Dissolution Enhancement and Controlled Release of Paclitaxel Drug via a Hybrid Nanocarrier Based on mPEG-PCL Amphiphilic Copolymer and Fe-BTC Porous Metal-Organic Framework


 ,
,  ,
,  ,
,  , , and
, , and 
Abstract
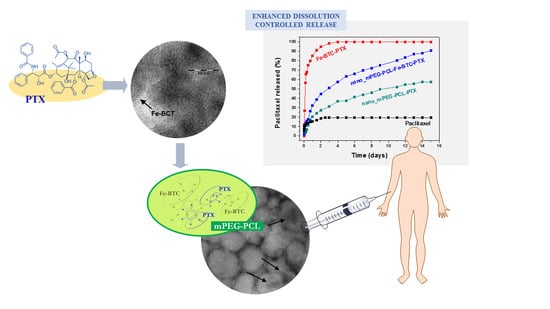
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of Poly(Ethylene Glycol) Methyl-Poly(Caprolactone) (mPEG-PCL)
2.2.1. Synthesis of mPEG-PCL
2.2.2. Characterization of mPEG-PCL
2.3. Nanoparticles Preparation and Characterization
2.3.1. Adsorption of Paclitaxel onto Fe Metal Ions and 1,3,5-Benzenetricarboxylate (Fe-BTC)
2.3.2. Nanoparticles Preparation
2.3.3. Nanoparticles Characterization
2.3.4. In Vitro Drug Release
2.3.5. High-Performance Liquid Chromatography (HPLC) Quantitative Analysis
2.4. Technical Details on the Computation
2.4.1. Structural and Vibrational Analysis
2.4.2. Interaction Energy
3. Results & Discussion
3.1. Synthesis and Characterization of mPEG-PCL
3.2. Characterization of Fe-BTC and Fe-BTC with Adsorbed Paclitaxel (Fe-BTC-PTX) Samples
3.3. Characterization of Nanoparticles
3.4. Computational Study
3.4.1. Structural Analysis
3.4.2. Vibrational Analysis
3.4.3. Intermolecular Interactions
3.5. In Vitro Drug Release
3.6. Modeling of the Release Kinetics
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nall, R. Medical News Today: What to Know about Cancer. Available online: https://www.medicalnewstoday.com/articles/323648 (accessed on 6 January 2020).
- Kar, S.; Katti, D.R.; Katti, K.S. Bone interface modulates drug resistance in breast cancer bone metastasis. Colloids Surf. B Biointerfaces 2020, 195, 111224. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Li, Y.; Cai, Q.; Tang, Z.; Yang, Y.; Yuan, C.; Xu, Y.; Zeng, B.; Luo, W.; Kuo, S. Tumor microenvironment-activated self-charge-generable metallosupramolecular polymer nanocapsules for photoacoustic imaging-guided targeted synergistic photothermal-chemotherapy. Chem. Eng. J. 2020, 405, 126690. [Google Scholar] [CrossRef]
- Obayemi, J.D.; Jusu, S.M.; Salifu, A.A.; Ghahremani, S.; Tadesse, M.; Uzonwanne, V.O.; Soboyejo, W.O. Degradable porous drug-loaded polymer scaffolds for localized cancer drug delivery and breast cell/tissue growth. Mater. Sci. Eng. C 2020, 112, 110794. [Google Scholar] [CrossRef] [PubMed]
- Nanaki, S.; Siafaka, P.I.; Zachariadou, D.; Nerantzaki, M.; Giliopoulos, D.J.; Triantafyllidis, K.S.; Kostoglou, M.; Nikolakaki, E.; Bikiaris, D.N. PLGA/SBA-15 mesoporous silica composite microparticles loaded with paclitaxel for local chemotherapy. Eur. J. Pharm. Sci. 2017, 99, 32–44. [Google Scholar] [CrossRef]
- Christodoulou, E.; Nerantzaki, M.; Nanaki, S.; Barmpalexis, P.; Anastasiou, A.D.; Bikiaris, D.N. Paclitaxel Magnetic Core-Shell Nanoparticles Based on Poly(lactic acid) Semitelechelic Novel Block Copolymers for Combined Hyperthermia and Chemotherapy Treatment of Cancer. Pharmaceutics 2019, 11, 213. [Google Scholar] [CrossRef] [Green Version]
