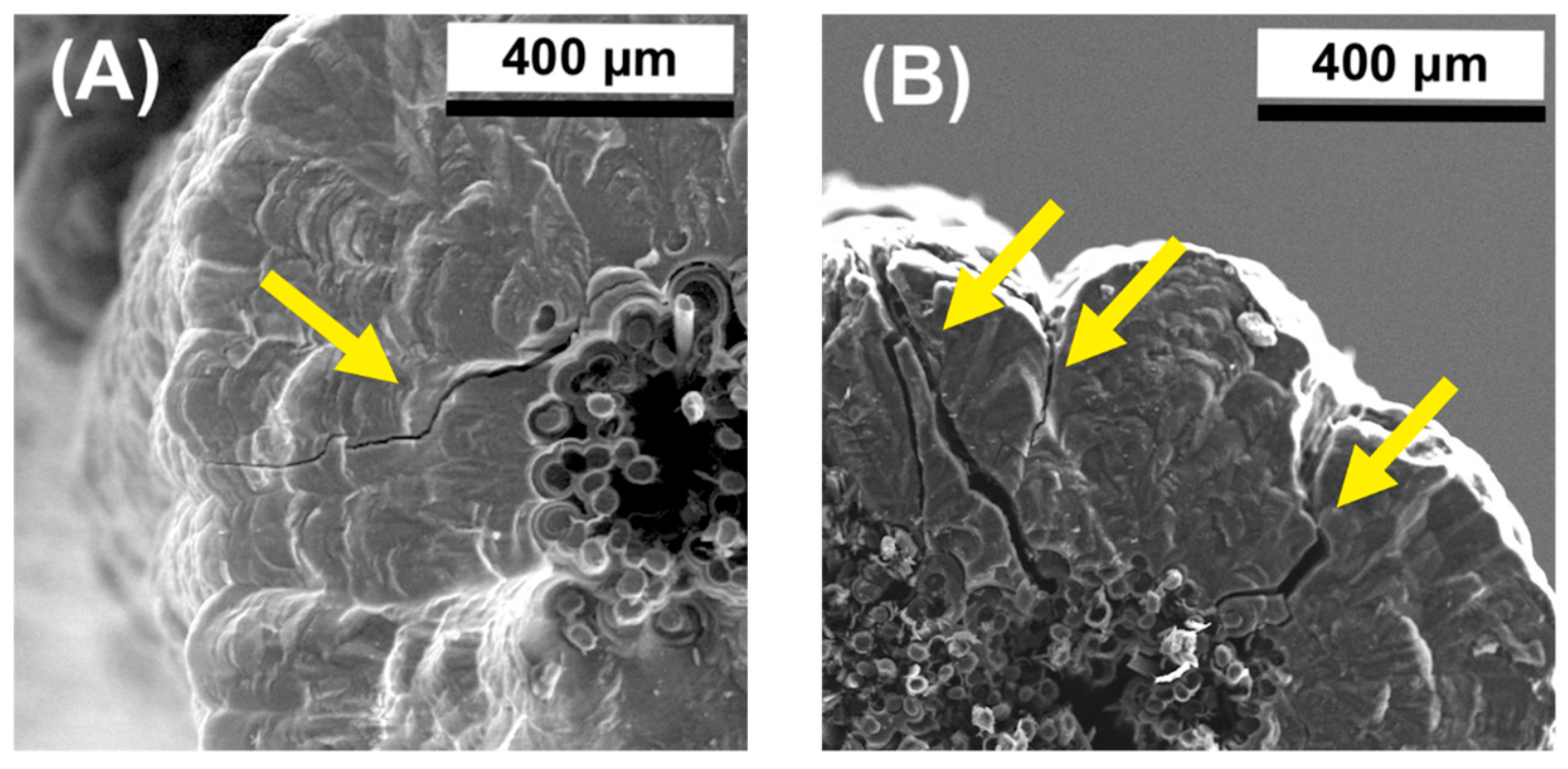
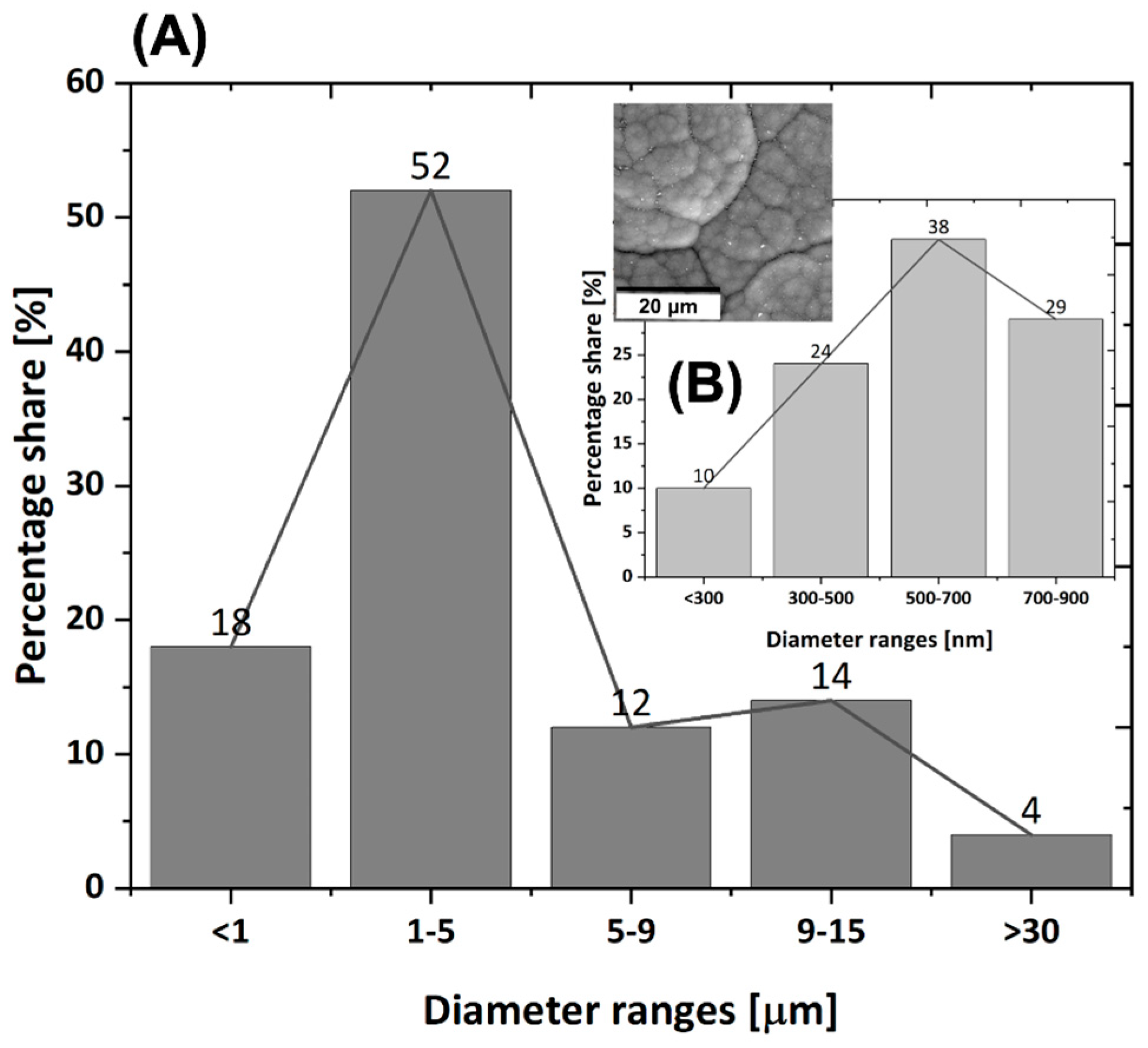
3.1. Morphology and Microstructure of Rod-Shaped C/C Composite
3.2. Structure of Rod-Shaped C/C Composite
3.3. Surface Chemistry of Rod-Shaped C/C Composite
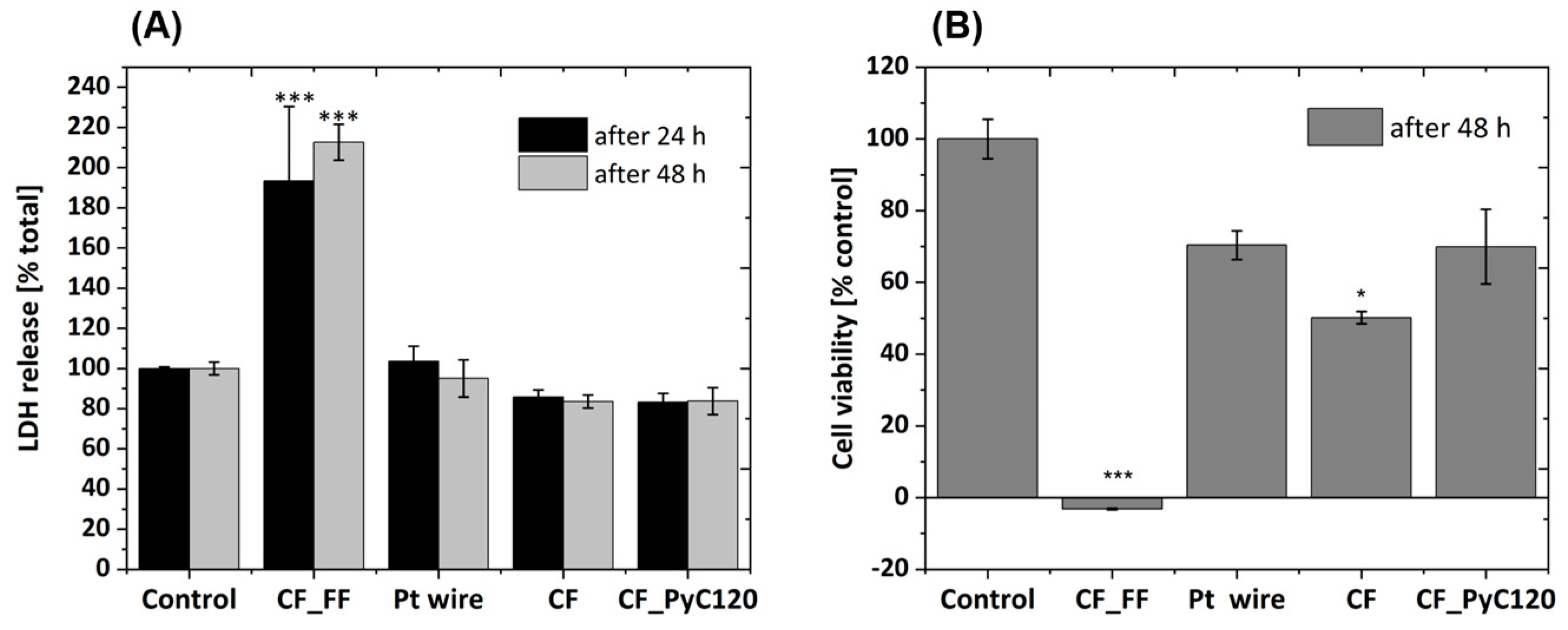
3.4. Biocompatibility of Rod-Shaped C/C Composite
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative Diseases: An Overview of Environmental Risk Factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- Checkoway, H.; Lundin, J.I.; Kelada, S.N. Neurodegenerative diseases. IARC Sci. Publ. 2011, 163, 407–419. [Google Scholar]
- Chi, H.; Chang, H.-Y.; Sang, T.-K. Neuronal Cell Death Mechanisms in Major Neurodegenerative Diseases. Int. J. Mol. Sci. 2018, 19, 3082. [Google Scholar] [CrossRef] [PubMed]
- Poddar, K.M.; Chakraborty, A.; Banerjee, S. Neurodegeneration: Diagnosis, Prevention, and Therapy. In Oxidoreductase; IntechOpen: London, UK, 2021. [Google Scholar]
- Hariz, M.; Blomstedt, P. Deep brain stimulation for Parkinson’s disease. J. Intern. Med. 2022, 292, 764–778. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Kim, S.; Kwon, Y.W.; Seo, H.; Kim, M.; Chung, W.G.; Park, W.; Song, H.; Lee, D.H.; Lee, J.; et al. Electrical stimulation for therapeutic approach. Interdiscip. Med. 2023, 1, e20230003. [Google Scholar] [CrossRef]
- Malek, N. Deep Brain Stimulation in Parkinson’s Disease. Neurol. India 2019, 67, 968. [Google Scholar] [CrossRef] [PubMed]
- Kolaya, E.; Firestein, B.L. Deep brain stimulation: Challenges at the tissue-electrode interface and current solutions. Biotechnol. Prog. 2021, 37, e3179. [Google Scholar] [CrossRef] [PubMed]
- Arcot Desai, S.; Gutekunst, C.-A.; Potter, S.M.; Gross, R.E. Deep brain stimulation macroelectrodes compared to multiple microelectrodes in rat hippocampus. Front. Neuroeng. 2014, 7, 16. [Google Scholar] [CrossRef]
- Hickey, P.; Stacy, M. Deep Brain Stimulation: A Paradigm Shifting Approach to Treat Parkinson’s Disease. Front. Neurosci. 2016, 10, 173. [Google Scholar] [CrossRef]
- Polikov, V.S.; Block, M.L.; Fellous, J.-M.; Hong, J.-S.; Reichert, W.M. In vitro model of glial scarring around neuroelectrodes chronically implanted in the CNS. Biomaterials 2006, 27, 5368–5376. [Google Scholar] [CrossRef]
- McConnell, G.C.; Rees, H.D.; Levey, A.I.; Gutekunst, C.-A.; Gross, R.E.; Bellamkonda, R.V. Implanted neural electrodes cause chronic, local inflammation that is correlated with local neurodegeneration. J. Neural Eng. 2009, 6, 056003. [Google Scholar] [CrossRef] [PubMed]
- Wellman, S.M.; Li, L.; Yaxiaer, Y.; McNamara, I.; Kozai, T.D.Y. Revealing Spatial and Temporal Patterns of Cell Death, Glial Proliferation, and Blood-Brain Barrier Dysfunction Around Implanted Intracortical Neural Interfaces. Front. Neurosci. 2019, 13, 493. [Google Scholar] [CrossRef] [PubMed]
