
Behavior of Silicon Carbide Materials under Dry to Hydrothermal Conditions


Abstract
:1. Introduction
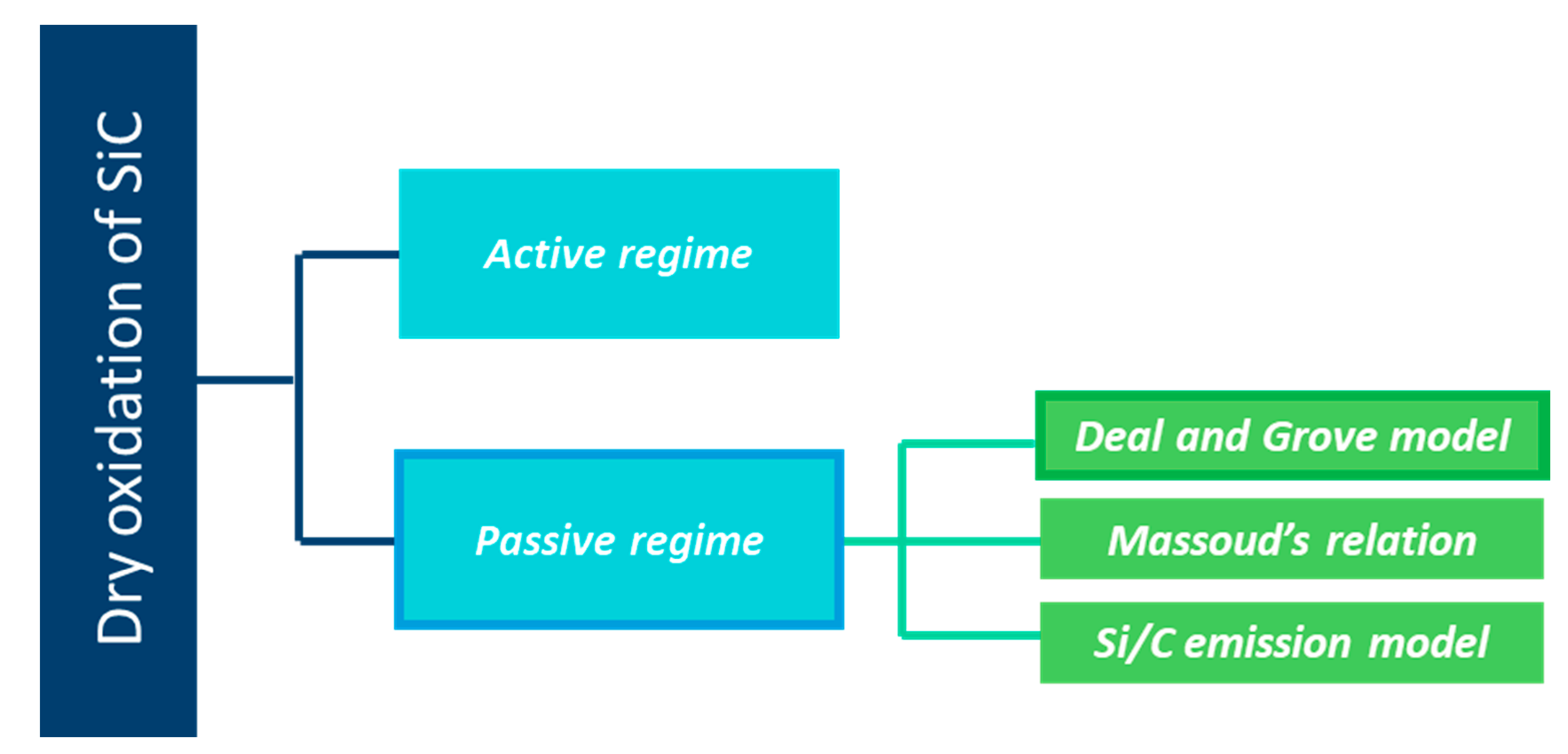
2. Dry Oxidation of Silicon Carbide Materials
2.1. The Passive Oxidation Regime
2.2. Kinetic Models for Si and SiC Oxidation
