Exfoliation Energy as a Descriptor of MXenes Synthesizability and Surface Chemical Activity




Abstract
:1. Introduction
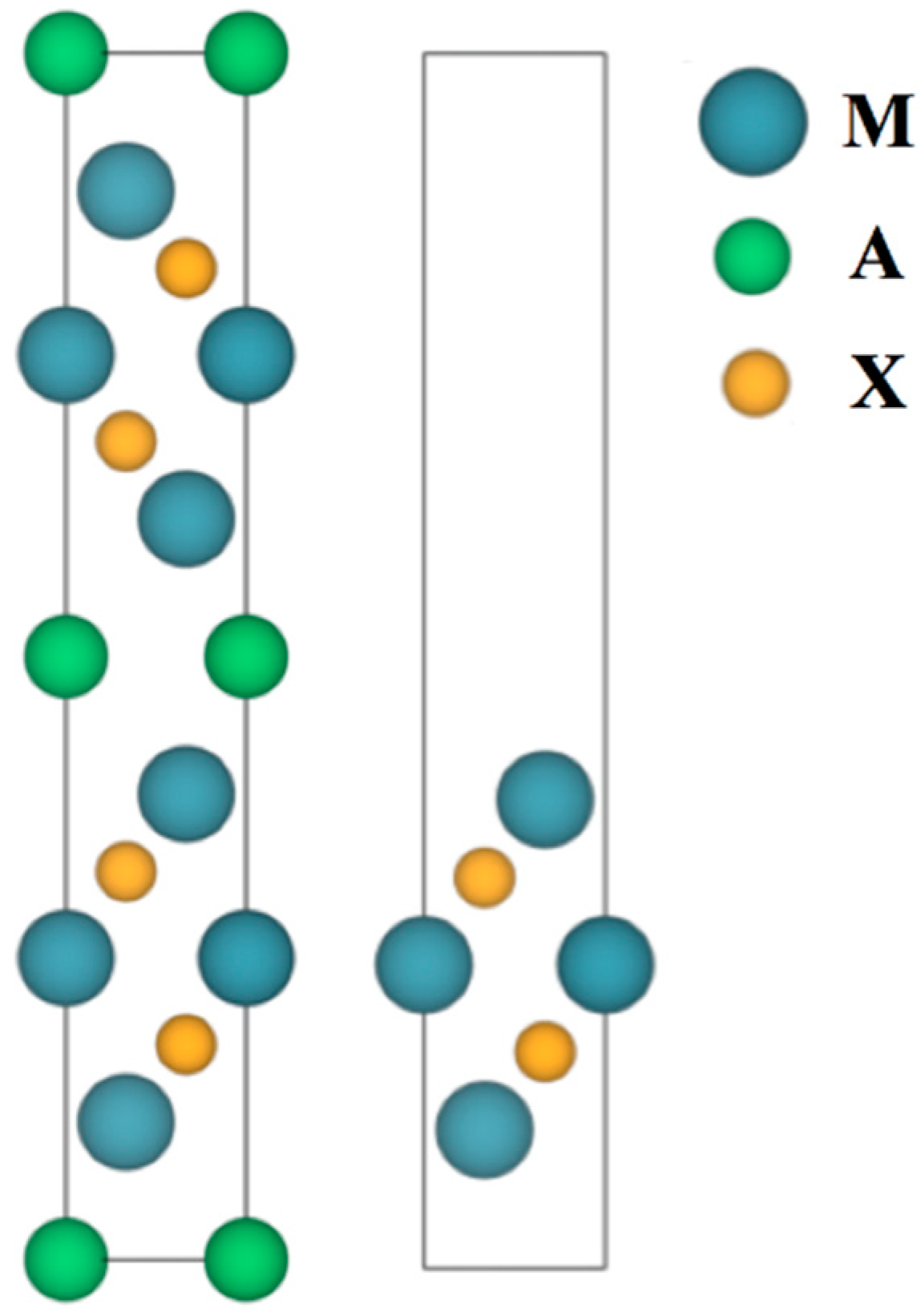
2. Computational Details
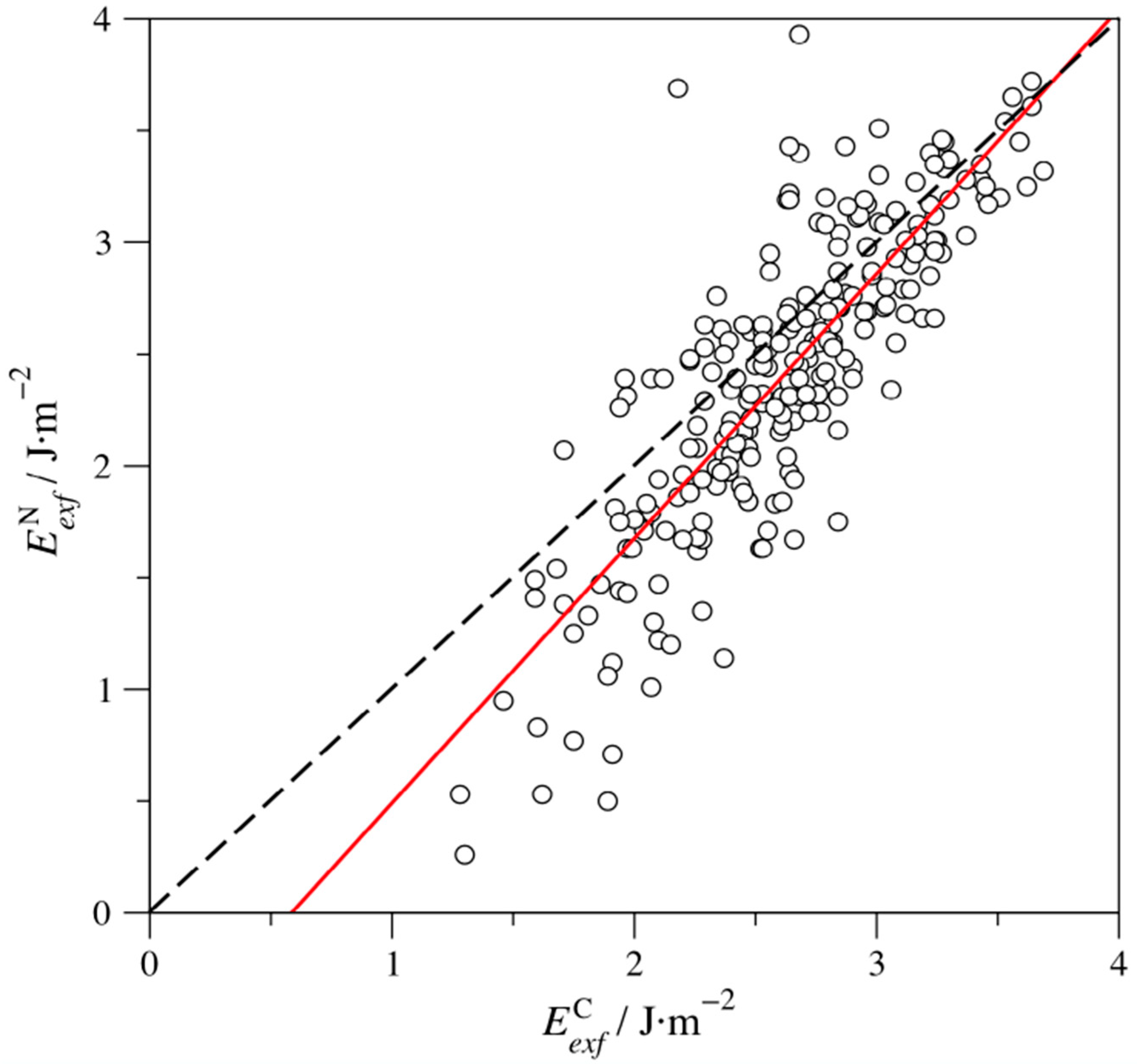
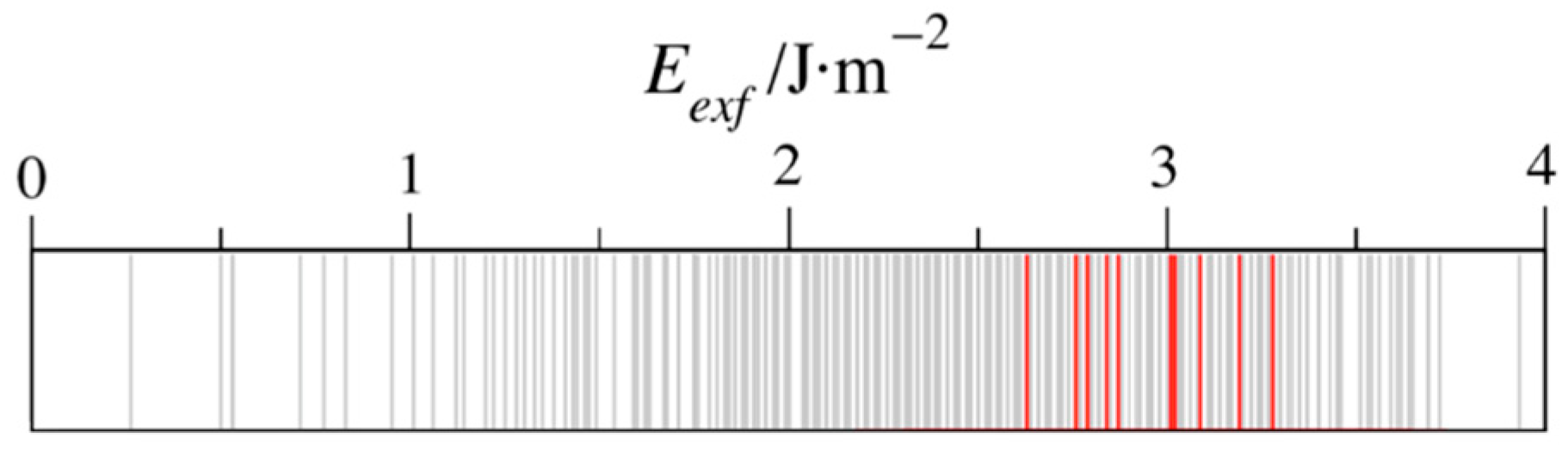
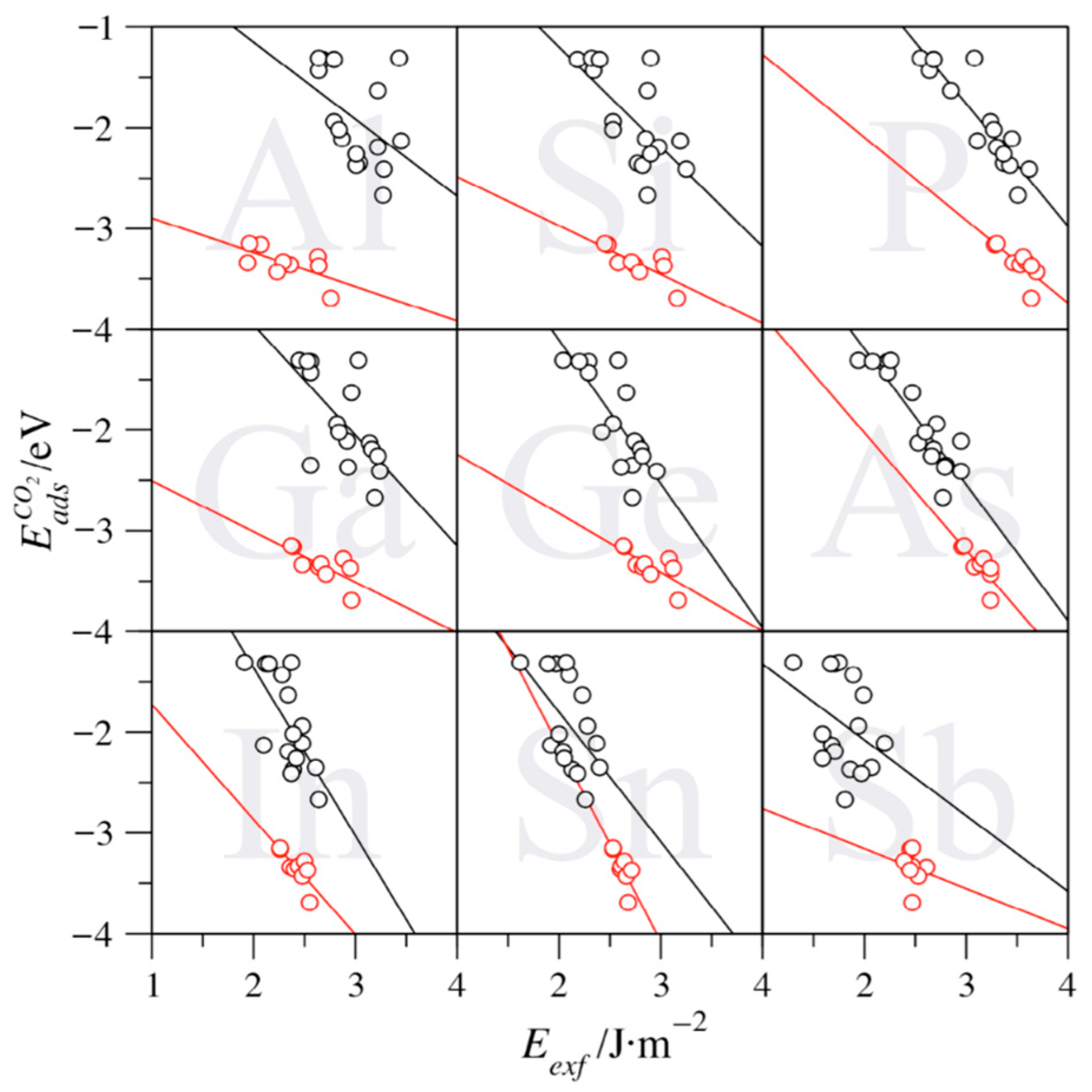
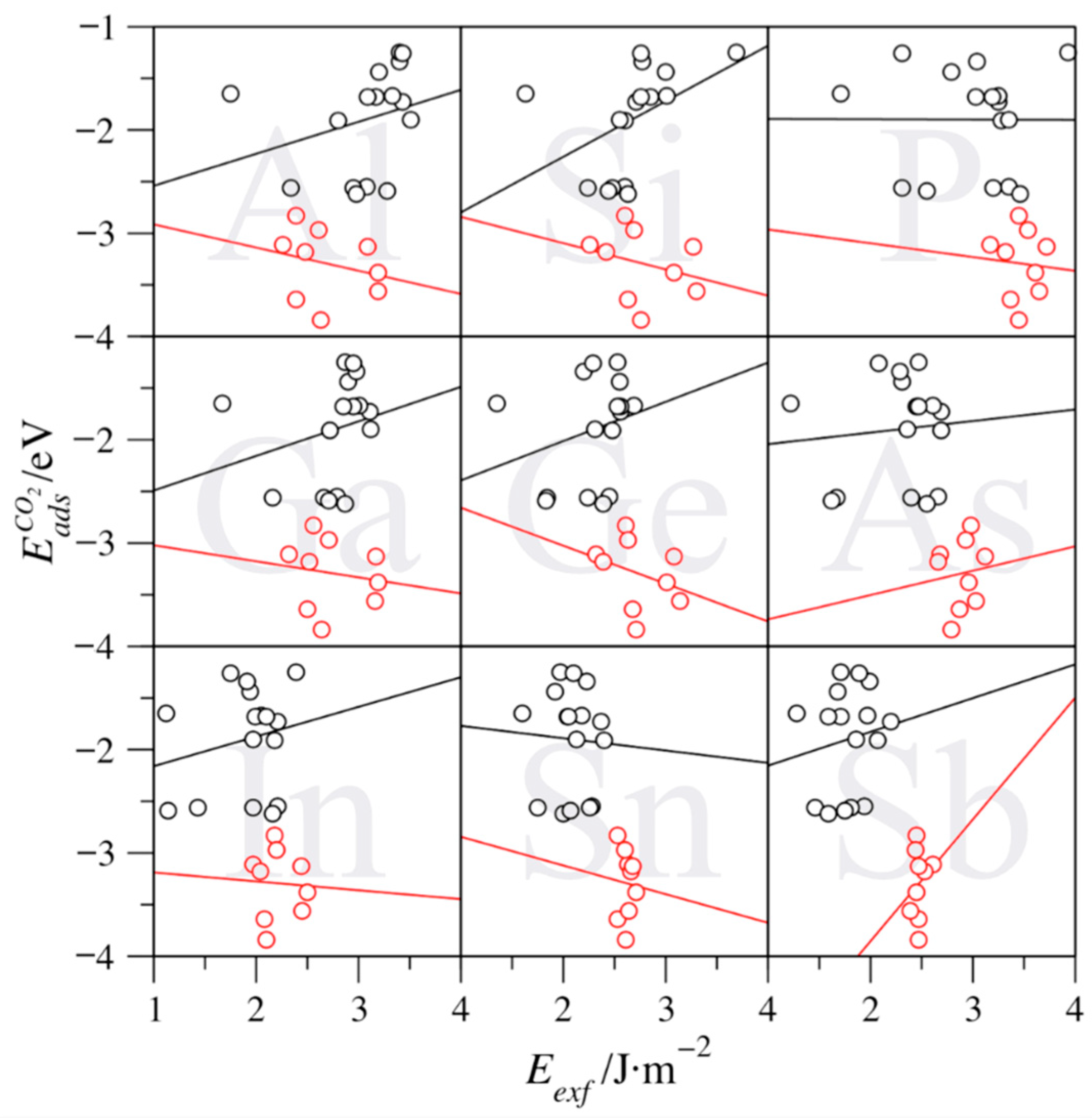
3. Results
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Naguib, M.; Kurtoglu, M.; Presser, V.; Lu, J.; Niu, J.; Heon, M.; Hultman, L.; Gogotsi, Y.; Barsoum, M.W. Two-Dimensional Nanocrystals: Two-Dimensional Nanocrystals Produced by Exfoliation of Ti3AlC2. Adv. Mater. 2011, 23, 4248–4253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lukatskaya, M.R.; Mashtalir, O.; Ren, C.E.; Dall’Agnese, Y.; Rozier, P.; Taberna, P.L.; Naguib, M.; Simon, P.; Barsoum, M.W.; Gogotsi, Y. Cation Intercalation and High Volumetric Capacitance of Two-Dimensional Titanium Carbide. Science 2013, 341, 1502–1505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jing, Y.; Zhou, Z.; Cabrera, C.R.; Chen, Z. Graphene, Inorganic Graphene Analogs and their Composites for Lithium Ion Batteries. J. Mater. Chem. A 2014, 2, 12104–12122. [Google Scholar] [CrossRef]
- Wu, N.; Bai, X.; Pan, D.; Dong, B.; Wei, R.; Naik, N.; Patil, R.R.; Guo, Z. Recent Advances of Asymmetric Supercapacitors. Adv. Mater. Interfaces 2020, 2001710. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Wang, C.; Liu, H.; Zhang, J.; Lin, J.; Fan, J.; Ding, T.; Ryu, J.E.; Guo, Z. Significantly Enhanced Ultrathin NiCo-based MOF Nanosheet Electrodes Hybrided with Ti3C2Tx MXene for High Performance Asymmetric Supercapacitor. Eng. Sci. 2020, 9, 50–59. [Google Scholar] [CrossRef]