- Usoro, J.O.; Sturgill, B.S.; Musselman, K.C.; Capadona, J.R.; Pancrazio, J.J. Intracortical Microelectrode Array Unit Yield under Chronic Conditions: A Comparative Evaluation. Micromachines 2021, 12, 972. [Google Scholar] [CrossRef]
- Cherry, J.D.; Olschowka, J.A.; O’Banion, M.K. Neuroinflammation and M2 microglia: The good, the bad, and the inflamed. J. Neuroinflamm. 2014, 11, 98. [Google Scholar] [CrossRef] [PubMed]
- Karumbaiah, L.; Saxena, T.; Carlson, D.; Patil, K.; Patkar, R.; Gaupp, E.A.; Betancur, M.; Stanley, G.B.; Carin, L.; Bellamkonda, R.V. Relationship between intracortical electrode design and chronic recording function. Biomaterials 2013, 34, 8061–8074. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, M.; Ivica, N.; Bjartmarz, H.; Thorbergsson, P.T.; Pettersson, L.M.E.; Thelin, J.; Schouenborg, J. Microelectrode clusters enable therapeutic deep brain stimulation without noticeable side-effects in a rodent model of Parkinson’s disease. J. Neurosci. Methods 2022, 365, 109399. [Google Scholar] [CrossRef]
- Tian, G.; Yang, D.; Chen, C.; Duan, X.; Kim, D.-H.; Chen, H. Simultaneous Presentation of Dexamethasone and Nerve Growth Factor via Layered Carbon Nanotubes and Polypyrrole to Interface Neural Cells. ACS Biomater. Sci. Eng. 2023, 9, 5015–5027. [Google Scholar] [CrossRef]
- Rodrigues, A.F.; Tavares, A.P.M.; Simões, S.; Silva, R.P.F.F.; Sobrino, T.; Figueiredo, B.R.; Sales, G.; Ferreira, L. Engineering graphene-based electrodes for optical neural stimulation. Nanoscale 2023, 15, 687–706. [Google Scholar] [CrossRef]
- Lim, J.; Lee, S.; Kim, J.; Hong, J.; Lim, S.; Kim, K.; Kim, J.; Yang, S.; Yang, S.; Ahn, J.-H. Hybrid graphene electrode for the diagnosis and treatment of epilepsy in free-moving animal models. NPG Asia Mater. 2023, 15, 7. [Google Scholar] [CrossRef]
- Nekounam, H.; Samadian, H.; Golmohammadi, H.; Asghari, F.; Shokrgozar, M.A.; Ahadian, S.; Majidi, R.F. Carbon nanofibers fabrication, surface modifications, and application as the innovative substrate for electrical stimulation of neural cell differentiation. Surf. Interfaces 2023, 40, 102926. [Google Scholar] [CrossRef]
- Hejazi, M.A.; Tong, W.; Stacey, A.; Soto-Breceda, A.; Ibbotson, M.R.; Yunzab, M.; Maturana, M.I.; Almasi, A.; Jung, Y.J.; Sun, S.; et al. Hybrid diamond/ carbon fiber microelectrodes enable multimodal electrical/chemical neural interfacing. Biomaterials 2020, 230, 119648. [Google Scholar] [CrossRef]
- Dresvyanina, E.N.; Tagandurdyyeva, N.A.; Kodolova-Chukhontseva, V.V.; Dobrovol’skaya, I.P.; Kamalov, A.M.; Nashchekina, Y.A.; Nashchekin, A.V.; Ivanov, A.G.; Yukina, G.Y.; Yudin, V.E. Structure and Properties of Composite Fibers Based on Chitosan and Single-Walled Carbon Nanotubes for Peripheral Nerve Regeneration. Polymers 2023, 15, 2860. [Google Scholar] [CrossRef]