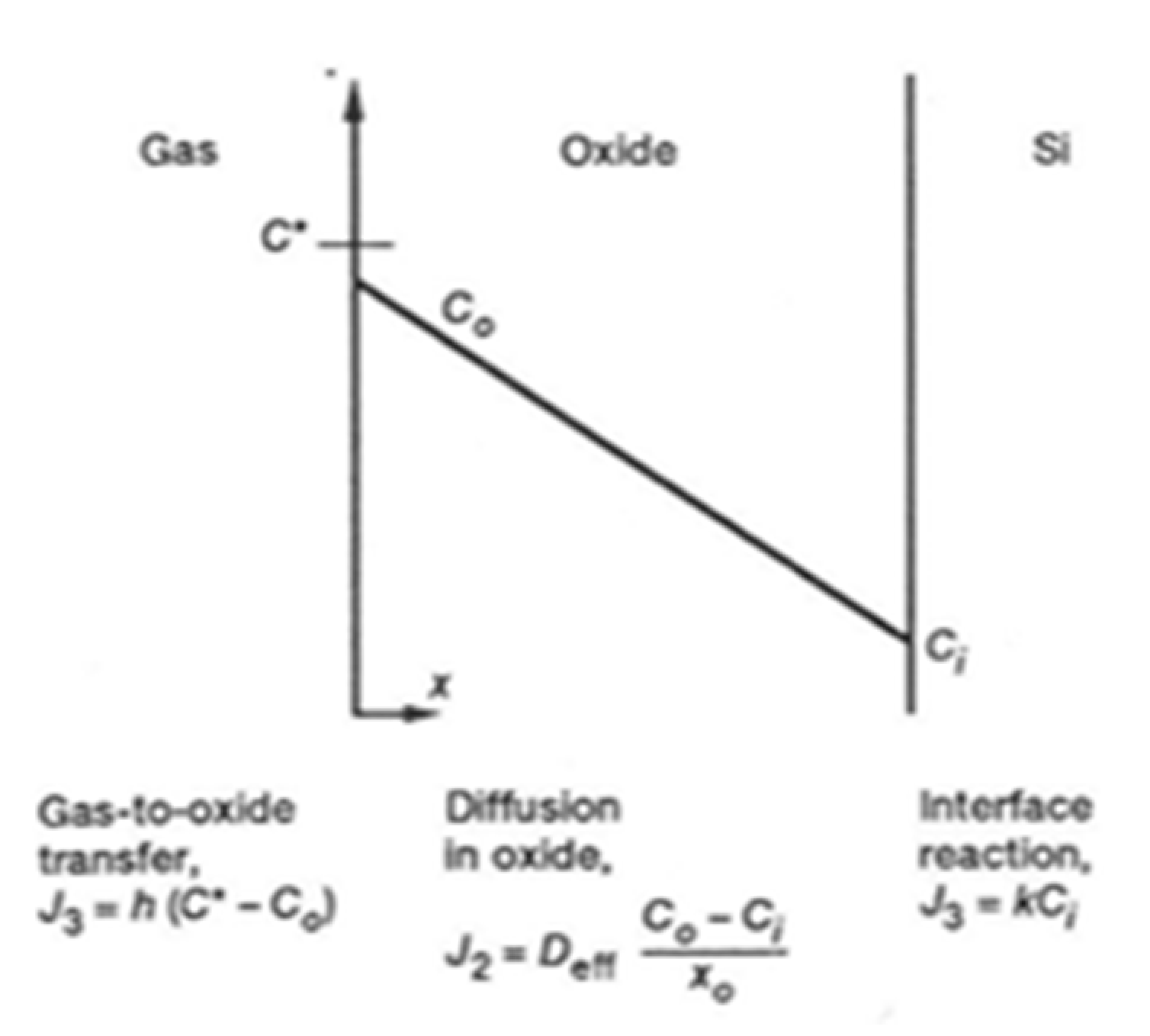
2.2.1. The Deal and Grove Model
2.2.2. Massoud Empirical Relation
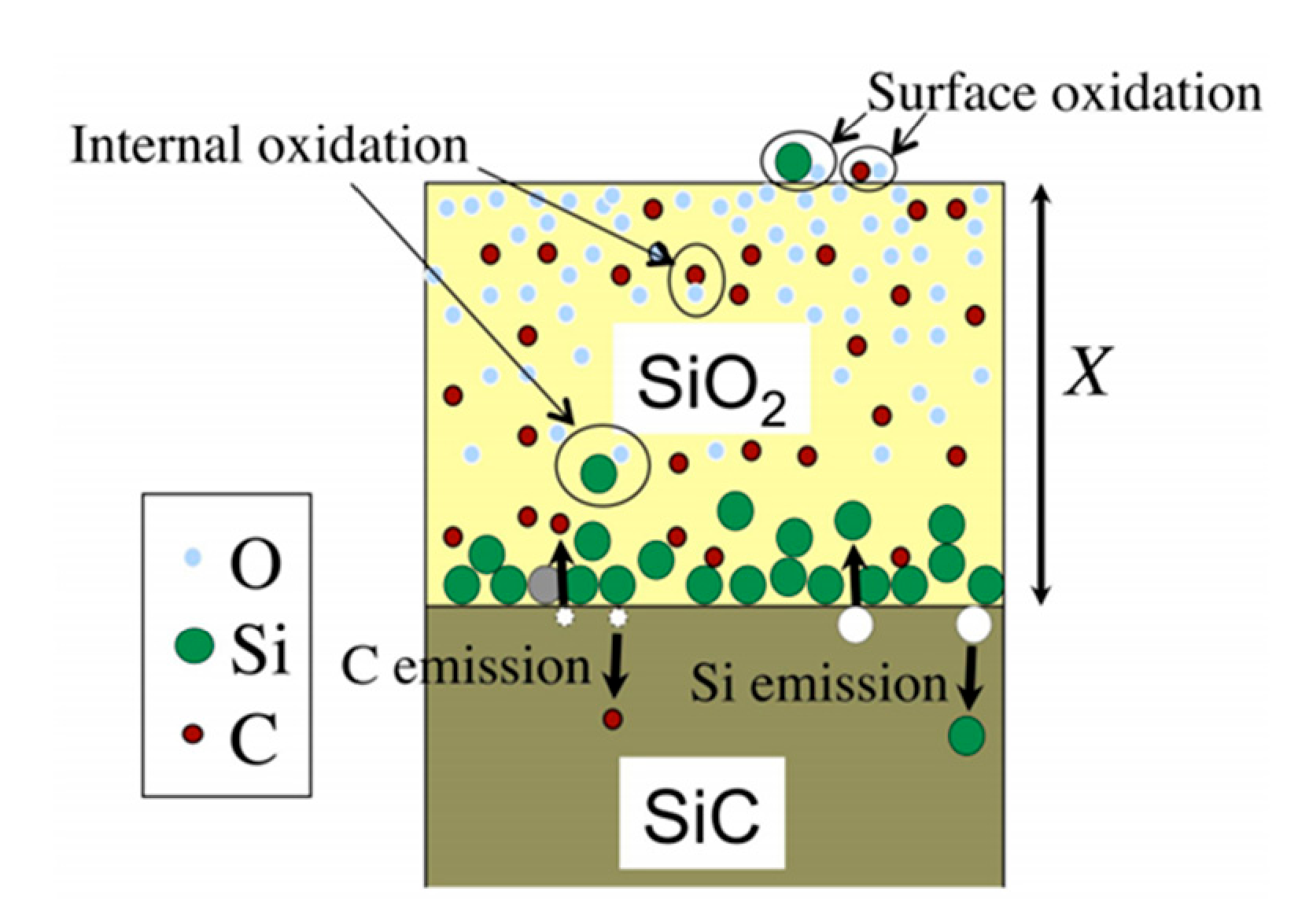
2.2.3. Si and C Emission Model
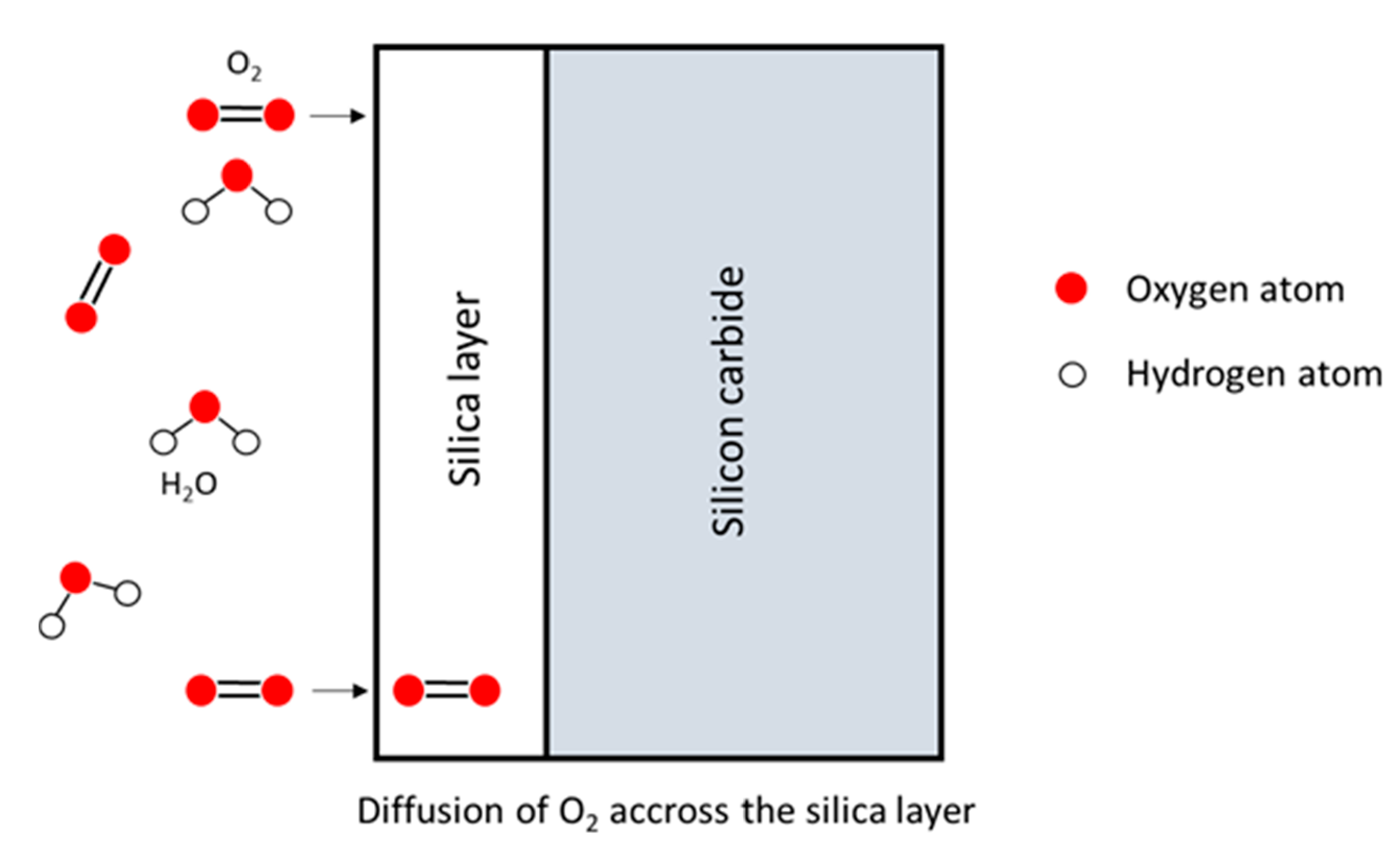
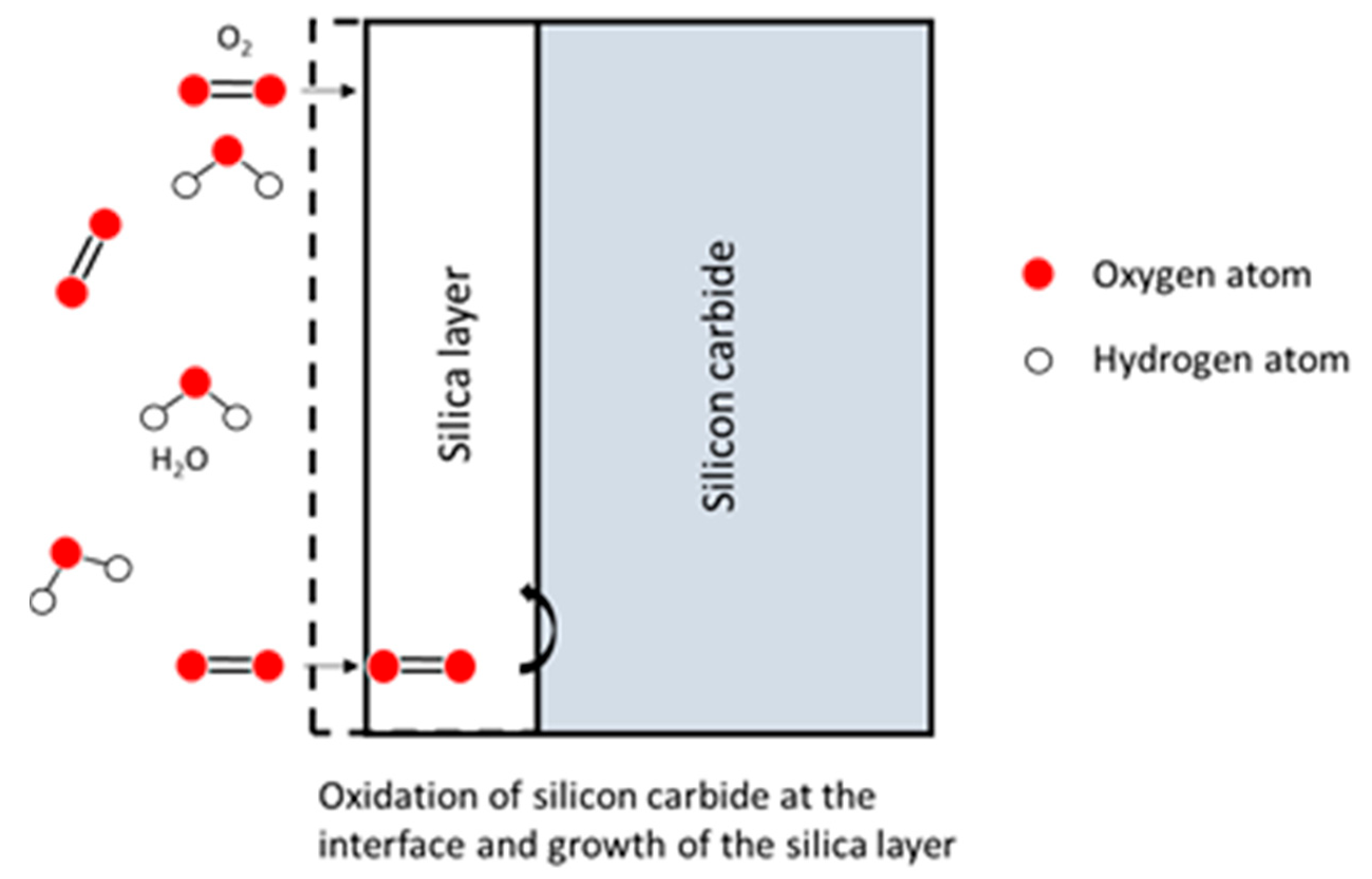