- Shahzad, F.; Alhalbeb, M.; Hatter, C.B.; Anasori, B.; Hong, S.M.; Koo, C.M.; Gogotsi, Y. Electromagnetic interference shielding with 2D transition metal carbides (MXenes). Science 2016, 353, 1137–1140. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Zhang, H.B.; Sun, R.H.; Liu, Y.F.; Liu, Z.S.; Zhou, A.G.; Yu, Z.Z. Hydrophobic, Flexible, and Lightweight MXene Foams for High-Performance Electromagnetic-Interference Shielding. Adv. Mater. 2017, 29, 1702367. [Google Scholar] [CrossRef]
- Zhao, B.; Deng, J.; Zhang, R.; Liang, L.; Fan, B.; Bai, Z.; Shao, G.; Park, C.B. Recent Advances on the Electromagnetic Wave Absorption Properties of Ni Based Materials. Eng. Sci. 2018, 3, 5–40. [Google Scholar] [CrossRef] [Green Version]
- Azofra, L.M.; Li, N.; MacFarlane, D.R.; Sun, C. Promising prospects for 2D d2-d4 M3C2 transition metal carbides (MXenes) in N2 capture and conversion into ammonia. Energy Environ. Sci. 2016, 9, 2545–2549. [Google Scholar] [CrossRef]
- Gouveia, J.D.; Morales-García, Á.; Viñes, F.; Gomes, J.R.B.; Illas, F. Facile Heterogeneously Catalyzed Nitrogen Fixation by MXenes. ACS Catal. 2020, 10, 5049–5056. [Google Scholar] [CrossRef]
- Morales-García, A.; Calle-Vallejo, F.; Illas, F. MXenes: New Horizons in Catalysis. ACS Catal. 2020, 10, 13487–13503. [Google Scholar] [CrossRef]
- Rakhi, R.B.; Nayak, P.; Xia, C.; Alshareef, H.N. Novel amperometric glucose biosensor based on MXene nanocomposites. Sci. Rep. 2016, 6, 36422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouveia, J.D.; Novell-Leruth, G.; Reis, P.M.L.S.; Viñes, F.; Illas, F.; Gomes, J.R.B. First-Principles Calculations on the Adsorption Behavior of Amino Acids on a Titanium Carbide MXene. ACS Appl. Bio Mater. 2020, 3, 5913–5921. [Google Scholar] [CrossRef]
- Fu, Y.; Pei, X.; Dai, Y.; Mo, D.; Lyu, S. Three-Dimensional Graphene-Like Carbon Prepared from CO2 as Anode Material for High-Performance Lithium-Ion Batteries. ES Energy Environ. 2019, 4, 66–73. [Google Scholar]