- Pi, W.; Zhang, Y.; Li, L.; Li, C.; Zhang, M.; Zhang, W.; Cai, Q.; Zhang, P. Polydopamine-coated polycaprolactone/carbon nanotube fibrous scaffolds loaded with brain-derived neurotrophic factor for peripheral nerve regeneration. Biofabrication 2022, 14, 035006. [Google Scholar] [CrossRef]
- Hejazi, M.; Tong, W.; Ibbotson, M.R.; Prawer, S.; Garrett, D.J. Advances in Carbon-Based Microfiber Electrodes for Neural Interfacing. Front. Neurosci. 2021, 15, 658703. [Google Scholar] [CrossRef] [PubMed]
- Devi, M.; Vomero, M.; Fuhrer, E.; Castagnola, E.; Gueli, C.; Nimbalkar, S.; Hirabayashi, M.; Kassegne, S.; Stieglitz, T.; Sharma, S. Carbon-based neural electrodes: Promises and challenges. J. Neural Eng. 2021, 18, 041007. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, P.; Goe, A. Carbon Fibres: Production, Properties and Potential Use. Mater. Sci. Res. India 2017, 14, 52–57. [Google Scholar] [CrossRef]
- Gillis, W.F.; Lissandrello, C.A.; Shen, J.; Pearre, B.W.; Mertiri, A.; Deku, F.; Cogan, S.; Holinski, B.J.; Chew, D.J.; White, A.E.; et al. Carbon fiber on polyimide ultra-microelectrodes. J. Neural Eng. 2018, 15, 016010. [Google Scholar] [CrossRef] [PubMed]
- Manciu, F.S.; Oh, Y.; Barath, A.; Rusheen, A.E.; Kouzani, A.Z.; Hodges, D.; Guerrero, J.; Tomshine, J.; Lee, K.H.; Bennet, K.E. Analysis of Carbon-Based Microelectrodes for Neurochemical Sensing. Materials 2019, 12, 3186. [Google Scholar] [CrossRef]
- Lee, Y.; Kong, C.; Chang, J.W.; Jun, S.B. Carbon-Fiber Based Microelectrode Array Embedded with a Biodegradable Silk Support for In Vivo Neural Recording. J. Korean Med. Sci. 2019, 34, e24. [Google Scholar] [CrossRef]
- Dunn, J.F.; Tuor, U.I.; Kmech, J.; Young, N.A.; Henderson, A.K.; Jackson, J.C.; Valentine, P.A.; Teskey, G.C. Functional brain mapping at 9.4T using a new MRI-compatible electrode chronically implanted in rats. Magn. Reson. Med. 2009, 61, 222–228. [Google Scholar] [CrossRef]
- Cruttenden, C.E.; Taylor, J.M.; Hu, S.; Zhang, Y.; Zhu, X.-H.; Chen, W.; Rajamani, R. Carbon nano-structured neural probes show promise for magnetic resonance imaging applications. Biomed. Phys. Eng. Express 2017, 4, 015001. [Google Scholar] [CrossRef] [PubMed]
- Huffman, M.L.; Venton, B.J. Carbon-fiber microelectrodes for in vivo applications. Analyst 2009, 134, 18–24. [Google Scholar] [CrossRef]
- Letner, J.G.; Patel, P.R.; Hsieh, J.-C.; Smith Flores, I.M.; della Valle, E.; Walker, L.A.; Weiland, J.D.; Chestek, C.A.; Cai, D. Post-explant profiling of subcellular-scale carbon fiber intracortical electrodes and surrounding neurons enables modeling of recorded electrophysiology. J. Neural Eng. 2023, 20, 026019. [Google Scholar] [CrossRef]
- Zhao, S.; Li, G.; Tong, C.; Chen, W.; Wang, P.; Dai, J.; Fu, X.; Xu, Z.; Liu, X.; Lu, L.; et al. Full activation pattern mapping by simultaneous deep brain stimulation and fMRI with graphene fiber electrodes. Nat. Commun. 2020, 11, 1788. [Google Scholar] [CrossRef]