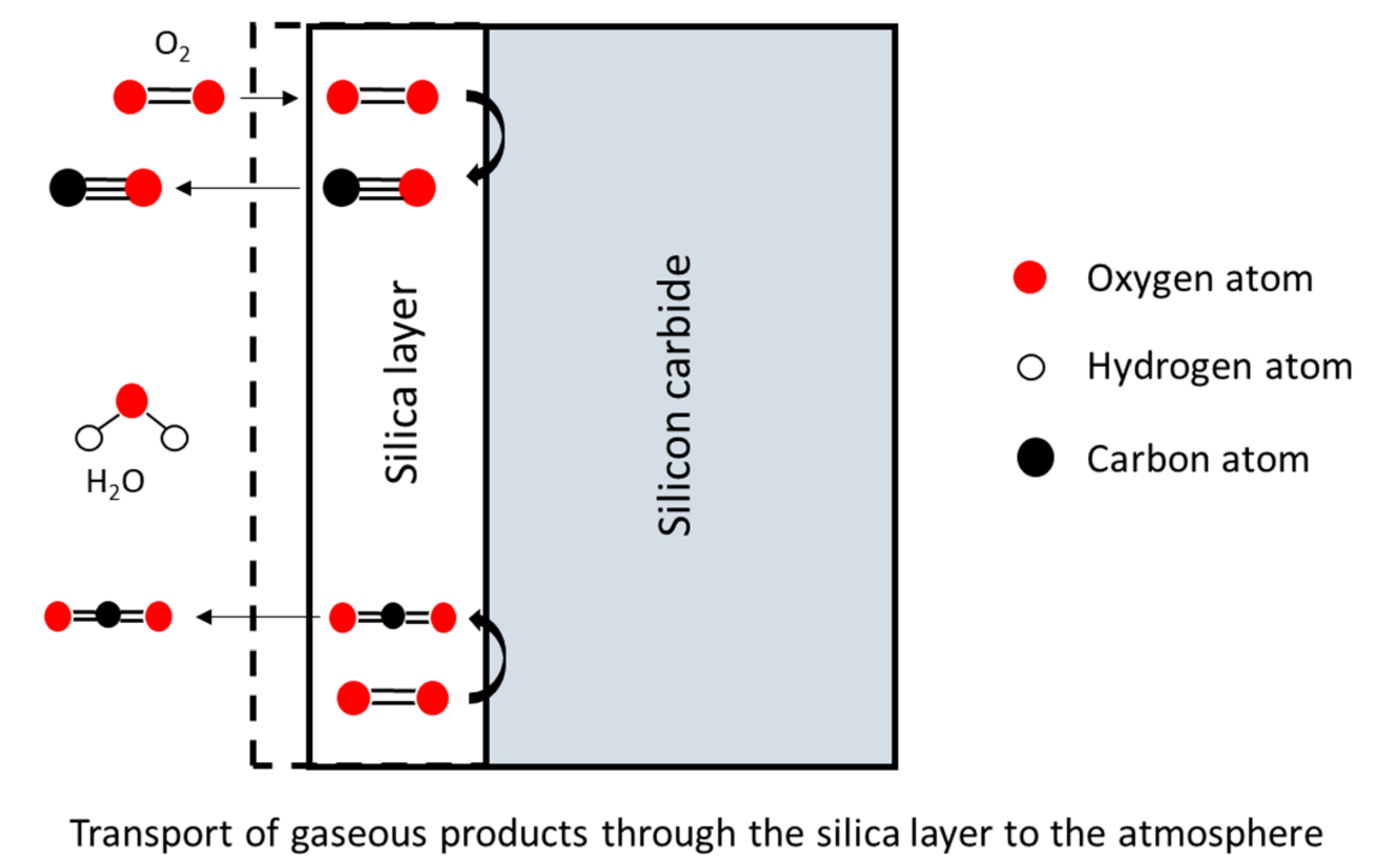
- If the oxide layer is thin enough, the Si atoms can diffuse through it and instantly react with the oxidant atmosphere,
- The Si atoms can also encounter oxidant molecules in the oxide layer itself, and react with it.