- Filippousi, M.; Turner, S.; Leus, K.; Siafaka, P.I.; Tseligka, E.D.; Vandichel, M.; Nanaki, S.G.; Vizirianakis, I.S.; Bikiaris, D.N.; Van Der Voort, P. Biocompatible Zr-based nanoscale MOFs coated with modified poly (ε-caprolactone) as anticancer drug carriers. Int. J. Pharm. 2016, 509, 208–218. [Google Scholar] [CrossRef]
- Karavelidis, V.; Bikiaris, D.; Avgoustakis, K. New thermosensitive nanoparticles prepared by biocompatible pegylated aliphatic polyester block copolymers for local cancer treatment. J. Pharm. Pharmacol. 2015, 67, 215–230. [Google Scholar] [CrossRef]
- Liu, M.; Fu, M.; Yang, X.; Jia, G.; Shi, X.; Ji, J.; Liu, X.; Zhai, G. Paclitaxel and quercetin co-loaded functional mesoporous silica nanoparticles overcoming multidrug resistance in breast cancer. Colloids Surf. B Biointerfaces 2020, 196, 111284. [Google Scholar] [CrossRef]
- Gao, J.; Fan, K.; Jin, Y.; Zhao, L.; Wang, Q.; Tang, Y.; Xu, H.; Liu, Z.; Wang, S.; Lin, J.; et al. European Journal of Pharmaceutical Sciences PEGylated lipid bilayer coated mesoporous silica nanoparticles co-delivery of paclitaxel and curcumin leads to increased tumor site drug accumulation and reduced tumor burden. Eur. J. Pharm. Sci. 2019, 140, 105070. [Google Scholar] [CrossRef]
- Li, Y.; Hou, H.; Zhang, P.; Zhang, Z. Co-delivery of doxorubicin and paclitaxel by reduction/pH dual responsive nanocarriers for osteosarcoma therapy. Drug Deliv. 2020, 27, 1044–1053. [Google Scholar] [CrossRef]
- Uthappa, U.T.; Arvind, O.R.; Sriram, G.; Losic, D.; Kigga, M.; Kurkuri, M.D. Nanodiamonds and their surface modification strategies for drug delivery applications. J. Drug Deliv. Sci. Technol. 2020, 60, 101993. [Google Scholar] [CrossRef]
- Lim, D.G.; Jung, J.H.; Ko, H.W.; Kang, E.; Jeong, S.H. Paclitaxel-nanodiamond nanocomplexes enhance aqueous dispersibility and drug retention in cells. ACS Appl. Mater. Interfaces 2016, 8, 23558–23567. [Google Scholar] [CrossRef]
- Bazzazzadeh, A.; Dizaji, B.F.; Kianinejad, N.; Nouri, A.; Irani, M. Fabrication of poly (acrylic acid) grafted-chitosan/polyurethane/magnetic MIL-53 metal organic framework composite core-shell nanofibers for co-delivery of temozolomide and paclitaxel against glioblastoma cancer cells. Int. J. Pharm. 2020, 587, 119674. [Google Scholar] [CrossRef] [PubMed]
- Mehnath, S.; Chitra, K.; Karthikeyan, K.; Jeyaraj, M. Localized delivery of active targeting micelles from nanofibers patch for effective breast cancer therapy. Int. J. Pharm. 2020, 584, 119412. [Google Scholar] [CrossRef] [PubMed]
- Kotzabasaki, M.; Froudakis, G.E. Review of computer simulations on anti-cancer drug delivery in MOFs. Inorg. Chem. Front. 2018, 5, 1255–1272. [Google Scholar] [CrossRef]
- Giliopoulos, D.; Zamboulis, A.; Giannakoudakis, D.; Bikiaris, D.; Triantafyllidis, K. Polymer/metal organic framework (MOF) nanocomposites for biomedical applications. Molecules 2020, 25, 185. [Google Scholar] [CrossRef] [Green Version]
