
Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties



Abstract
:1. Introduction
2. Materials and Methods
2.1. Parent Substrates and Silver Impregnation
2.1.1. Synthesis of Parent SBA-15
2.1.2. Other Substrates
2.1.3. Preparation of Silver-Impregnated Materials
2.2. Characterization Techniques
2.3. Iodine Adsorption Tests
2.3.1. I2 adsorption Tests
2.3.2. CH3I Adsorption Tests
3. Results and Discussion
3.1. Characterization of Silver-Impregnated Adsorbents
3.1.1. Silver Content
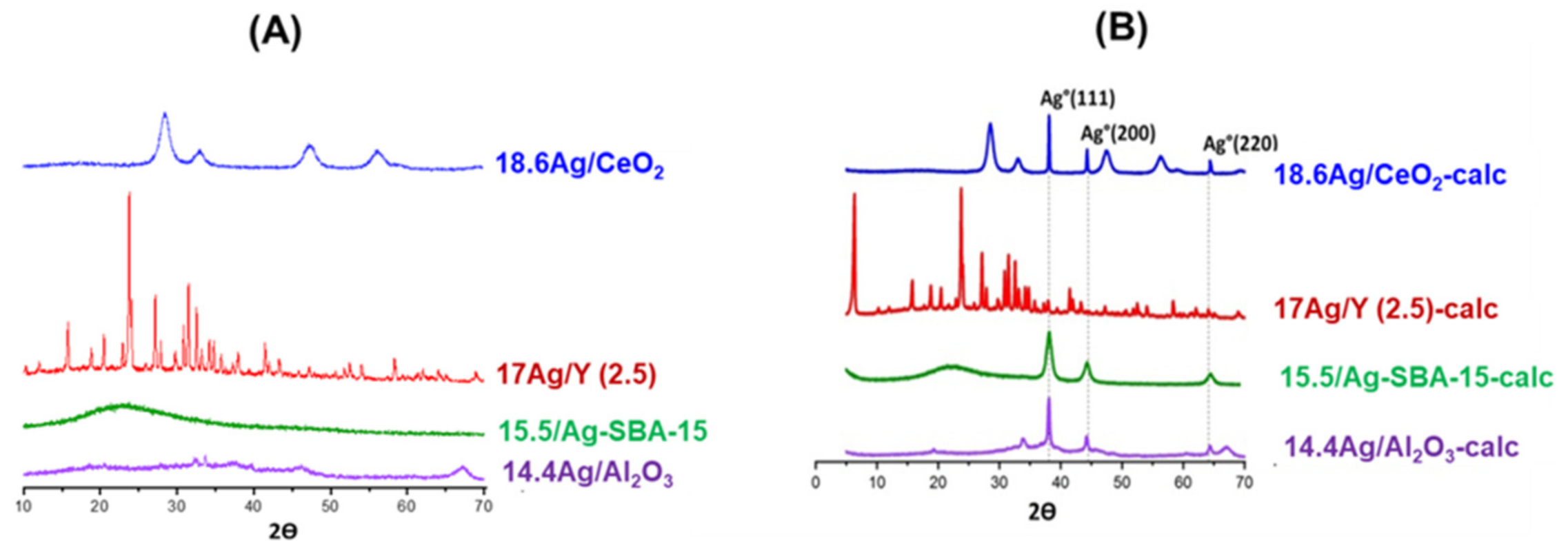
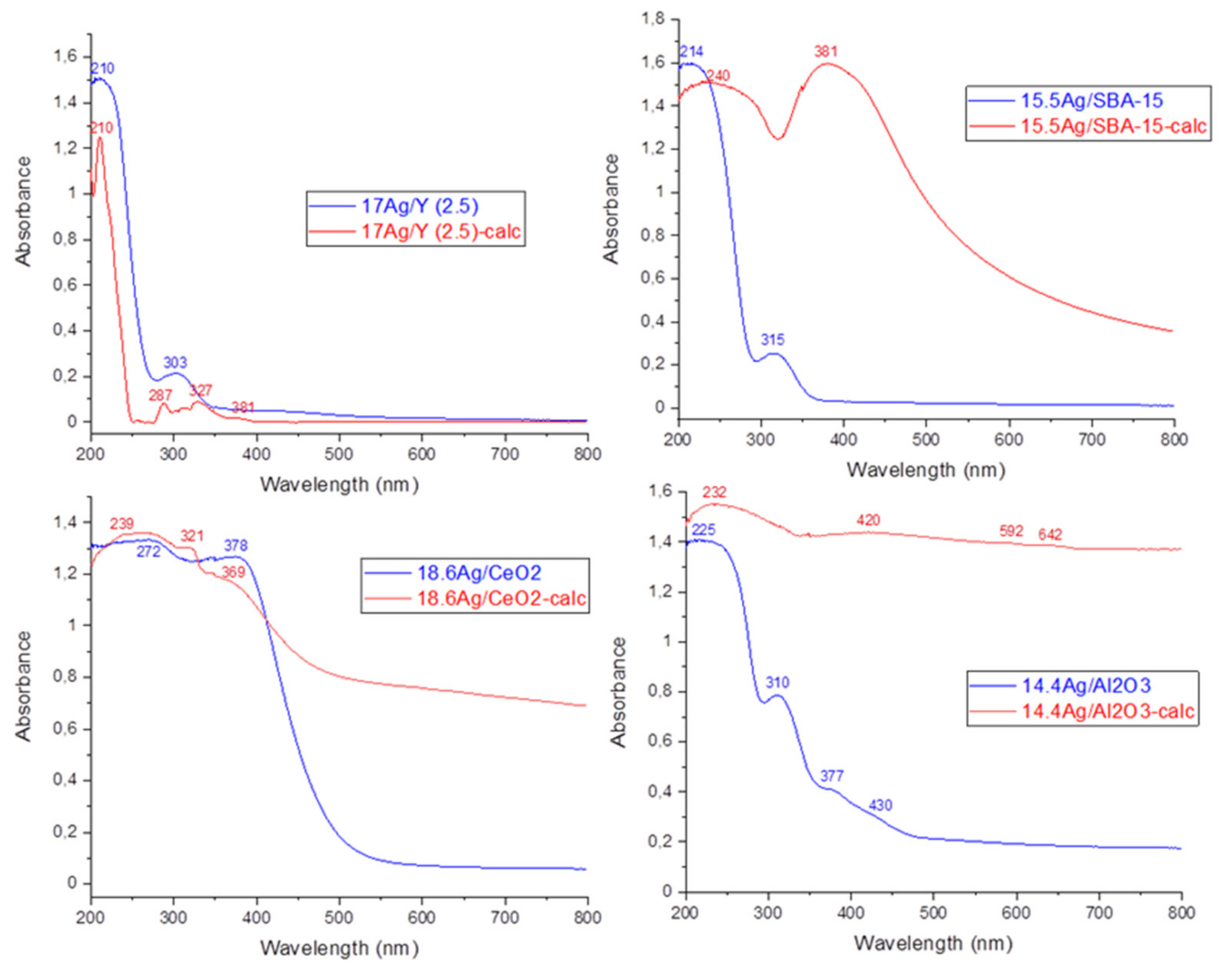
3.1.2. Study of Silver Speciation and Dispersion before and after Calcination by X-Ray Diffraction (XRD) and DR-UV-Visible Spectroscopy (DRS-UV-Vis)
3.2. Adsorption Properties for Molecular Iodine and Iodomethane
Comparison of Adsorption Capacities
3.3. Characterizations Performed on the Spent Adsorbents after I2 and CH3I Tests
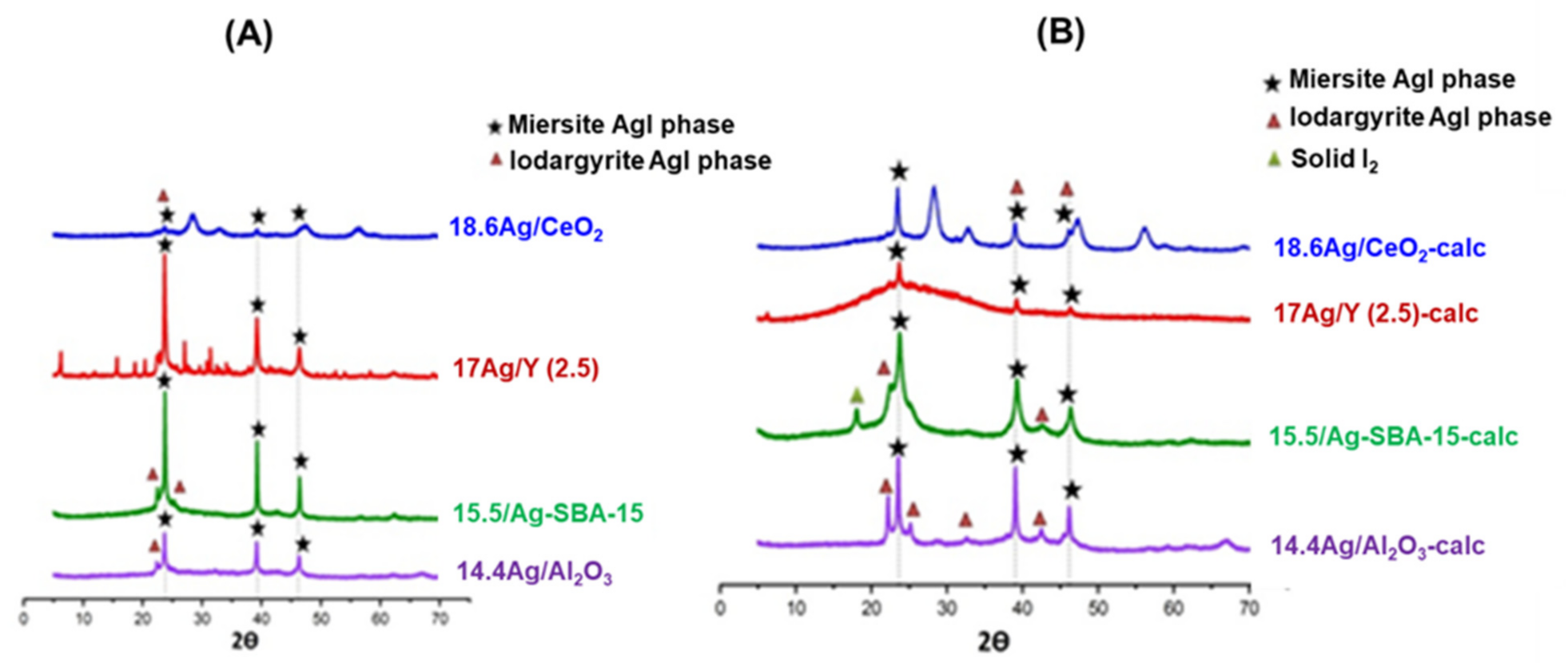
3.3.1. X-Ray Diffraction Analyses of AgI Precipitates
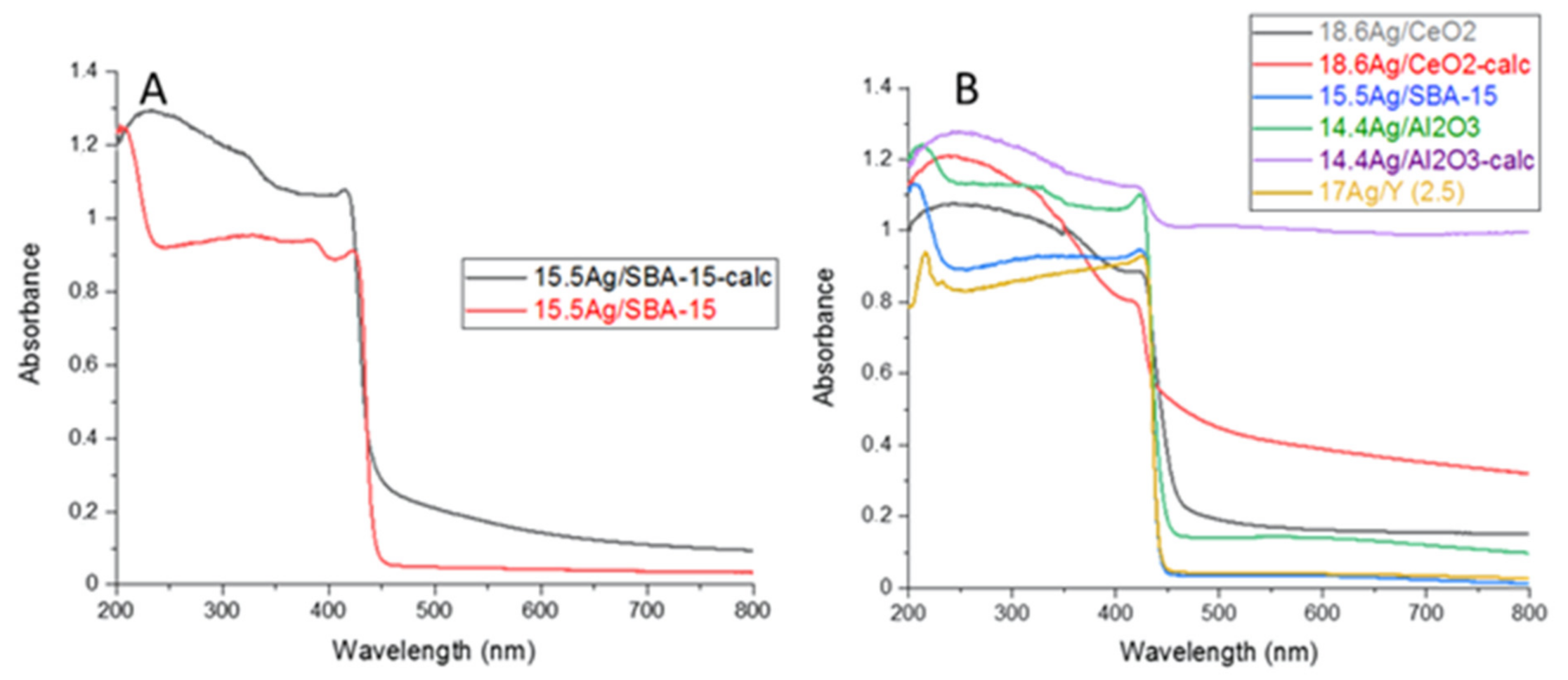
3.3.2. DRS-UV-Vis and TEM Analyses of AgI Precipitates
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clement, B.; Cantrel, L.; Ducros, G.; Funke, F.; Herranz, L.; Rydl, A.; Weber, G.; Wren, C. State of the art report on iodine chemistry. In OECD Report, NEA/CSNI/R; OECD: Paris, France, 2007. [Google Scholar]
- Haefner, D.R.; Tranter, T.J. Methods of Gas Phase Capture of Iodine from Fuel Reprocessing Off-Gas: A Literature Survey. In INL/EXT-07-12299; Idaho National Laboratory: Idaho Falls, ID, USA, 2007. [Google Scholar]
- Cantrel, L.; Herranz, L.E.; Guieu, S.; Albiol, T.; Collet, R.; Lind, T.; Karkela, T.; Mun, C.; Jacquemain, D.; Chebbi, M. Overview of ongoing and planned R&D works on delayed releases and FCVS efficiencies. In Proceedings of the ICAPP 2015, Nice, France, 3–6 May 2015. [Google Scholar]