- Bennet, K.E.; Tomshine, J.R.; Min, H.-K.; Manciu, F.S.; Marsh, M.P.; Paek, S.B.; Settell, M.L.; Nicolai, E.N.; Blaha, C.D.; Kouzani, A.Z.; et al. A Diamond-Based Electrode for Detection of Neurochemicals in the Human Brain. Front. Hum. Neurosci. 2016, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- More, R.B.; Haubold, A.D.; Bokros, J.C. Pyrolytic Carbon for Long-Term Medical Implants. In Biomaterials Science; Elsevier: Amsterdam, The Netherlands, 2013; pp. 209–222. [Google Scholar]
- Li, A.; Norinaga, K.; Zhang, W.; Deutschmann, O. Modeling and simulation of materials synthesis: Chemical vapor deposition and infiltration of pyrolytic carbon. Compos. Sci. Technol. 2008, 68, 1097–1104. [Google Scholar] [CrossRef]
- Forti, S.; Lunelli, L.; Della Volpe, C.; Siboni, S.; Pasquardini, L.; Lui, A.; Canteri, R.; Vanzetti, L.; Potrich, C.; Vinante, M.; et al. Hemocompatibility of pyrolytic carbon in comparison with other biomaterials. Diam. Relat. Mater. 2011, 20, 762–769. [Google Scholar] [CrossRef]
- Daecke, W.; Veyel, K.; Wieloch, P.; Jung, M.; Lorenz, H.; Martini, A.-K. Osseointegration and Mechanical Stability of Pyrocarbon and Titanium Hand Implants in a Load-Bearing In Vivo Model for Small Joint Arthroplasty. J. Hand Surg. Am. 2006, 31, 90–97. [Google Scholar] [CrossRef]
- Stanley, J.; Klawitter, J.; More, R. Replacing joints with pyrolytic carbon. In Joint Replacement Technology; Elsevier: Amsterdam, The Netherlands, 2008; pp. 631–656. [Google Scholar]
- Norinaga, K.; Deutschmann, O.; Saegusa, N.; Hayashi, J. Analysis of pyrolysis products from light hydrocarbons and kinetic modeling for growth of polycyclic aromatic hydrocarbons with detailed chemistry. J. Anal. Appl. Pyrolysis 2009, 86, 148–160. [Google Scholar] [CrossRef]
- Drescher, M.; Hüttinger, K.J.; Dormann, E. Pyrolytic carbon layers—An electron spin resonance analysis. Carbon 2003, 41, 773–783. [Google Scholar] [CrossRef]
- He, Y.-G.; Li, K.-Z.; Li, H.-J.; Wei, J.-F.; Fu, Q.-G.; Zhang, D.-S. Effect of interface structures on the fracture behavior of two-dimensional carbon/carbon composites by isothermal chemical vapor infiltration. J. Mater. Sci. 2010, 45, 1432–1437. [Google Scholar] [CrossRef]
- Oku, T. Carbon/Carbon Composites and Their Properties. In Carbon Alloys; Elsevier: Amsterdam, The Netherlands, 2003; pp. 523–544. [Google Scholar]
- Reznik, B.; Gerthsen, D.; Hüttinger, K.J. Micro- and nanostructure of the carbon matrix of infiltrated carbon fiber felts. Carbon 2001, 39, 215–229. [Google Scholar] [CrossRef]
- Xu, X.; Ouyang, T.; Zeng, L.; Chai, L. Study on the Pyrolytic Carbon Generated by the Electric Heating CVD Method. J. Wuhan Univ. Technol. Sci. Ed. 2018, 33, 409–413. [Google Scholar] [CrossRef]
- Kovalevich, J.; Langford, D. Considerations for the Use of SH-SY5Y Neuroblastoma Cells in Neurobiology. Methods Mol. Biol. 2013, 1078, 9–21. [Google Scholar] [PubMed]