- Li, N.; Zhang, F.; Hou, H.W.S. Catalytic Degradation of 4-Nitrophenol in Polluted Water by Three-Dimensional Gold Nanoparticles/Reduced Graphene Oxide Microspheres. Eng. Sci. 2019, 7, 72–79. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, S.; Liu, X.; Wang, H.; Wang, H.; Chen, Y.; Ni, Y. XTe (X = Ge, Sn, Pb) Monolayers: Promising Thermoelectric Materials with Ultralow Lattice Thermal Conductivity and High-power Factor. ES Energy Environ. 2020, 10, 59–65. [Google Scholar]
- Morales-García, Á.; Fernández-Fernández, A.; Viñes, F.; Illas, F. CO2 Abatement Using Two-Dimensional MXene Carbides. J. Mater. Chem. A 2018, 6, 3381–3385. [Google Scholar] [CrossRef]
- Persson, I.; Halim, J.; Lind, H.; Hansen, T.W.; Wagner, J.B.; Näslund, L.-Å.; Darakchieva, V.; Palisaitis, J.; Rosen, J.; Persson, P.O.Å. 2D Transition Metal Carbides (MXenes) for Carbon Capture. Adv. Mater. 2019, 31, 1805472. [Google Scholar] [CrossRef]
- Morales-García, Á.; Mayans-Llorach, M.; Viñes, F.; Illas, F. Thickness Biased Capture of CO2 on Carbide MXenes. Phys. Chem. Chem. Phys. 2019, 21, 23136–23142. [Google Scholar] [CrossRef]
- Hwu, H.H.; Chen, J.G. Surface Chemistry of Transition Metal Carbides. Chem. Rev. 2005, 105, 185–212. [Google Scholar] [CrossRef]
- Quesne, M.G.; Roldán, A.; de Leeuw, N.H.; Catlow, C.R.A. Bulk and surface properties of metal carbides: Implications for catalysis. Phys. Chem. Chem. Phys. 2018, 20, 6905–6916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, H.; Tkalych, A.J.; Carter, E.A. Surface Energy as a Descriptor of Catalytic Activity. J. Phys. Chem. C 2016, 120, 23698–23706. [Google Scholar] [CrossRef]
- Vega, L.; Martínez, B.; Viñes, F.; Illas, F. Robustness of surface activity electronic structure-based descriptors of transition metals. Phys. Chem. Chem. Phys. 2018, 20, 20548–20554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, B.; Nørskov, J.K. Electronic factors determining the reactivity of metal surfaces. Surf. Sci. 1995, 343, 211–220. [Google Scholar] [CrossRef]