- Ikemoto, T.; Magara, Y. Measures against impacts of nuclear disaster on drinking water supply systems in Japan. Water Pract. Technol. 2011, 6. [Google Scholar] [CrossRef]
- Maeck, W.J.; Pence, D.T.; Keller, J.H. A Highly Efficient Inorganic Adsorber for Airborne Iodine Species (Silver Zeolites Development Studies); Idaho Nuclear Corporation: Idaho Falls, ID, USA, 1969. [Google Scholar]
- Pence, D.T.; Duce, F.A.; Maeck, W.J.; First, M.W. Developments in the Removal of Airborne Iodine Species with Metal Substituted Zeolites. In Proceedings of the 12th AEC Air Cleaning Conference, Oak Ridge, TN, USA, 28–31 August 1972; AEC: Washington, DC, USA, 1972. [Google Scholar]
- Thomas, T.R.; Staples, B.A.; Murphy, L.P.; Nichols, J.T. Airborne Elemental Iodine Loading Capacities of Metal Exchanged Zeolites and a Method for Recycling Silver Zeolites; Idaho National Engineering Laboratory: Idaho Falls, ID, USA, 1977. [Google Scholar]
- Jubin, R.T. A Literature Survey of Methods to Remove Iodine from Off-Gas Streams Using Solid Sorbents; Report ORNL/TM-6607; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1979. [Google Scholar]
- Herranz, L.E.; Lind, T.; Dieschbourg, K.; Riera, E.; Morandi, S.; Rantanen, P.; Chebbi, M.; Losch, N. State of the Art Report: Technical Bases for Experimentation on Source Term Mitigation Systems. In Proceedings of the 10th International Topical Meeting on Nuclear Thermal Hydraulics, Operation and Safety (NUTHOS-10), Okinawa, Japan, 14–21 December 2014. [Google Scholar]
- Huve, J.; Ryzhikov, A.; Nouali, H.; Lalia, V.; Augé, G.; Daou, T.J. Porous sorbents for the capture of radioactive iodine compounds: A review. RSC Adv. 2018, 8, 29248–29273. [Google Scholar] [CrossRef] [Green Version]