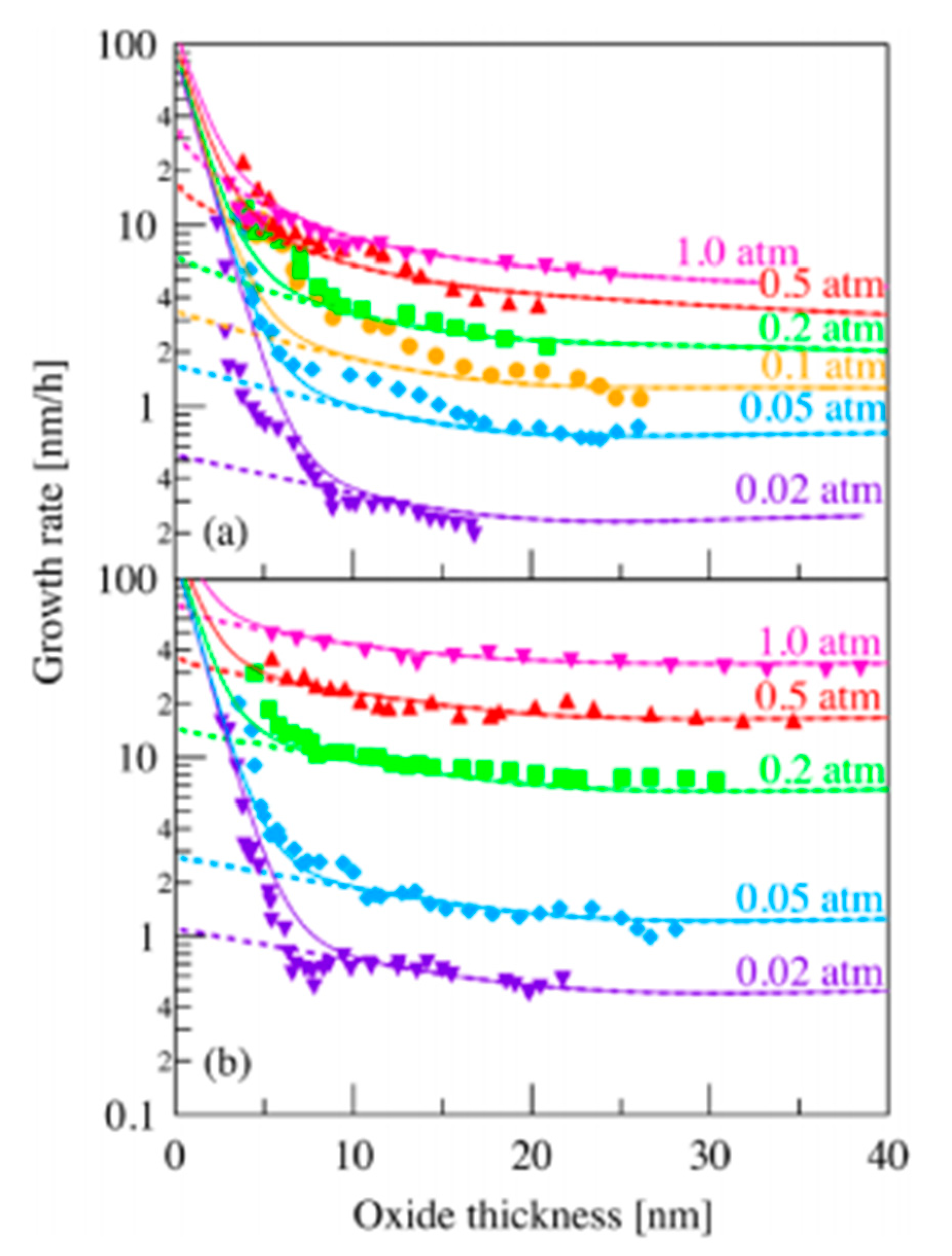
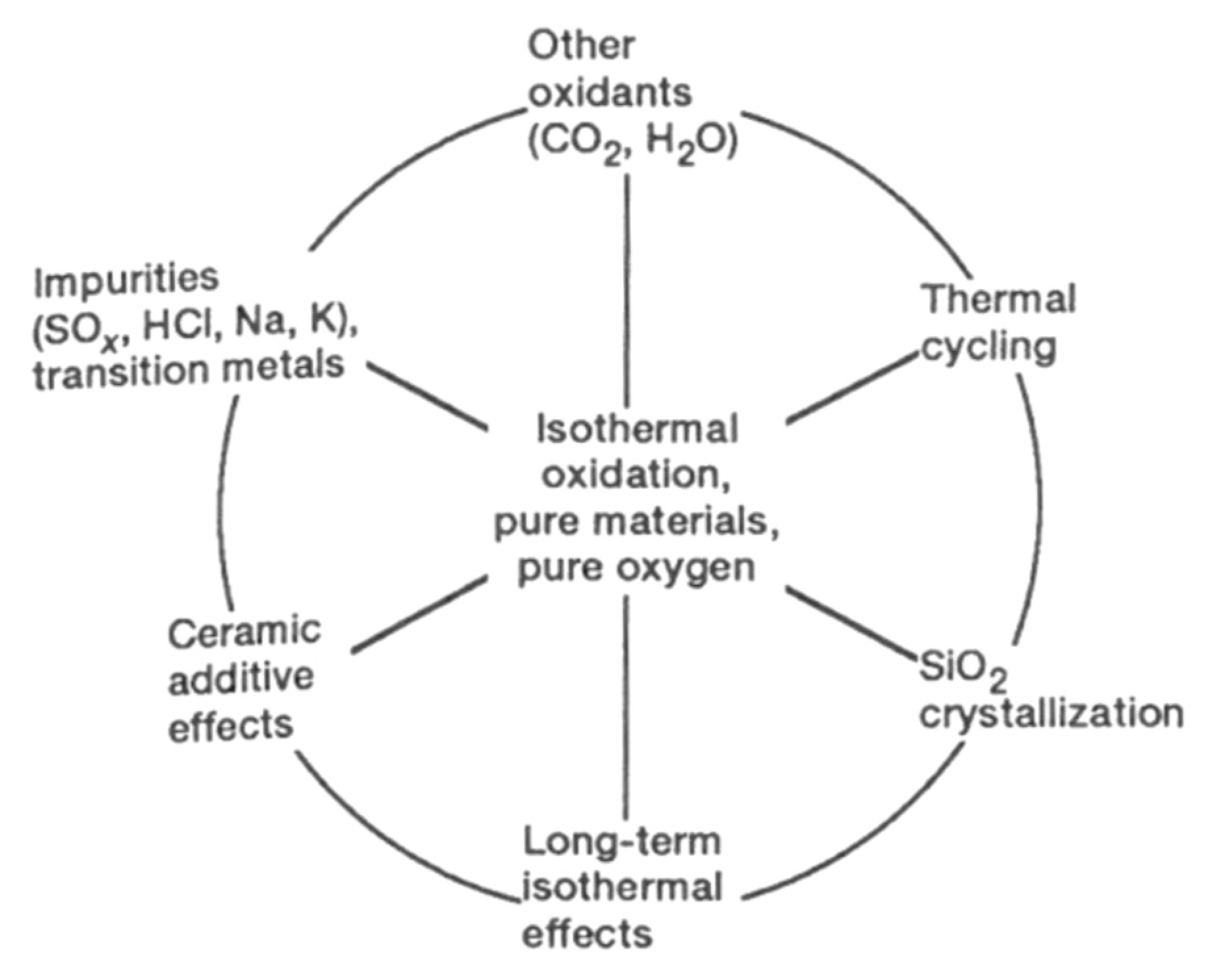
2.3. Parameters Which Can Influence the Oxidation of SiC Materials
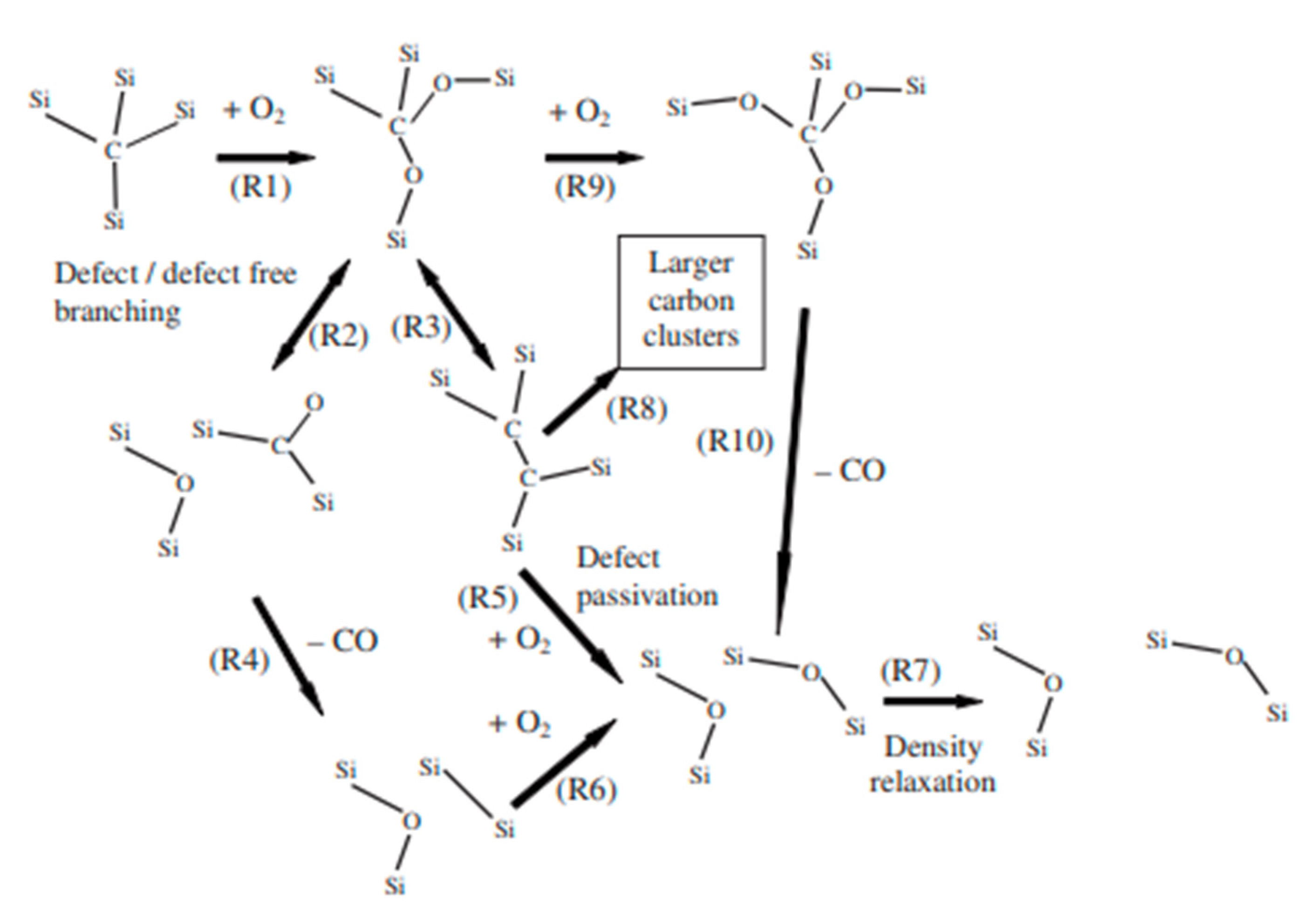
2.3.1. Interpretation of the Activation Energy Values
- Their data differ depending on the fitting of experimental values and the nature of SiC samples,
- Their data differ due to the presence of impurities from either the sample or the apparatus, or the gas phases present. Thus, the oxidation rate is determined by the nature and concentration of impurities as well as other physicochemical parameters,