- Jin, D.; Johnson, L.R.; Raman, A.S.; Ming, X.; Gao, Y.; Du, F.; Wei, Y.; Chen, G.; Vojvodic, A.; Gogotsi, Y.; et al. Computational Screening of 2D Ordered Double Transition-Metal Carbides (MXenes) as Electrocatalysts for Hydrogen Evolution Reaction. J. Phys. Chem. C 2020, 124, 10584–10592. [Google Scholar] [CrossRef]
- Deysher, G.; Shuck, C.E.; Hantanasirisakul, K.; Frey, N.C.; Foucher, A.C.; Maleski, K.; Sarycheva, A.; Shenoy, V.B.; Stach, E.A.; Anasori, B.; et al. Synthesis of Mo4VAlC4 MAX phase and two-dimensional Mo4VC4 MXene with five atomic layers of transition metals. ACS Nano 2020, 14, 204–217. [Google Scholar] [CrossRef]
- Khazaei, M.; Ranjbar, A.; Esfarjani, K.; Bogdanovski, D.; Dronskowski, R.; Yunoki, S. Insights into Exfoliation Possibility of MAX Phases to MXenes. Phys. Chem. Chem. Phys. 2018, 20, 8579–8592. [Google Scholar] [CrossRef] [Green Version]
- Naguib, M.; Gogotsi, Y. Synthesis of two-dimensional materials by selective extraction. Acc. Chem. Res. 2015, 48, 128–135. [Google Scholar] [CrossRef]
- Naguib, M.; Mochalin, V.N.; Barsoum, M.W.; Gogotsi, Y. 25th Anniversary Article: MXenes: A new family of two-dimensional materials. Adv. Mater. 2014, 26, 992–1005. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, J. Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Politi, J.d.R.S.; Viñes, F.; Rodriguez, J.A.; Illas, F. Atomic and Electronic Structure of Molybdenum Carbide Phases: Bulk and Low Miller-Index Surfaces. Phys. Chem. Chem. Phys. 2013, 145, 12617–12625. [Google Scholar] [CrossRef] [PubMed]
- Blöchl, P.E. Projector Augmented-Wave Method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.J.; Pack, J.D. Special Points for Brillouin-Zone Integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Jain, A.; Ong, S.P.; Hautier, G.; Chen, W.; Richards, W.D.; Dacek, S.; Cholia, S.; Gunter, D.; Skinner, D.; Ceder, G.; et al. The Materials Project: A materials genome approach to accelerating materials innovation. APL Mater. 2013, 1, 011002. [Google Scholar] [CrossRef] [Green Version]