- Chebbi, M. Piégeage D’espèces Iodées Volatiles sur des Adsorbants Poreux de Type Zéolithique dans le Contexte d’un Accident Nucléaire Grave. Ph.D. Thesis, Université de Lorraine, Lorraine, France, 2016. [Google Scholar]
- Chebbi, M.; Azambre, B.; Cantrel, L.; Koch, A. A combined DRIFTS and DR-UV–Vis spectroscopic in situ study on the trapping of CH3I by silver-exchanged faujasite zeolite. J. Phys. Chem. C 2016, 120, 18694–18706. [Google Scholar] [CrossRef]
- Chebbi, M.; Chibani, S.; Paul, J.F.; Cantrel, L.; Badawi, M. Evaluation of volatile iodine trapping in presence of contaminants: A periodic DFT study on cation exchanged-faujasite. Microporous Mesoporous Mater. 2017, 239, 111–122. [Google Scholar] [CrossRef]
- Chebbi, M.; Azambre, B.; Cantrel, L.; Huvé, M.; Albiol, T. Influence of structural, textural and chemical parameters of silver zeolites on the retention of methyl iodide. Microporous Mesoporous Mater. 2017, 244, 137–150. [Google Scholar] [CrossRef]
- Azambre, B.; Chebbi, M. Evaluation of silver zeolites sorbents toward their ability to promote stable CH3I storage as AgI Precipitates. ACS Appl. Mater. Interfaces 2017, 9, 25194–25203. [Google Scholar] [CrossRef] [PubMed]
- Azambre, B.; Chebbi, M.; Leroy, O.; Cantrel, L. Effects of zeolitic parameters and irradiation on the retention properties of silver zeolites exposed to molecular iodine. Ind. Eng. Chem. Res. 2018, 57, 1468–1479. [Google Scholar] [CrossRef]
- Azambre, B.; Chebbi, M.; Hijazi, A. Effects of the cation and Si/Al ratio on CH3I adsorption by faujasite zeolites. Chem. Eng. J. 2020, 379, 122308. [Google Scholar] [CrossRef]