- Huxford, R.C.; Della Rocca, J.; Lin, W. Metal–organic frameworks as potential drug carriers. Curr. Opin. Chem. Biol. 2010, 14, 262–268. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Zheng, M.; Xie, Z. Nanoscale metal–Organic frameworks for drug delivery: a conventional platform with new promise. J. Mater. Chem. B 2018, 6, 707–717. [Google Scholar] [CrossRef]
- Sun, Y.; Zheng, L.; Yang, Y.; Qian, X.; Fu, T.; Li, X.; Yang, Z.; Yan, H.; Cui, C.; Tan, W. Metal–Organic Framework Nanocarriers for Drug Delivery in Biomedical Applications. Nano-Micro Lett. 2020, 12, 1–29. [Google Scholar] [CrossRef]
- Martínez, F.; Leo, P.; Orcajo, G.; Díaz-García, M.; Sanchez-Sanchez, M.; Calleja, G. Sustainable Fe-BTC catalyst for efficient removal of mehylene blue by advanced fenton oxidation. Catal. Today 2018, 313, 6–11. [Google Scholar] [CrossRef]
- Sun, D.T.; Gasilova, N.; Yang, S.; Oveisi, E.; Queen, W.L. Rapid, selective extraction of trace amounts of gold from complex water mixtures with a metal–organic framework (MOF)/polymer composite. J. Am. Chem. Soc. 2018, 140, 16697–16703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Li, N.; Xiao, T.; Tang, W.; Xiu, G. Removal of antimonite and antimonate from water using Fe-based metal-organic frameworks: The relationship between framework structure and adsorption performance. J. Environ. Sci. 2019, 86, 213–224. [Google Scholar] [CrossRef]
- Gascón, V.; Jiménez, M.B.; Blanco, R.M.; Sanchez-Sanchez, M. Semi-crystalline Fe-BTC MOF material as an efficient support for enzyme immobilization. Catal. Today 2018, 304, 119–126. [Google Scholar] [CrossRef] [Green Version]
- Nagarjun, N.; Dhakshinamoorthy, A. Aerobic Oxidation of Benzylic Hydrocarbons by Iron-Based Metal Organic Framework as Solid Heterogeneous Catalyst. ChemistrySelect 2018, 3, 12155–12162. [Google Scholar] [CrossRef]
- Adhikari, C.; Mishra, A.; Nayak, D.; Chakraborty, A. Metal organic frameworks modified mesoporous silica nanoparticles (MSN): A nano-composite system to inhibit uncontrolled chemotherapeutic drug delivery from bare-msn. J. Drug Deliv. Sci. Technol. 2018, 47, 1–11. [Google Scholar] [CrossRef]
- Nanaki, S.G.; Pantopoulos, K.; Bikiaris, D.N. Synthesis of biocompatible poly(e-caprolactone)- block-poly(propylene adipate) copolymers appropriate for drug nanoencapsulation in the form of core-shell nanoparticles. Int. J. Nanomed. 2011, 6, 2981–2995. [Google Scholar]
- Solomon, O.F.; Ciutǎ, I.Z. Détermination de la viscosité intrinsèque de solutions de polymères par une simple détermination de la viscosité. J. Appl. Polym. Sci. 1962, 6, 683–686. [Google Scholar] [CrossRef]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, e01493. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1 − 86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Hidalgo, T.; Giménez-Marqués, M.; Bellido, E.; Avila, J.; Asensio, M.C.; Salles, F.; Lozano, M.V.; Guillevic, M.; Simón-Vázquez, R.; González-Fernández, A. Chitosan-coated mesoporous MIL-100 (Fe) nanoparticles as improved bio-compatible oral nanocarriers. Sci. Rep. 2017, 7, 43099. [Google Scholar] [CrossRef]