- Gouveia, J.D.; Viñes, F.; Illas, F.; Gomes, J.R.B. MXenes atomic layer stacking phase transitions and their chemical activity consequences. Phys. Rev. Mat. 2020, 4, 054003. [Google Scholar] [CrossRef]
- Verger, L.; Xu, C.; Natu, V.; Cheng, H.-M.; Ren, W.; Barsoum, M.W. Overview of the Synthesis of MXenes and Other Ultrathin 2D Transition Metal Carbides and Nitrides. Curr. Opin. Solid State Mater. Sci. 2019, 23, 149–163. [Google Scholar] [CrossRef]
- Morales-Salvador, R.; Morales-García, Á.; Viñes, F.; Illas, F. Two-Dimensional Nitrides as Highly Efficient Potential Candidates for CO2 Capture and Activation. Phys. Chem. Chem. Phys. 2018, 20, 17117–17124. [Google Scholar] [CrossRef] [Green Version]
- Prats, H.; McAloone, H.; Viñes, F.; Illas, F. Ultra-high selectivity biogas upgrading through porous MXenes. J. Mater. Chem. A 2020, 8, 12296–12300. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, S. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Kamysbayev, V.; Filatov, A.S.; Hu, H.; Rui, X.; Lagunas, F.; Wang, D.; Klie, R.F.; Talapin, D.V. Covalent surface modifications and superconductivity of two-dimensional metal carbide MXenes. Science 2020, 369, 979–983. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
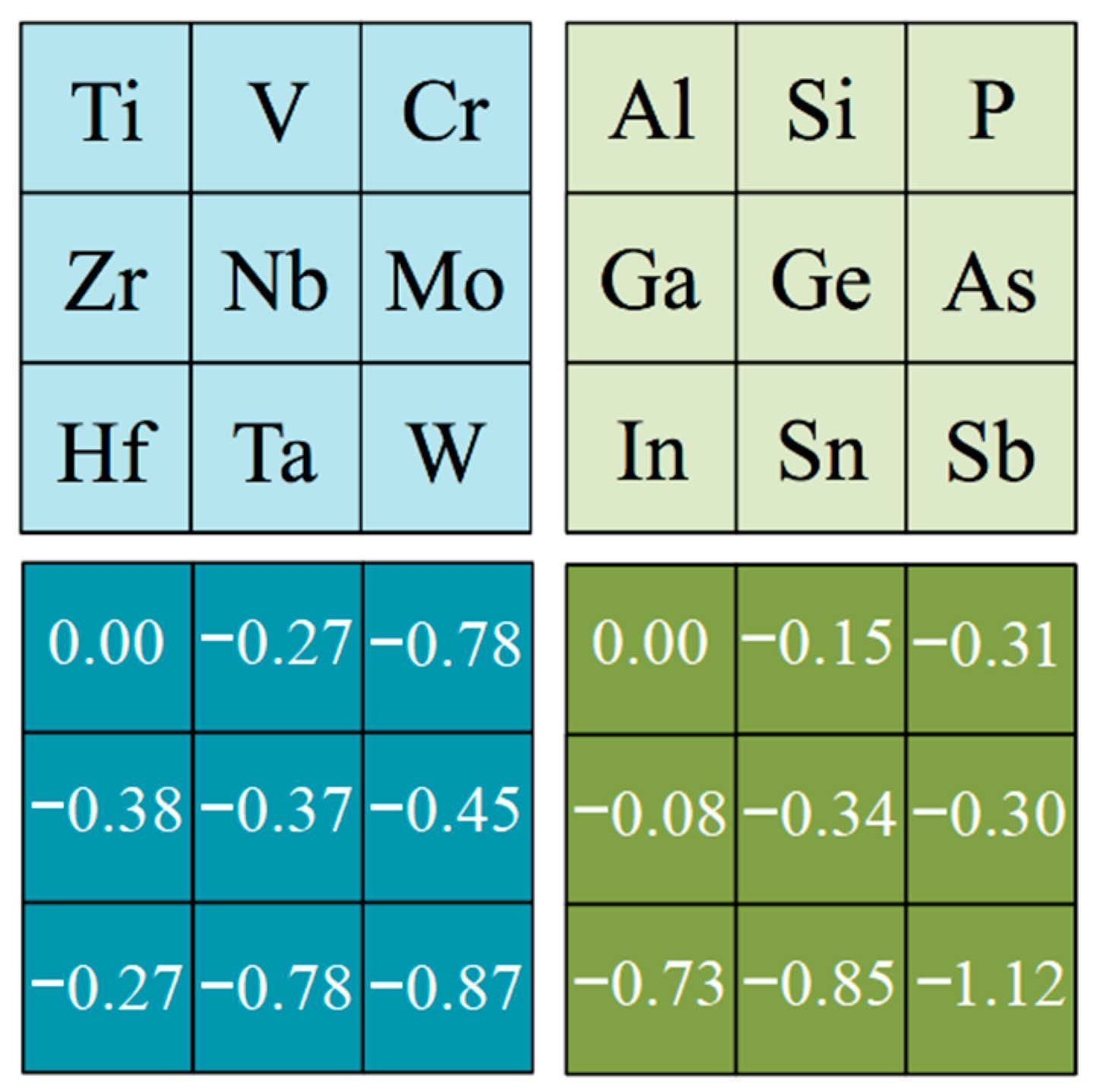
| Eexf M2AX/J·m−2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| X | M/A | Al | Si | P | Ga | Ge | As | In | Sn | Sb | |
| C | d2 | Ti | 2.76 | 3.16 | 3.64 | 2.96 | 3.17 | 3.24 | 2.55 | 2.68 | 2.47 |
| Zr | 2.07 | 2.48 | 3.28 | 2.39 | 2.64 | 2.96 | 2.26 | 2.53 | 2.45 | ||
| Hf | 2.36 | 2.74 | 3.53 | 2.64 | 2.82 | 3.08 | 2.40 | 2.60 | 2.44 | ||
| d3 | V | 3.28 | 3.25 | 3.62 | 3.24 | 2.96 | 2.95 | 2.37 | 2.18 | 1.97 | |
| Nb | 2.87 | 2.85 | 3.45 | 2.92 | 2.74 | 2.95 | 2.48 | 2.37 | 2.20 | ||
| Ta | 3.01 | 2.82 | 3.43 | 2.93 | 2.61 | 2.79 | 2.39 | 2.13 | 1.86 | ||
| d4 | Cr | 3.45 | 3.19 | 3.11 | 3.14 | 2.76 | 2.53 | 2.10 | 1.92 | 1.68 | |