- Chebbi, M.; Azambre, B.; Monsanglant-Louvet, C.; Marcillaud, B.; Roynette, A.; Cantrel, L. Effects of water vapour and temperature on the retention of radiotoxic CH3I by silver faujasite zeolites. J. Hazard. Mater. 2021, 409, 124947. [Google Scholar] [CrossRef] [PubMed]
- Jacquemain, D.; Guentay, S.; Basu, S.; Sonnenkalb, M.; Lebel, L.; Allelein, H.J.; Liebana, B.; Eckardt, B.; Ammirabile, L. Status Report on Filtered Containment Venting. In OECD/NEA/CSNI, Report NEA/ CSNI/R; OECD: Paris, France, 2014. [Google Scholar]
- Herrmann, F.J.; Herrmann, B.; Hoeflich, V.; Beyer, C.H.; Furrer, J. Removal Efficiency of Silver Impregnated Filter Materials and Performance of Iodine Filters in of the Off-Gases of the Karlsruhe Reprocessing Plant WAK. In Proceedings of the 24th DOE/NRC Nuclear Air Cleaning and Treatment Conference, Portland, OR, USA, 15–18 July 1996. [Google Scholar]
- Fukasawa, T.; Funabashi, K.; Kondo, Y. Influences of impurities on iodine removal efficiency. In Proceedings of the 24th DOE/NRC Nuclear Air Cleaning Conference and Air Treatment, Portland, OR, USA, 15–18 July 1996. [Google Scholar]
- Wilhelm, J.G.; Furrer, J. Head-end iodine removal from a reprocessing plant with a solid sorbent. In Proceedings of the ERDA 14th Air Cleaning Conference, CONF720823, Springfield, VA, USA, 28–31 August 1977. [Google Scholar]
- IAEA Report. Treatment, Conditioning and Disposal of Iodine-129; Technical Reports Series No. 276; International Atomic Energy Agency: Vienna, Austria, 1987. [Google Scholar]
- Matyáš, J.; Fryxell, G.E.; Busche, B.J.; Wallace, K.; Fifield, L.S. Ceramic Materials for Energy Applications: Ceramic Engineering and Science; Lin, H.-T., Katoh, Y., Fox, K.M., Belharouak, I., Widjaja, S., Singh, D., Eds.; Wiley-American Ceramic Society: Daytona Beach, FL, USA, 2011; pp. 23–33. [Google Scholar]