- Jantas, D.; Chwastek, J.; Malarz, J.; Stojakowska, A.; Lasoń, W. Neuroprotective Effects of Methyl Caffeate against Hydrogen Peroxide-Induced Cell Damage: Involvement of Caspase 3 and Cathepsin D Inhibition. Biomolecules 2020, 10, 1530. [Google Scholar] [CrossRef]
- Ruffels, J.; Griffin, M.; Dickenson, J.M. Activation of ERK1/2, JNK and PKB by hydrogen peroxide in human SH-SY5Y neuroblastoma cells: Role of ERK1/2 in H2O2-induced cell death. Eur. J. Pharmacol. 2004, 483, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Jantas, D.; Piotrowski, M.; Lason, W. An Involvement of PI3-K/Akt Activation and Inhibition of AIF Translocation in Neuroprotective Effects of Undecylenic Acid (UDA) Against Pro-Apoptotic Factors-Induced Cell Death in Human Neuroblastoma SH-SY5Y Cells. J. Cell. Biochem. 2015, 116, 2882–2895. [Google Scholar] [CrossRef] [PubMed]
- Reznik, B.; Hüttinger, K. On the terminology for pyrolytic carbon. Carbon 2002, 40, 621–624. [Google Scholar] [CrossRef]
- Meadows, P.J.; López-Honorato, E.; Xiao, P. Fluidized bed chemical vapor deposition of pyrolytic carbon—II. Effect of deposition conditions on anisotropy. Carbon 2009, 47, 251–262. [Google Scholar] [CrossRef]
- Meier, R.J. On art and science in curve-fitting vibrational spectra. Vib. Spectrosc. 2005, 39, 266–269. [Google Scholar] [CrossRef]
- Cançado, L.G.; Takai, K.; Enoki, T.; Endo, M.; Kim, Y.A.; Mizusaki, H.; Jorio, A.; Coelho, L.N.; Magalhães-Paniago, R.; Pimenta, M.A. General equation for the determination of the crystallite size La of nanographite by Raman spectroscopy. Appl. Phys. Lett. 2006, 88, 163106. [Google Scholar] [CrossRef]
- Tiab, D.; Donaldson, E.C. Wettability. In Petrophysics; Elsevier: Amsterdam, The Netherlands, 2012; pp. 371–418. [Google Scholar]
- Qiu, S.; Fuentes, C.A.; Zhang, D.; Van Vuure, A.W.; Seveno, D. Wettability of a Single Carbon Fiber. Langmuir 2016, 32, 9697–9705. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fuentes, C.A.; Zhang, D.; Wang, X.; Van Vuure, A.W.; Seveno, D. Wettability of carbon fibres at micro- and mesoscales. Carbon 2017, 120, 438–446. [Google Scholar] [CrossRef]
- Yuan, Y.; Lee, T.R. Contact Angle and Wetting Properties. In Surface Science Techniques; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3–34. [Google Scholar]
- Fraczek-Szczypta, A.; Jantas, D.; Ciepiela, F.; Grzonka, J. Graphene oxide-conductive polymer nanocomposite coatings obtained by the EPD method as substrates for neurite outgrowth. Diam. Relat. Mater. 2020, 102, 107663. [Google Scholar] [CrossRef]
- Jantas, D.; Malarz, J.; Le, T.N.; Stojakowska, A. Neuroprotective Properties of Kempferol Derivatives from Maesa membranacea against Oxidative Stress-Induced Cell Damage: An Association with Cathepsin D Inhibition and PI3K/Akt Activation. Int. J. Mol. Sci. 2021, 22, 10363. [Google Scholar] [CrossRef]
- StatSoft. Statistica, Version 13.3; Tibco Software Inc.: Palo Alto, CA, USA, 2017.