| Mo | 3.22 | 2.87 | 2.85 | 2.96 | 2.66 | 2.47 | 2.34 | 2.23 | 1.99 | ||
| W | 3.43 | 2.90 | 3.08 | 3.03 | 2.58 | 2.26 | 2.37 | 2.07 | 1.75 | ||
| N | d2 | Ti | 3.09 | 3.27 | 3.72 | 3.17 | 3.08 | 3.12 | 2.44 | 2.31 | 2.16 |
| Zr | 2.39 | 2.60 | 3.45 | 2.56 | 2.61 | 2.98 | 2.18 | 2.28 | 2.15 | ||
| Hf | 2.61 | 2.69 | 3.54 | 2.71 | 2.63 | 2.93 | 2.20 | 2.15 | 1.91 | ||
| d3 | V | 3.33 | 3.01 | 3.25 | 3.01 | 2.69 | 2.61 | 2.05 | 1.86 | 1.63 | |
| Nb | 3.43 | 2.71 | 3.25 | 3.11 | 2.56 | 2.69 | 2.21 | 2.12 | 1.96 | ||
| Ta | 3.51 | 2.55 | 3.35 | 3.12 | 2.31 | 2.36 | 1.97 | 1.71 | 1.47 | ||
| d4 | Cr | 3.20 | 3.00 | 2.79 | 2.90 | 2.55 | 2.31 | 1.94 | 1.81 | 1.54 | |
| Mo | 3.40 | 2.77 | 3.04 | 2.98 | 2.20 | 2.29 | 1.91 | 1.88 | 1.63 | ||
| W | 3.28 | 2.44 | 2.55 | 2.71 | 1.83 | 1.62 | 1.14 | 1.01 | 0.77 | ||
| Eexf M3AX2/J·m−2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| X | M/A | Al | Si | P | Ga | Ge | As | In | Sn | Sb | |
| C | d2 | Ti | 2.63 | 3.01 | 3.56 | 2.88 | 3.08 | 3.17 | 2.50 | 2.64 | 2.39 |
| Zr | 1.96 | 2.45 | 3.30 | 2.37 | 2.63 | 2.98 | 2.26 | 2.53 | 2.47 | ||
| Hf | 2.29 | 2.71 | 3.59 | 2.66 | 2.84 | 3.14 | 2.44 | 2.61 | 2.47 | ||
| d3 | V | 3.22 | 2.98 | 3.30 | 3.16 | 2.80 | 2.68 | 2.34 | 2.04 | 1.71 | |
| Nb | 3.04 | 2.77 | 3.37 | 3.04 | 2.72 | 2.80 | 2.61 | 2.40 | 2.07 | ||
| Ta | 3.27 | 2.87 | 3.51 | 3.19 | 2.72 | 2.77 | 2.64 | 2.26 | 1.81 | ||
| d4 | Cr | 3.06 | 2.77 | 2.84 | 2.84 | 2.47 | 2.28 | 1.97 | 1.75 | 1.46 | |
| Mo | 2.68 | 2.18 | 2.68 | 2.56 | 2.29 | 2.23 | 2.12 | 1.97 | 1.71 | ||
| W | 2.64 | 2.32 | 2.55 | 2.45 | 2.04 | 1.94 | 1.91 | 1.62 | 1.30 | ||
| N | d2 | Ti | 3.19 | 3.30 | 3.65 | 3.16 | 3.14 | 3.03 | 2.45 | 2.37 | 2.00 |
| Zr | 2.39 | 2.63 | 3.37 | 2.50 | 2.68 | 2.87 | 2.08 | 2.32 | 2.08 | ||
| Hf | 2.63 | 2.76 | 3.45 | 2.64 | 2.71 | 2.79 | 2.10 | 2.23 | 1.84 | ||
| d3 | V | 3.17 | 2.85 | 3.19 | 2.95 | 2.56 | 2.45 | 1.99 | 1.71 | 1.38 | |