- Mnasri, N.; Charnay, C.; Ménoval, L.; Moussaoui, Y.; Elaloui, E.; Zajac, J. Silver nanoparticle-containing submicron-in-size mesoporous silica-based systems for iodine entrapment and immobilization from gas phase. Microporous Mesoporous Mater. 2014, 196, 305–313. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hijazi, A.; Azambre, B.; Finqueneisel, G.; Vibert, F.; Blin, J.L. High iodine adsorption by polyethyleneimine impregnated nanosilica sorbents. Microporous Mesoporous Mater. 2019, 288, 109586. [Google Scholar] [CrossRef]
- Trovarelli, A. Catalytic Properties of Ceria and CeO2-Containing Materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Flanigen, E.M. Introduction to Zeolite Science and Practice, Studies in Surface Science and Catalysis; Van Bekkum, H., Flanigen, E.M., Jansen, J.C., Eds.; Elsevier: Amsterdam, The Netherlands, 1991; p. 58. [Google Scholar]
- Bartolomeu, R.; Azambre, B.; Westermann, A.; Fernandes, A.; Bertolo, R.; Issa Hamoud, H.; Henriques, C.; Da Costa, P. Investigation of the nature of silver species on different Ag-containing NOx reduction catalysts: On the effect of the support. Appl. Catal. B Environ. 2014, 150–151, 204–217. [Google Scholar] [CrossRef]
- Salles, N. Etude des Différents Polymorphes de L’Alumine et des Phases Transitoires Apparaissant lors des Premiers Stades D’Oxydation de L’Aluminium: Simulation à L’Échelle Atomique par un Modèle à Charges Variables en Liaisons Fortes. Ph.D. Thesis, Université de Bourgogne, Dijon, France, 2014. [Google Scholar]