- The oxidation period seems to affect the oxidation kinetics:
- ➢
- For short oxidation times, a thin amorphous oxide film is created, and the kinetics of the oxide growth follow a linear regime, which implies that this mechanism is surface-controlled,
- ➢
- ➢
- Finally, the rate determining step of the oxidation is thought to be either the inward diffusion of oxygen or the outward diffusion of CO (i.e., product gases).
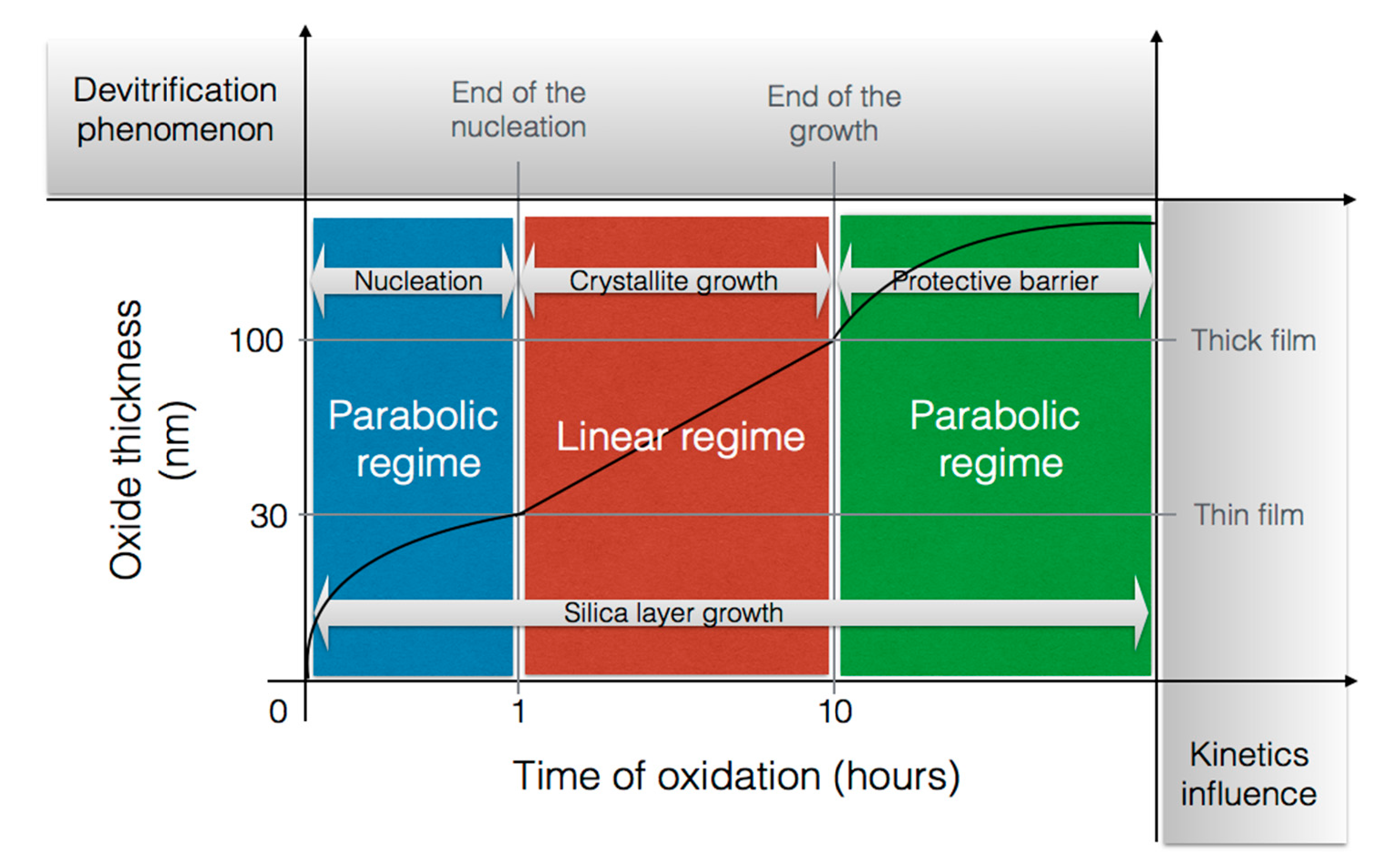
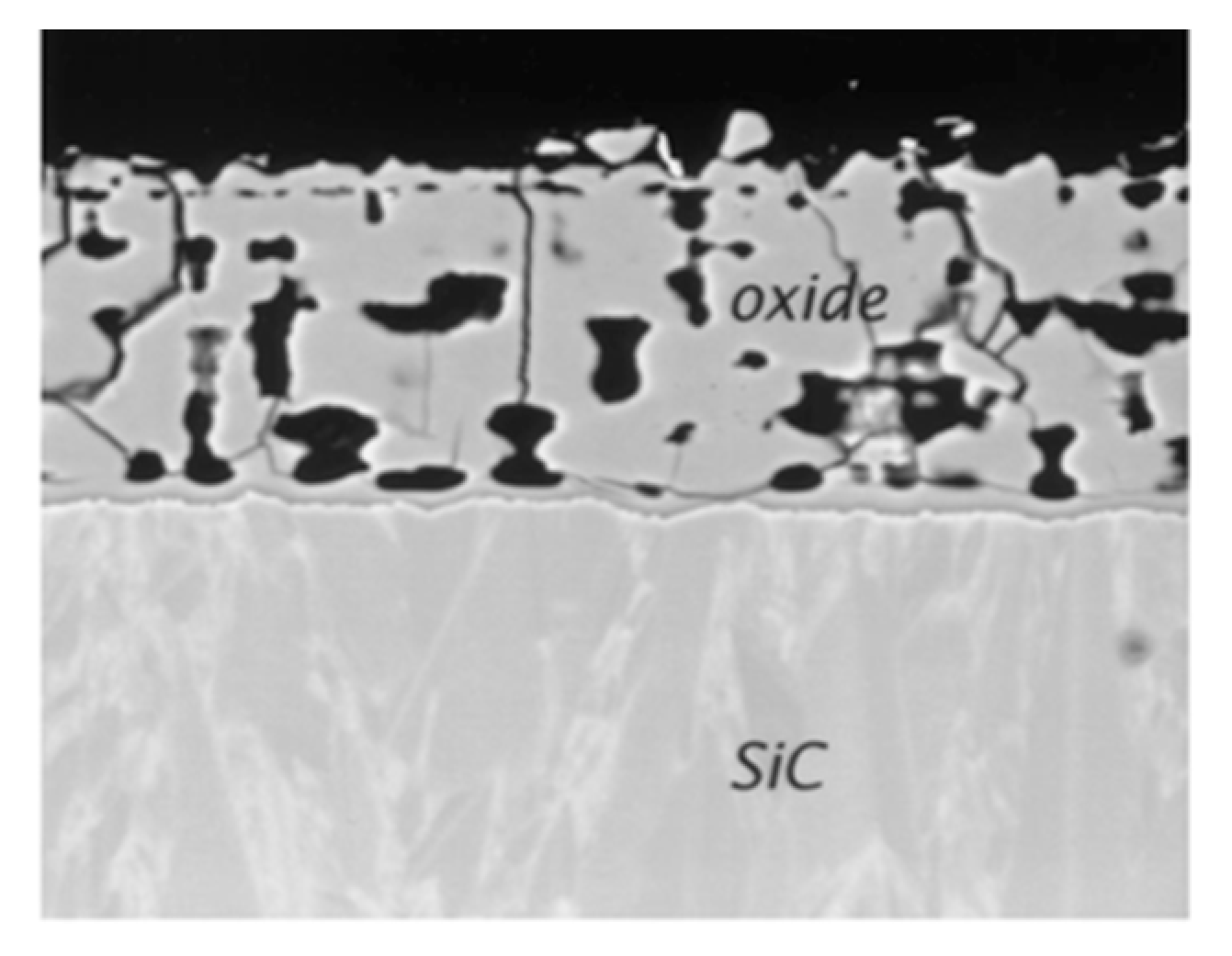
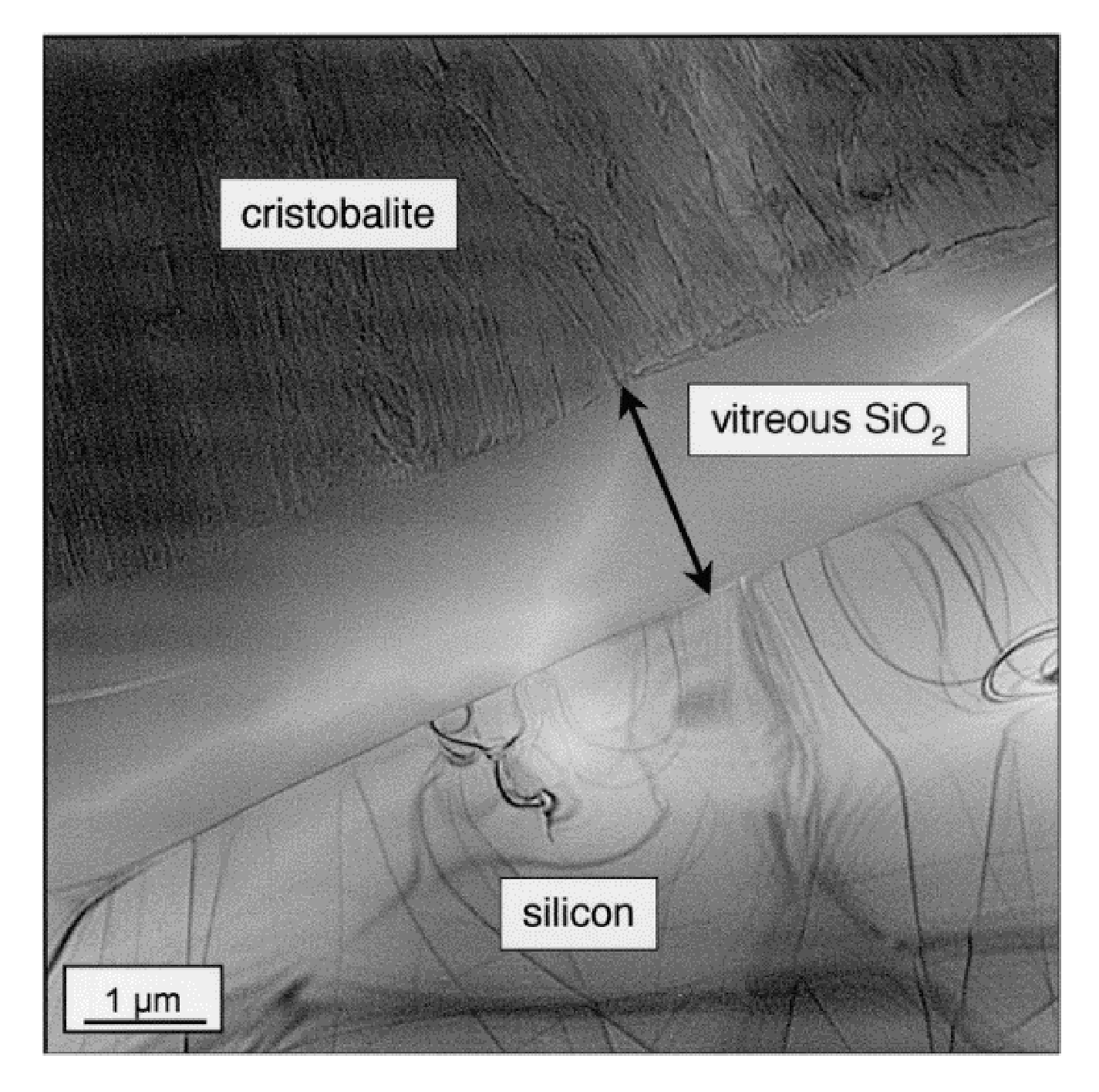
2.3.2. Nature of the Silica Layer
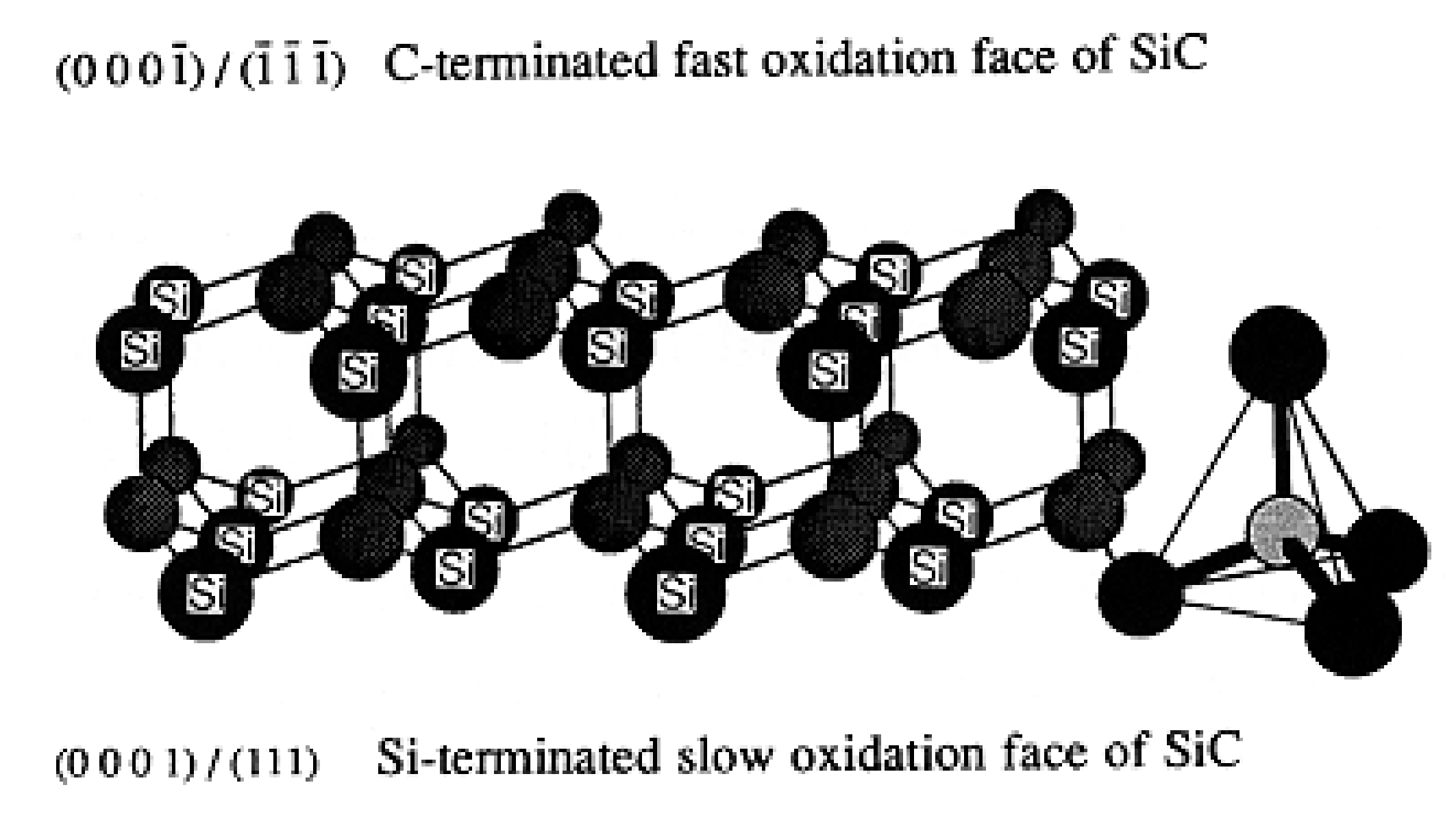