| Nb | 2.80 | 2.60 | 3.28 | 2.72 | 2.48 | 2.69 | 2.18 | 2.05 | 1.79 | ||
| Ta | 2.95 | 2.48 | 3.20 | 2.66 | 2.24 | 2.40 | 1.97 | 1.68 | 1.33 | ||
| d4 | Cr | 2.34 | 2.24 | 2.31 | 2.16 | 1.84 | 1.67 | 1.43 | 1.25 | 0.95 | |
| Mo | 3.40 | 3.69 | 3.93 | 2.87 | 2.53 | 2.47 | 2.39 | 2.31 | 2.07 | ||
| W | 3.22 | 2.42 | 2.52 | 2.63 | 1.75 | 1.44 | 0.71 | 0.53 | 0.26 | ||
| Eexf M4AX3/J·m−2 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| X | M/A | Al | Si | P | Ga | Ge | As | In | Sn | Sb | |
| C | d2 | Ti | 2.64 | 3.03 | 3.64 | 2.95 | 3.12 | 3.24 | 2.53 | 2.71 | 2.45 |
| Zr | 1.94 | 2.58 | 3.46 | 2.48 | 2.76 | 3.12 | 2.36 | 2.63 | 2.61 | ||
| Hf | 2.23 | 2.79 | 3.69 | 2.71 | 2.90 | 3.24 | 2.48 | 2.66 | 2.53 | ||
| d3 | V | 3.01 | 2.90 | 3.37 | 3.22 | 2.82 | 2.66 | 2.42 | 2.05 | 1.59 | |
| Nb | 2.79 | 2.53 | 3.24 | 2.82 | 2.53 | 2.71 | 2.48 | 2.28 | 1.94 | ||
| Ta | 2.84 | 2.53 | 3.27 | 2.84 | 2.42 | 2.60 | 2.39 | 2.00 | 1.59 | ||
| d4 | Cr | 2.84 | 2.52 | 2.55 | 2.66 | 2.28 | 2.10 | 1.91 | 1.60 | 1.28 | |
| Mo | 2.64 | 2.34 | 2.64 | 2.56 | 2.29 | 2.23 | 2.28 | 2.10 | 1.89 | ||
| W | 2.79 | 2.40 | 2.68 | 2.53 | 2.20 | 2.08 | 2.15 | 1.89 | 1.67 | ||
| N | d2 | Ti | 3.19 | 3.08 | 3.61 | 3.19 | 3.01 | 2.96 | 2.50 | 2.32 | 1.88 |
| Zr | 2.26 | 2.26 | 3.17 | 2.32 | 2.32 | 2.68 | 1.97 | 2.04 | 1.84 | ||
| Hf | 2.48 | 2.42 | 3.32 | 2.52 | 2.39 | 2.66 | 2.04 | 1.94 | 1.63 | ||
| d3 | V | 3.09 | 2.76 | 3.03 | 2.85 | 2.53 | 2.47 | 2.10 | 1.83 | 1.49 | |
| Nb | 3.08 | 2.60 | 3.35 | 2.79 | 2.45 | 2.66 | 2.21 | 1.94 | 1.75 | ||
| Ta | 2.98 | 2.63 | 3.46 | 2.87 | 2.39 | 2.55 | 2.16 | 1.76 | 1.41 | ||
| d4 | Cr | 1.75 | 1.63 | 1.71 | 1.67 | 1.35 | 1.22 | 1.12 | 0.83 | 0.53 | |
| Mo | 3.43 | 2.76 | 2.31 | 2.95 | 2.29 | 2.08 | 1.75 | 1.47 | 1.06 | ||
| W | 3.20 | 2.34 | 2.39 | 2.56 | 1.67 | 1.30 | 1.20 | 0.50 | −0.18 | ||