- Reznik, B.; Norinaga, K.; Gerthsen, D.; Deutschmann, O. The effect of cooling rate on hydrogen release from a pyrolytic carbon coating and its resulting morphology. Carbon 2006, 44, 1330–1334. [Google Scholar] [CrossRef]
- Ren, J.; Li, K.; Zhang, S.; Yao, X.; Tian, S. Preparation of carbon/carbon composite by pyrolysis of ethanol and methane. Mater. Des. 2015, 65, 174–178. [Google Scholar] [CrossRef]
- López-Honorato, E.; Meadows, P.J.; Xiao, P.; Marsh, G.; Abram, T.J. Structure and mechanical properties of pyrolytic carbon produced by fluidized bed chemical vapor deposition. Nucl. Eng. Des. 2008, 238, 3121–3128. [Google Scholar] [CrossRef]
- Tezcan, J.; Ozcan, S.; Gurung, B.; Filip, P. Measurement and analytical validation of interfacial bond strength of PAN-fiber-reinforced carbon matrix composites. J. Mater. Sci. 2008, 43, 1612–1618. [Google Scholar] [CrossRef]
- Schierholz, R.; Kröger, D.; Weinrich, H.; Gehring, M.; Tempel, H.; Kungl, H.; Mayer, J.; Eichel, R.-A. The carbonization of polyacrylonitrile-derived electrospun carbon nanofibers studied by in situ transmission electron microscopy. RSC Adv. 2019, 9, 6267–6277. [Google Scholar] [CrossRef]
- Zambrzycki, M.; Piech, R.; Raga, S.R.; Lira-Cantu, M.; Fraczek-Szczypta, A. Hierarchical carbon nanofibers/carbon nanotubes/NiCo nanocomposites as novel highly effective counter electrode for dye-sensitized solar cells: A structure-electrocatalytic activity relationship study. Carbon 2023, 203, 97–110. [Google Scholar] [CrossRef]
- Boehm, R.D.; Jin, C.; Narayan, R.J. Carbon and Diamond. In Comprehensive Biomaterials II; Elsevier: Amsterdam, The Netherlands, 2017; pp. 145–164. [Google Scholar]
- López-Honorato, E.; Meadows, P.J.; Xiao, P. Fluidized bed chemical vapor deposition of pyrolytic carbon—I. Effect of deposition conditions on microstructure. Carbon 2009, 47, 396–410. [Google Scholar] [CrossRef]
- Hu, Z.J.; Zhang, W.G.; Hüttinger, K.J.; Reznik, B.; Gerthsen, D. Influence of pressure, temperature and surface area/volume ratio on the texture of pyrolytic carbon deposited from methane. Carbon 2003, 41, 749–758. [Google Scholar] [CrossRef]
- Dong, G.L.; Hüttinger, K.J. Consideration of reaction mechanisms leading to pyrolytic carbon of different textures. Carbon 2002, 40, 2515–2528. [Google Scholar] [CrossRef]
- De Pauw, V.; Collin, A.; Send, W.; Hawecker, J.; Gerthsen, D.; Pfrang, A.; Schimmel, T. Deposition rates during the early stages of pyrolytic carbon deposition in a hot-wall reactor and the development of texture. Carbon 2006, 44, 3091–3101. [Google Scholar] [CrossRef]
- Hu, Z.J.; Hüttinger, K.J. Mechanisms of carbon deposition—A kinetic approach. Carbon 2002, 40, 624–628. [Google Scholar] [CrossRef]
- Vignoles, G.L.; Langlais, F.; Descamps, C.; Mouchon, A.; Le Poche, H.; Reuge, N.; Bertrand, N. CVD and CVI of pyrocarbon from various precursors. Surf. Coatings Technol. 2004, 188–189, 241–249. [Google Scholar] [CrossRef]