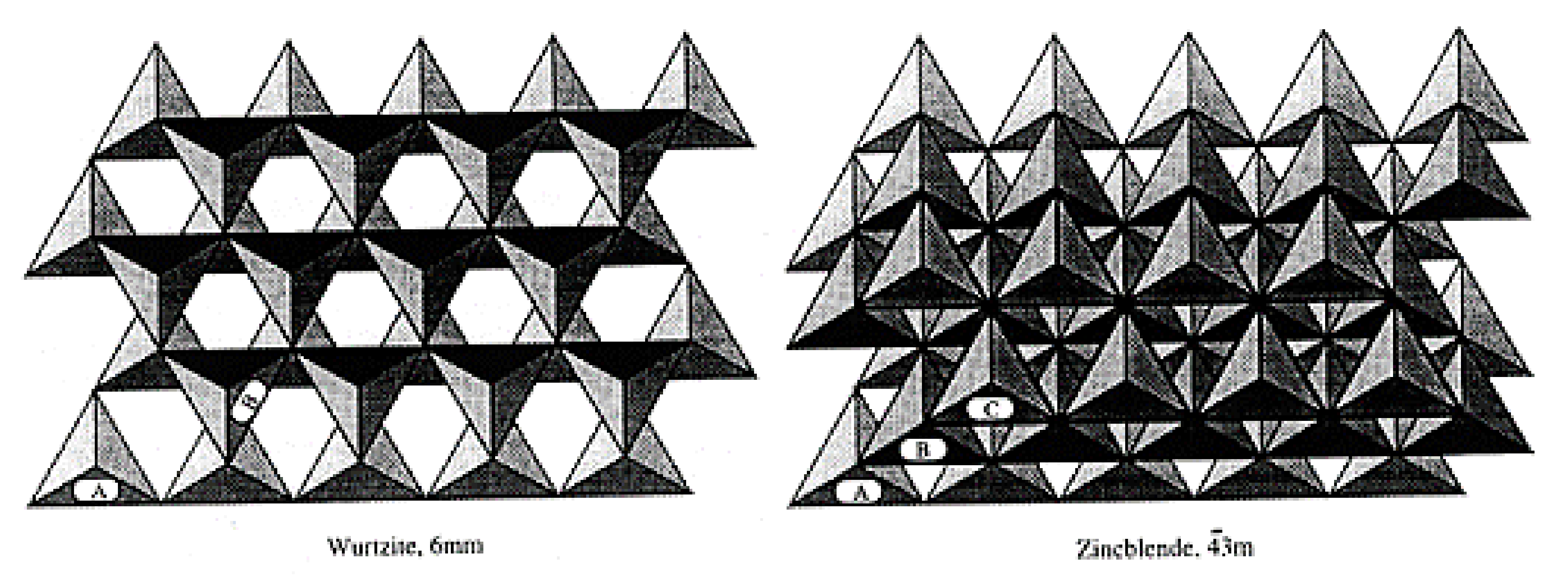
2.3.3. Crystal Faces Effects
2.3.4. Oxidation Rate-Determining Step
- The penetration of oxygen into the silica scale:
- 2.
- Reaction of oxygen with SiC:
- 3.
- Passivation reactions of carbon defect by O2:
- 4.
- Density relaxation process:
2.4. Conclusions
- At short oxidation times, a gas diffusion mechanism is dominant (parabolic regime) whereas at long times, a surface-reaction mechanism is dominant (linear regime),
- For the gas diffusion mechanism, the temperature plays an important role: at low temperatures, the oxygen diffusion is molecular, whereas above 1350 °C, the diffusing species is ionic oxygen. Therefore, a C-rich inner oxide layer is created on the Si-faces,
- The oxidation behavior becomes complicated when the crystallization of amorphous silica takes part in the oxidation process. This reduces the oxidant transport and leads to the decrease of oxidation rates.
- Finally, the presence of impurities is not negligible and may have an impact in all these studies. A high degree of impurities will enhance both the crystallization of the scale and the formation of defects, which allows for faster oxygen diffusion.