| X | Al | Si | P | Ga | Ge | As | In | Sn | Sb | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| C | IV | a | −0.34 | −0.49 | −0.82 | −0.61 | −0.70 | −1.17 | −1.14 | −1.94 | −0.40 |
| b | −2.56 | −2.00 | 0.45 | −1.72 | −1.34 | 0.31 | −0.58 | 1.73 | −2.36 | ||
| R | 0.65 | 0.76 | 0.76 | 0.78 | 0.79 | 0.78 | 0.77 | 0.74 | 0.15 | ||
| V + VI | a | −0.76 | −0.99 | −1.22 | −1.10 | −1.43 | −1.35 | −1.67 | −1.29 | −0.75 | |
| b | 0.38 | 0.78 | 1.89 | 1.23 | 1.75 | 1.50 | 1.97 | 0.77 | 0.57 | ||
| R | 0.45 | 0.67 | 0.91 | 0.65 | 0.79 | 0.89 | 0.68 | 0.55 | 0.35 | ||
| N | IV | a | −0.22 | −0.25 | −0.13 | −0.16 | −0.37 | 0.24 | −0.09 | −0.28 | 1.18 |
| b | −2.69 | −2.59 | −2.83 | −2.87 | −2.29 | −3.97 | −3.10 | −2.57 | −6.21 | ||
| R | 0.25 | 0.28 | 0.00 | 0.16 | 0.32 | 0.11 | 0.05 | 0.05 | 0.22 | ||
| V + VI | a | 0.31 | 0.54 | 0.00 | 0.33 | 0.38 | 0.11 | 0.29 | −0.12 | 0.33 | |
| b | −2.85 | −3.34 | −1.89 | −2.83 | −2.77 | −2.15 | −2.45 | −1.65 | −2.48 | ||
| R | 0.28 | 0.45 | 0.07 | 0.24 | 0.26 | 0.09 | 0.21 | 0.05 | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dolz, D.; Morales-García, Á.; Viñes, F.; Illas, F. Exfoliation Energy as a Descriptor of MXenes Synthesizability and Surface Chemical Activity. Nanomaterials 2021, 11, 127. https://doi.org/10.3390/nano11010127
Dolz D, Morales-García Á, Viñes F, Illas F. Exfoliation Energy as a Descriptor of MXenes Synthesizability and Surface Chemical Activity. Nanomaterials. 2021; 11(1):127. https://doi.org/10.3390/nano11010127
Chicago/Turabian StyleDolz, Daniel, Ángel Morales-García, Francesc Viñes, and Francesc Illas. 2021. "Exfoliation Energy as a Descriptor of MXenes Synthesizability and Surface Chemical Activity" Nanomaterials 11, no. 1: 127. https://doi.org/10.3390/nano11010127
APA StyleDolz, D., Morales-García, Á., Viñes, F., & Illas, F. (2021). Exfoliation Energy as a Descriptor of MXenes Synthesizability and Surface Chemical Activity. Nanomaterials, 11(1), 127. https://doi.org/10.3390/nano11010127