- Vitillo, J.G.; Bhan, A.; Cramer, C.J.; Lu, C.C.; Gagliardi, L. Quantum Chemical Characterization of Structural Single Fe (II) Sites in MIL-Type Metal–Organic Frameworks for the Oxidation of Methane to Methanol and Ethane to Ethanol. ACS Catal. 2019, 9, 2870–2879. [Google Scholar] [CrossRef]
- Mali, G.; Mazaj, M.; Arcon, I.; Hanzel, D.; Arcon, D.; Jaglicić, Z. Unraveling the Arrangement of Al and Fe within the Framework Explains the Magnetism of Mixed-Metal MIL-100 (Al, Fe). J. Phys. Chem. Lett. 2019, 10, 1464–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spicher, S.; Grimme, S. Robust Atomistic Modeling of Materials, Organometallic, and Biochemical Systems. Angew. Chem. 2020, 132, 15795–15803. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Johnson, E.R.; Becke, A.D. A post-Hartree–Fock model of intermolecular interactions. J. Chem. Phys. 2005, 123, 24101. [Google Scholar] [CrossRef] [PubMed]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hyun, H.; Park, S.H.; Kwon, D.Y.; Khang, G.; Lee, H.B.; Kim, M.S. Thermo-responsive injectable MPEG-polyester diblock copolymers for sustained drug release. Polymers (Basel) 2014, 6, 2670–2683. [Google Scholar] [CrossRef] [Green Version]
- Koulouktsi, C.; Nanaki, S.; Barmpalexis, P.; Kostoglou, M. Bikiaris, D. Preparation and characterization of Alendronate depot microspheres based on novel poly (-ε-caprolactone )/Vitamin E TPGS copolymers. Int. J. Pharm. X 2019, 1, 100014. [Google Scholar]
- Al Haydar, M.; Abid, H.; Sunderland, V.; Wang, S. Multimetal organic frameworks as drug carriers: Aceclofenac as a drug candidate. Drug Des. Dev. Ther. 2018, 13, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Horcajada, P.; Serre, C.; Maurin, G.; Ramsahye, N.; Balas, F.; Vallet-Regí, M.; Sebban, M.; Taulelle, F.; Férey, G. Flexible Porous Metal-Organic Frameworks for a Controlled Drug Delivery. J. Am. Chem. Soc. 2008, 130, 6774–6780. [Google Scholar] [CrossRef]
- Vu, H.T.; Nguyen, M.B.; Vu, T.M.; Le, G.H.; Pham, T.T.T.; Nguyen, T.D.; Vu, T.A. Synthesis and Application of Novel Nano Fe-BTC/GO Composites as Highly Efficient Photocatalysts in the Dye Degradation. Top. Catal. 2020, 63, 1046–1055. [Google Scholar] [CrossRef]
- Akarçay, Ö.; Kurtoğlu, S.F.; Uzun, A. Ammonia decomposition on a highly-dispersed carbon-embedded iron catalyst derived from Fe-BTC: Stable and high performance at relatively low temperatures. Int. J. Hydrogen Energy 2020, 45, 28664–28681. [Google Scholar] [CrossRef]
- Sanchez-Sanchez, M.; de Asua, I.; Ruano, D.; Diaz, K. Direct synthesis, structural features, and enhanced catalytic activity of the basolite F300-like semiamorphous Fe-BTC framework. Cryst. Growth Des. 2015, 15, 4498–4506. [Google Scholar] [CrossRef]
- Matzger, A.; Suresh, D.K. Enhanced Drug Delivery by Dissolution of Amorphous Drug Encapsulated in a Water Unstable MOF. Angew. Chem. 2019, 131. [Google Scholar] [CrossRef]
- Nanaki, S.G.; Spyrou, K.; Bekiari, C.; Veneti, P.; Baroud, T.N.; Karouta, N.; Grivas, I.; Papadopoulos, G.C.; Gournis, D.; Bikiaris, D.N. Hierarchical Porous Carbon—PLLA and PLGA Hybrid Nanoparticles for Intranasal Delivery of Galantamine for Alzheimer’s Disease Therapy. Pharmaceutics 2020, 12, 227. [Google Scholar] [CrossRef] [Green Version]