- Kanipandian, N.; Kannan, S.; Ramesh, R.; Subramanian, P.; Thirumurugan, R. Characterization, antioxidant and cytotoxicity evaluation of green synthesized silver nanoparticles using Cleistanthus collinus extract as surface modifier. Mater. Res. 2014, 49, 494–502. [Google Scholar] [CrossRef]
- Sayah, E.; Brouri, D.; Wu, Y.; Musi, A.; Da Costa, P.; Massiani, P. A comparative in situ TEM and UV—Visible spectroscopic study of the thermal evolution of Ag species dispersed on Al2O3 and NaX zeolite supports. Appl. Catal. A Gen. 2011, 406, 94–101. [Google Scholar] [CrossRef]
- Aspromonte, S.G.; Mizrahi, M.D.; Schneeberger, F.A.; Lopez, J.M.R.; Boix, A.V. Study of the nature and location of Silver in Ag-exchanged mordenite catalysts. Characterization by spectroscopic techniques. J. Phys. Chem. C 2013, 117, 25433–25442. [Google Scholar] [CrossRef] [Green Version]
- Shibata, J.; Takada, Y.; Shichi, A.; Satokawa, S.; Satsuma, A.; Hattori, T. Influence of zeolite support on activity enhancement by addition of hydrogen for SCR of NO by propane over Ag-zeolites. Appl. Catal. B Environ. 2004, 54, 137–144. [Google Scholar] [CrossRef]
- Baker, M.D.; Ozin, G.A.; Godber, J. Far-infrared studies of silver atoms, silver ions, and silver clusters in zeolites A and Y. J. Phys. Chem. 1985, 89, 305–311. [Google Scholar] [CrossRef]
- Funke, F.; Greger, G.-U.; Bleier, A.; Hellmann, S.; Morell, W. The Reaction between Iodine and Silver under Severe PWR Accident Conditions. An Experimental Parameter Study. In PSI-97-02, 28035729 Report; IAEA: Vienna, Austria, 1996. [Google Scholar]
- Andryushechkin, B.V.; Zhidomirov, G.M.; Eltsov, K.N.; Hladchanka, Y.V.; Korlyukov, A.A. Local structure of the Ag(100) surface reacting with molecular iodine: Experimental and theoretical study. Phys. Rev. B 2009, 80, 125409. [Google Scholar] [CrossRef]