- Sadezky, A.; Muckenhuber, H.; Grothe, H.; Niessner, R.; Pöschl, U. Raman microspectroscopy of soot and related carbonaceous materials: Spectral analysis and structural information. Carbon 2005, 43, 1731–1742. [Google Scholar] [CrossRef]
- Zambrzycki, M.; Łoś, S.; Fraczek-Szczypta, A. Structure and electrical transport properties of carbon nanofibres/carbon nanotubes 3D hierarchical nanocomposites: Impact of the concentration of acetylacetonate catalyst. Ceram. Int. 2021, 47, 4020–4033. [Google Scholar] [CrossRef]
- Zambrzycki, M.; Jeleń, P.; Fraczek-Szczypta, A. Structure and electrical transport properties of electrospun carbon nanofibers/carbon nanotubes 3D hierarchical nanocomposites: Effect of the CCVD synthesis conditions. J. Mater. Sci. 2022, 57, 9334–9356. [Google Scholar] [CrossRef]
- Ferrari, A.C.; Basko, D.M. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Schuepfer, D.B.; Badaczewski, F.; Guerra-Castro, J.M.; Hofmann, D.M.; Heiliger, C.; Smarsly, B.; Klar, P.J. Assessing the structural properties of graphitic and non-graphitic carbons by Raman spectroscopy. Carbon 2020, 161, 359–372. [Google Scholar] [CrossRef]
- Ma, B.; Rodriguez, R.D.; Ruban, A.; Pavlov, S.; Sheremet, E. The correlation between electrical conductivity and second-order Raman modes of laser-reduced graphene oxide. Phys. Chem. Chem. Phys. 2019, 21, 10125–10134. [Google Scholar] [CrossRef]
- Ferrari, A.C. Raman spectroscopy of graphene and graphite: Disorder, electron–phonon coupling, doping and nonadiabatic effects. Solid State Commun. 2007, 143, 47–57. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, G.; Zhan, Z.; An, J.; Zheng, L.; Xie, E. Probing structure and strain transfer in dry-spun carbon nanotube fibers by depth-profiled Raman spectroscopy. Appl. Phys. Lett. 2013, 103, 031912. [Google Scholar] [CrossRef]
- Torrisi, L.; Scolaro, C. Blood Wettability of Haemocompatible Carbon-based Materials. J. Adv. Chem. Eng. 2017, 7. [Google Scholar] [CrossRef]
- Vigano, G.; Ten Brink, G.; Pollack, D.K.M.; Mariani, M.A.; Kooi, B.J. Wettability Properties of Standard Pyrolytic Carbon Bileaflet Mechanical Heart Valve Prostheses. Struct. Hear. 2020, 4, 41. [Google Scholar] [CrossRef]
- Xie, J.; Xin, D.; Cao, H.; Wang, C.; Zhao, Y.; Yao, L.; Ji, F.; Qiu, Y. Improving carbon fiber adhesion to polyimide with atmospheric pressure plasma treatment. Surf. Coatings Technol. 2011, 206, 191–201. [Google Scholar] [CrossRef]
- An, F.; Lu, C.; Guo, J.; He, S.; Lu, H.; Yang, Y. Preparation of vertically aligned carbon nanotube arrays grown onto carbon fiber fabric and evaluating its wettability on effect of composite. Appl. Surf. Sci. 2011, 258, 1069–1076. [Google Scholar] [CrossRef]
- Contact Angle of Water on Smooth Surfaces and Wettability. Available online: http://www.uskino.com/articleshow_113.html (accessed on 9 August 2023).