- Danafar, H. Synthesis and characterization of mpeg-PCL copolymers as a polymersomes for delivery of enalapril as a model hydrophilic drug. Iran. J. Pharm. Sci. 2018, 14, 25–38. [Google Scholar]
- Behl, A.; Parmar, V.; Malhotra, S.; Chhillar, A. Biodegradable diblock copolymeric PEG-PCLnanoparticles: Synthesis, characterization and applications as anticancer drug delivery agents. Polymers (Guildf) 2020, 122901. [Google Scholar] [CrossRef]
- Horcajada, P.; Chalati, T.; Serre, C.; Gillet, B.; Sébrié, C.; Baati, T.; Eubank, J.; Heurtaux, D.; Clayette, P.; Kreuz, C.; et al. Porous metal-organic-framework nanoscale carriers as a potential platform for drug delivery and imaging. Nat. Mater. 2009, 9, 172–178. [Google Scholar] [CrossRef]
- DeStefano, V.; Khan, S.; Tabada, A. Applications of PLA in modern medicine. Eng. Regen. 2020, 1, 76–87. [Google Scholar] [CrossRef]
- Liu, S.; Qin, S.; He, M.; Zhou, D.; Qin, Q.; Wang, H. Current applications of poly(lactic acid) composites in tissue engineering and drug delivery. Compos. Part B Eng. 2020, 199, 108238. [Google Scholar] [CrossRef]
- Pawar, R.; Tekale, S.; Shisodia, S.; Totre, J.; Domb, A. Biomedical Applications of Poly(Lactic Acid). Rec. Pat. Regen. Med. 2014, 4, 40–51. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Bikiaris, D.; Avgoustaκis, K. Microwave-induced enhancement of the dissolution rate of poorly water-soluble Tibolone from poly(ethylene glycol) solid dispersions. J. Appl. Polym. Sci. 2008, 108, 1249–1258. [Google Scholar] [CrossRef]
- Nivetha, R.; Gothandapani, K.; Raghavan, V.; Jacob, G.; Sellappan, R.; Bhardwaj, P.; Pitchaimuthu, S.; Kannan, A.N.M.; Jeong, S.K.; Grace, A.N. Highly Porous MIL-100(Fe) for the Hydrogen Evolution Reaction (HER) in Acidic and Basic Media. ACS Omega 2020, 5, 18941–18949. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.A.; Anggraeni, E.; Soetaredjo, F.E.; Santoso, S.P.; Irawaty, W.; Thanh, T.C.; Hartono, S.B.; Yuliana, M.; Ismadji, S. Hydrothermal Synthesize of HF-Free MIL-100(Fe) for Isoniazid-Drug Delivery. Sci. Rep. 2019, 9, 16907. [Google Scholar] [CrossRef] [PubMed]
- Oveisi, A.; Khorramabadi-zad, A.; Daliran, S. Iron-based metal-organic framework, Fe(BTC): An effective dual-functional catalyst for oxidative cyclization of bisnaphthols and tandem synthesis of quinazolin-4(3H)-ones. RSC Adv. 2015, 6, 1136–1142. [Google Scholar] [CrossRef]
- Filippousi, M.; Papadimitriou, S.; Bikiaris, D.; Pavlidou, E.; Angelakeris, M.; Zamboulis, D.; Tian, H.; van Tendeloo, G. Novel core–shell magnetic nanoparticles for Taxol encapsulation in biodegradable and biocompatible block copolymers: Preparation, characterization and release properties. Int. J. Pharm. 2013, 448, 221–230. [Google Scholar] [CrossRef]
- Dordunoo, S.; Burt, H. Solubility and stability of taxol: Effects of buffers and cyclodextrins. Int. J. Pharm. 1996, 133, 191–201. [Google Scholar] [CrossRef]