- Bruffey, S.H.; Jubin, R.T.; Jordan, J.A. Capture of elemental and organic iodine from dilute gas streams by silver-exchanged mordenite. Proc. Chem. 2016, 21, 293–299. [Google Scholar] [CrossRef]
- Zhou, X.-L.; Solymosi, F.; Blass, P.M.; Cannon, K.C.; White, J.M. Interactions of methyl halides (Cl, Br, and I) with Ag(111). Surf. Sci. 1989, 219, 294–316. [Google Scholar] [CrossRef]
- Chapman, K.W.; Chupas, P.J.; Nenoff, T.M. Radioactive iodine capture in silver-containing mordenites through nanoscale silver iodide formation. J. Am. Chem. Soc. 2010, 132, 8897–8899. [Google Scholar] [CrossRef] [PubMed]
- Kodaira, T.; Ikeda, T.; Takeo, H. Optical and X-ray diffraction study of AgI clusters incorporated into zeolite LTA. Eur. Phys. J. D 1999, 9, 601–604. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Adsorbent | %Ag | Silver Speciation |
|---|---|---|
| 14.4 Ag/Al2O3 | 14.4 | Ag+, Agnδ+, Ag°m, traces of Ag° |
| 14.4 Ag/Al2O3-calc | 14.4 | Ag° (27 nm) |
| 15.5 Ag/SBA-15 | 15.5 | Ag+, Agnδ+, Ag°m, traces of Ag° |
| 15.5Ag/SBA-15-calc | 15.5 | Ag° (9 nm) |
| 17 Ag/Y (2.5) | 17 | Ag+, Agnδ+, Ag°m, traces of Ag° |
| 17Ag/Y (2.5)-calc | 17 | Ag+, Agnδ+, Ag°m |
| 18.6 Ag/CeO2 | 18.6 | n.d. |
| 18.6Ag/CeO2-calc | 18.6 | Ag° (106 nm) |
| Adsorbent | QI2 (mg/g) | I/Ag | Color after I2 Exposure | Detected Phases by XRD | D AgI Miersite Phase (nm) |
|---|---|---|---|---|---|
| 14.4 Ag/Al2O3 | 177 | 1.04 | Yellow | AgI Miersite AgI iodargyrite | 37 |
| 14.4 Ag/Al2O3-calc | 196 | 1.16 | Black | AgI Miersite AgI iodargyrite | 49 |
| 15.5 Ag/SBA-15 | 159 | 0.86 | Yellow | AgI Miersite AgI iodargyrite | 64 |
| 15.5Ag/SBA-15-calc | 172 | 0.94 | Yellow | AgI Miersite AgI iodargyrite | 16 |
| 17 Ag/Y (2.5) | 205 | 1.02 | Pale yellow | AgI Miersite | 37 |