- Lopez-Suarez, L.; Al Awabdh, S.; Coumoul, X.; Chauvet, C. The SH-SY5Y human neuroblastoma cell line, a relevant in vitro cell model for investigating neurotoxicology in human: Focus on organic pollutants. Neurotoxicology 2022, 92, 131–155. [Google Scholar] [CrossRef]
- Xie, H.; Hu, L.; Li, G. SH-SY5Y human neuroblastoma cell line: In vitro cell model of dopaminergic neurons in Parkinson’s disease. Chin. Med. J. 2010, 123, 1086–1092. [Google Scholar]
- Majhy, B.; Priyadarshini, P.; Sen, A.K. Effect of surface energy and roughness on cell adhesion and growth—Facile surface modification for enhanced cell culture. RSC Adv. 2021, 11, 15467–15476. [Google Scholar] [CrossRef]
- Zhu, L.; Luo, D.; Liu, Y. Effect of the nano/microscale structure of biomaterial scaffolds on bone regeneration. Int. J. Oral Sci. 2020, 12, 6. [Google Scholar] [CrossRef] [PubMed]
- Giljean, S.; Bigerelle, M.; Anselme, K. Roughness statistical influence on cell adhesion using profilometry and multiscale analysis. Scanning 2014, 36, 2–10. [Google Scholar] [CrossRef]
- Robinson, D.; Efrat, M.; Mendes, D.G.; Halperin, N.; Nevo, Z. Implants composed of carbon fiber mesh and bone-marrow-derived, chondrocyte-enriched cultures for joint surface reconstruction. Bull. Hosp. Jt. Dis. 1993, 53, 75–82. [Google Scholar]
- Sengupta, B.; Gregory, W.E.; Zhu, J.; Dasetty, S.; Karakaya, M.; Brown, J.M.; Rao, A.M.; Barrows, J.K.; Sarupria, S.; Podila, R. Influence of carbon nanomaterial defects on the formation of protein corona. RSC Adv. 2015, 5, 82395–82402. [Google Scholar] [CrossRef] [PubMed]
- Atilhan, M.; Costa, L.T.; Aparicio, S. On the interaction between carbon nanomaterials and lipid biomembranes. J. Mol. Liq. 2019, 295, 111714. [Google Scholar] [CrossRef]
- Baoukina, S.; Monticelli, L.; Tieleman, D.P. Interaction of Pristine and Functionalized Carbon Nanotubes with Lipid Membranes. J. Phys. Chem. B 2013, 117, 12113–12123. [Google Scholar] [CrossRef] [PubMed]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | ID | IG | I2D | ID/IG | I2D/IG | La [nm] |
|---|---|---|---|---|---|---|
| CF | 6148 | 17,107 | 15,937 | 0.36 | 0.93 | 37.88 |
| CF_PyC120 | 7102 | 6844 | 3739 | 1.04 | 0.55 | 13.12 |
| Sample | Elemental Composition (%) | (-) | Deconvolution of the C1s Spectra (%) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C | O | O/C | 284.5 eV C=C (sp2) | 285.3 eV C-C (sp3) | 286.1 eV C-O, C-OH | 287.0 eV C=O, O-C-O | 288.5 eV O-C=O | 291.0 eV π→π* | |
| CF | 87.60 | 12.40 | 0.14 | 65.90 | 14.20 | 3.10 | 2.00 | 0.90 | 1.50 |
| CF_PyC120 | 90.30 | 9.70 | 0.11 | 56.10 | 23.10 | 2.80 | 3.40 | 1.60 | 3.30 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fraczek-Szczypta, A.; Kondracka, N.; Zambrzycki, M.; Gubernat, M.; Czaja, P.; Pawlyta, M.; Jelen, P.; Wielowski, R.; Jantas, D. Exploring CVD Method for Synthesizing Carbon–Carbon Composites as Materials to Contact with Nerve Tissue. J. Funct. Biomater. 2023, 14, 443. https://doi.org/10.3390/jfb14090443
Fraczek-Szczypta A, Kondracka N, Zambrzycki M, Gubernat M, Czaja P, Pawlyta M, Jelen P, Wielowski R, Jantas D. Exploring CVD Method for Synthesizing Carbon–Carbon Composites as Materials to Contact with Nerve Tissue. Journal of Functional Biomaterials. 2023; 14(9):443. https://doi.org/10.3390/jfb14090443
Chicago/Turabian StyleFraczek-Szczypta, Aneta, Natalia Kondracka, Marcel Zambrzycki, Maciej Gubernat, Pawel Czaja, Miroslawa Pawlyta, Piotr Jelen, Ryszard Wielowski, and Danuta Jantas. 2023. "Exploring CVD Method for Synthesizing Carbon–Carbon Composites as Materials to Contact with Nerve Tissue" Journal of Functional Biomaterials 14, no. 9: 443. https://doi.org/10.3390/jfb14090443
APA StyleFraczek-Szczypta, A., Kondracka, N., Zambrzycki, M., Gubernat, M., Czaja, P., Pawlyta, M., Jelen, P., Wielowski, R., & Jantas, D. (2023). Exploring CVD Method for Synthesizing Carbon–Carbon Composites as Materials to Contact with Nerve Tissue. Journal of Functional Biomaterials, 14(9), 443. https://doi.org/10.3390/jfb14090443