- Haydar, M.; Sunderland, V.; Abid, H.; Wang, S. Metal organic frameworks as a drug delivery system for flurbiprofen. Drug Des. Dev. Ther. 2017, 11, 2685–2695. [Google Scholar] [CrossRef] [Green Version]
- Papadimitriou, S.; Bikiaris, D. Dissolution rate enhancement of the poorly water-soluble drug Tibolone using PVP, SiO 2, and their nanocomposites as appropriate drug carriers. Drug Dev. Ind. Pharm. 2009, 35, 1128–1138. [Google Scholar] [CrossRef]
- Friedrich, H.; Fussnegger, B.; Kolter, K.; Bodmeier, R. Dissolution rate improvement of poorly water-soluble drugs obtained by adsorbing solutions of drugs in hydrophilic solvents onto high surface area carriers. Eur. J. Pharm. Biopharm. 2006, 62, 171–177. [Google Scholar] [CrossRef]
- Takeuchi, H.; Nagira, S.; Yamamoto, H.; Kawashima, Y. Solid dispersion particles of tolbutamide prepared with fine silica particles by the spray-drying method. Powder Technol. 2004, 141, 187–195. [Google Scholar] [CrossRef]
- Papadimitriou, S.; Bikiaris, D. Novel self-assembled core-shell nanoparticles based on crystalline amorphous moieties of aliphatic copolyesters for efficient controlled drug release. J. Control. Release 2009, 138, 177–184. [Google Scholar] [CrossRef]
- Vasileiou, A.; Papadimitriou, S.; Bikiaris, D.; Matthaiolampakis, G.; Avgoustaκis, K. Facile synthesis of polyester-PEG triblock copolymers and preparation of amphiphilic nanoparticles as drug carriers. J. Control. Release 2010, 148, 388–395. [Google Scholar] [CrossRef]
- Chu, B.; Zhang, L.; Qu, Y.; Chen, X.; Peng, J.; Huang, Y.; Qian, Z. Synthesis, characterization and drug loading property of Monomethoxy-Poly(ethylene glycol)-Poly(ε-caprolactone)-Poly(D,L-lactide) (MPEG-PCLA) copolymers. Sci. Rep. 2016, 6, 34069. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, A.; Jana, U.; Manna, P.; Mohanta, G. Synthesis and evaluation of MePEG-PCL diblock copolymers: Surface properties and controlled release behavior. Prog. Biomater. 2015, 4, 89–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bikiaris, D.N. Solid dispersions, Part I: Recent evolutions and future opportunities in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1501–1519. [Google Scholar] [CrossRef] [PubMed]
- Bikiaris, D.N. Solid dispersions, Part II: New strategies in manufacturing methods for dissolution rate enhancement of poorly water-soluble drugs. Expert Opin. Drug Deliv. 2011, 8, 1663–1680. [Google Scholar] [CrossRef] [PubMed]
- Kocbek, P.; Baumgartner, S.; Kristl, J. Preparation and evaluation of nanosuspensions for enhancing the dissolution of poorly soluble drugs. Int. J. Pharm. 2006, 312, 179–186. [Google Scholar] [CrossRef]
- Crank, J. The Mathematics of Diffusion; Oxford University Press: Oxford, UK, 1975. [Google Scholar]
- Tien, C. Adsorption Calculations and Modeling; Butterworth-Heinemann: Boston, MA, USA, 1994; ISBN 978-0-7506-9121-5. [Google Scholar]
- Korsmeyer, R.W.; Gumy, R.; Doelker, E.; Buri, P.; Peppas, N.A. Mechanisms of solute release from porous hydrophilic polymers. Int. J. Pharm. 1983, 15, 25–35. [Google Scholar] [CrossRef]