| 17Ag/Y (2.5)-calc | 208 | 1.03 | Pale yellow | AgI Miersite | 35 |
| 18.6 Ag/CeO2 | 224 | 1.02 | Yellow | Small peaks AgI Miersite AgI iodargyrite | 16 |
| 18.6Ag/CeO2-calc | 253 | 1.15 | Grey | AgI Miersite AgI iodargyrite | 26 |
| Adsorbent | QCH3I (mg/g) | I/Ag | Detected Phases by XRD | D AgI Miersite Phase (nm) | D Ag° (nm) |
|---|---|---|---|---|---|
| 14.4 Ag/Al2O3 | 122 | 0.64 | AgI Miersite AgI iodargyrite | 44 | **** |
| 14.4 Ag/Al2O3-calc | 38 | 0.2 | Ag° nanoparticles | Small AgI peaks | 28 |
| 15.5 Ag/SBA-15 | 143 | 0.7 | AgI Miersite AgI iodargyrite | 73 | **** |
| 15.5 Ag/SBA-15-calc | 61 | 0.3 | Ag° nanoparticles | Small AgI peaks | 30 |
| 17 Ag/Y (2.5) | 150 | 0.67 | AgI Miersite | 73 | **** |
| 17 Ag/Y (2.5)-calc | 144 | 0.64 | AgI Miersite | 58 | **** |
| 18.6 Ag/CeO2 | 189 | 0.77 | AgI Miersite AgI iodargyrite | 39 | **** |
| 18.6 Ag/CeO2-calc | 22 | 0.09 | Ag° nanoparticles | **** | 29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Azambre, B.; Chebbi, M.; Ibrahim, N. Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties. Nanomaterials 2021, 11, 1300. https://doi.org/10.3390/nano11051300
Azambre B, Chebbi M, Ibrahim N. Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties. Nanomaterials. 2021; 11(5):1300. https://doi.org/10.3390/nano11051300
Chicago/Turabian StyleAzambre, Bruno, Mouheb Chebbi, and Nagham Ibrahim. 2021. "Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties" Nanomaterials 11, no. 5: 1300. https://doi.org/10.3390/nano11051300
APA StyleAzambre, B., Chebbi, M., & Ibrahim, N. (2021). Structure–Activity Relationships between the State of Silver on Different Supports and Their I2 and CH3I Adsorption Properties. Nanomaterials, 11(5), 1300. https://doi.org/10.3390/nano11051300