- Stobiecka, M.; Jakiela, S.; Chalupa, A.; Bednarczyk, P. Biosensors and Bioelectronics Mitochondria—Based biosensors with piezometric and RELS transduction for potassium uptake and release investigations. Biosens. Bioelectron. 2016, 84, 1–8. [Google Scholar]
- Ratajczak, K.; Stobiecka, M. Ternary Interactions and Energy Transfer between Fluorescein Isothiocyanate, Adenosine Triphosphate, and Graphene Oxide Nanocarriers. J. Phys. Chem. B 2017, 121, 6822–6830. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. Software update: The ORCA program system, version 4.0. Wires Comput. Mol. Sci. 2018, 8, e1327. [Google Scholar] [CrossRef]





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | SSA a (m2/g) | Total Pore Volume b (cc/g) | Micropore Volume c (cc/g) | Micropore Area (m2/g) | Average Pore Diameter d (nm) |
|---|---|---|---|---|---|
| Fe-BTC | 1360 | 0.798 | 0.257 | 658.71 | 1.2 |
| Fe-BTC-PTX | 826 | 0.428 | 0.195 | 470.74 | 1.2 |
| Sample (Nanoparticles) | Average Size (nm) | Polydispersity Index (PDI) | Z-Potential (mV) |
|---|---|---|---|
| nano mPEG-PCL-PTX | 138 ± 0.54 | 0.24 ± 0.06 | −29.58 ± 0.04 |
| nano mPEG-PCL-Fe-BTX-PTX | 143 ± 0.59 | 0.23 ± 0.07 | −32.49 ± 0.08 |
| Sample | Nanoparticles Yield (%) | Drug Loading (%) | Entrapment Efficiency (%) |
|---|---|---|---|
| mPEG-PCL-PTX | 70.22 ± 0.09 | 7.48 ± 0.17 | 74.79 ± 0.07 |
| mPEG-PCL-Fe-BTX-PTX | 64.79 ± 0.15 | 3.92 ± 0.16 | 10.00 ± 0.03 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bikiaris, N.D.; Ainali, N.M.; Christodoulou, E.; Kostoglou, M.; Kehagias, T.; Papasouli, E.; Koukaras, E.N.; Nanaki, S.G. Dissolution Enhancement and Controlled Release of Paclitaxel Drug via a Hybrid Nanocarrier Based on mPEG-PCL Amphiphilic Copolymer and Fe-BTC Porous Metal-Organic Framework. Nanomaterials 2020, 10, 2490. https://doi.org/10.3390/nano10122490
Bikiaris ND, Ainali NM, Christodoulou E, Kostoglou M, Kehagias T, Papasouli E, Koukaras EN, Nanaki SG. Dissolution Enhancement and Controlled Release of Paclitaxel Drug via a Hybrid Nanocarrier Based on mPEG-PCL Amphiphilic Copolymer and Fe-BTC Porous Metal-Organic Framework. Nanomaterials. 2020; 10(12):2490. https://doi.org/10.3390/nano10122490
Chicago/Turabian StyleBikiaris, Nikolaos D., Nina Maria Ainali, Evi Christodoulou, Margaritis Kostoglou, Thomas Kehagias, Emilia Papasouli, Emmanuel N. Koukaras, and Stavroula G. Nanaki. 2020. "Dissolution Enhancement and Controlled Release of Paclitaxel Drug via a Hybrid Nanocarrier Based on mPEG-PCL Amphiphilic Copolymer and Fe-BTC Porous Metal-Organic Framework" Nanomaterials 10, no. 12: 2490. https://doi.org/10.3390/nano10122490
APA StyleBikiaris, N. D., Ainali, N. M., Christodoulou, E., Kostoglou, M., Kehagias, T., Papasouli, E., Koukaras, E. N., & Nanaki, S. G. (2020). Dissolution Enhancement and Controlled Release of Paclitaxel Drug via a Hybrid Nanocarrier Based on mPEG-PCL Amphiphilic Copolymer and Fe-BTC Porous Metal-Organic Framework. Nanomaterials, 10(12), 2490. https://doi.org/10.3390/nano10122490