3. Wet Oxidation of Silicon Carbide Materials
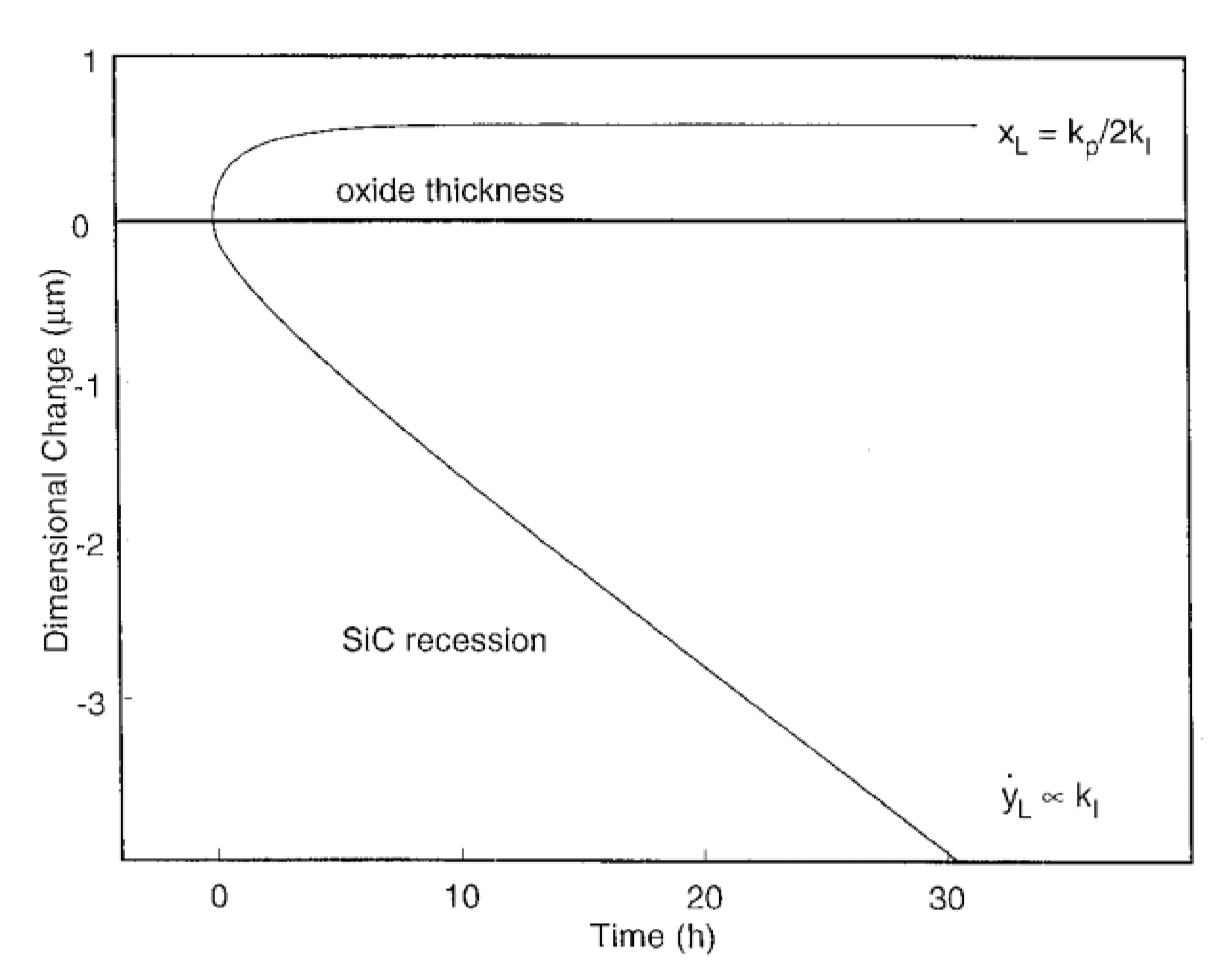
3.1. The Two Competitive Oxidation Regimes
3.2. Main Characteristics of Wet Oxidation of SiC Materials
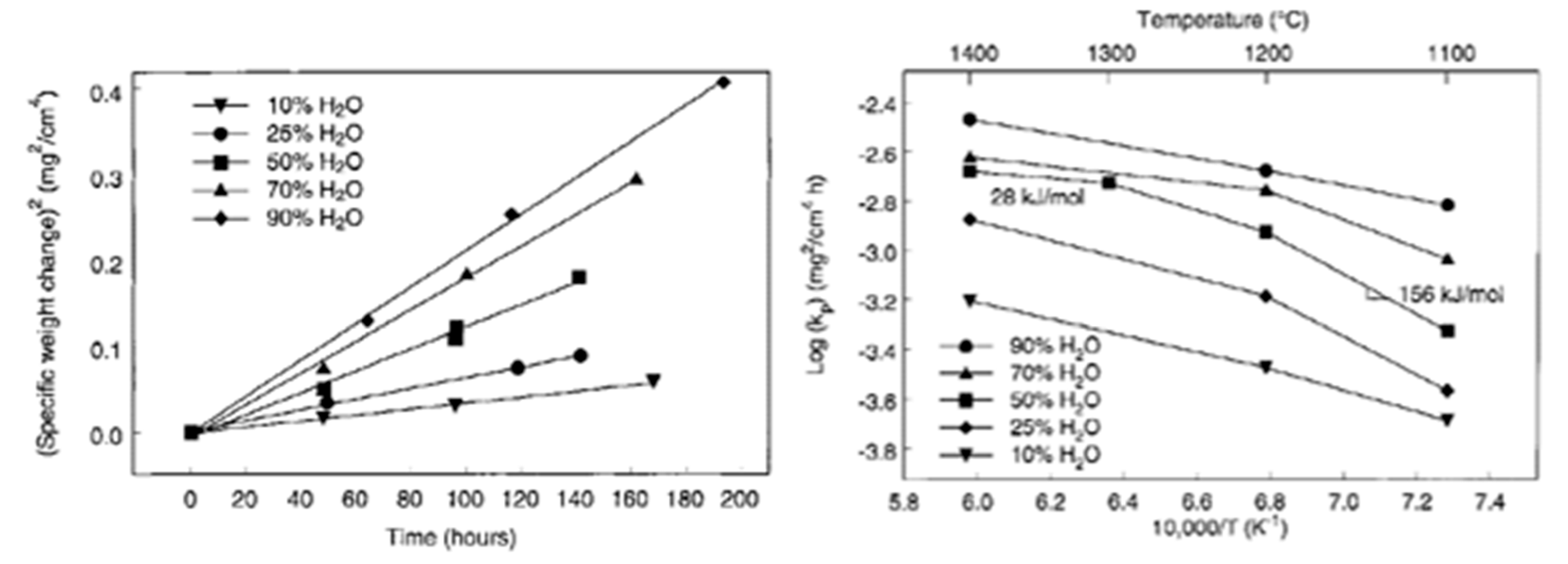
3.2.1. Activation Energy of SiC Wet Oxidation
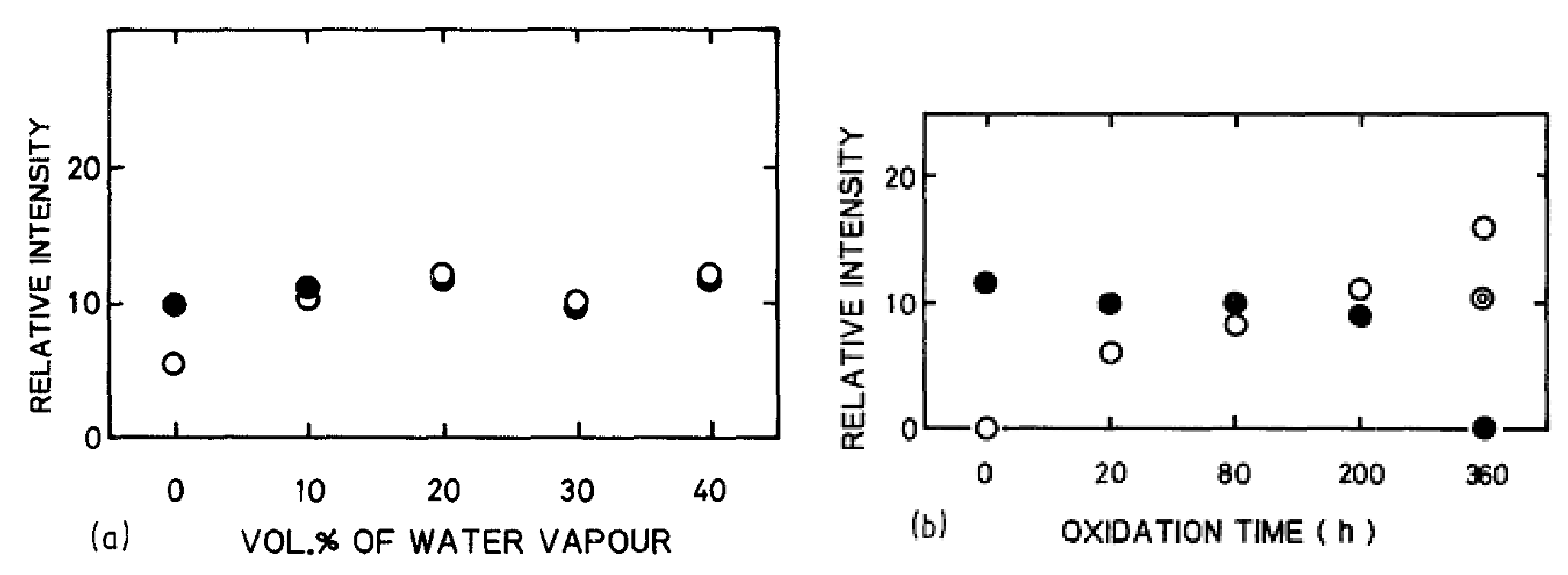
3.2.2. Impurities Effect
3.2.3. Nature of the Oxidant Species
3.2.4. Nature of the Silica Layer
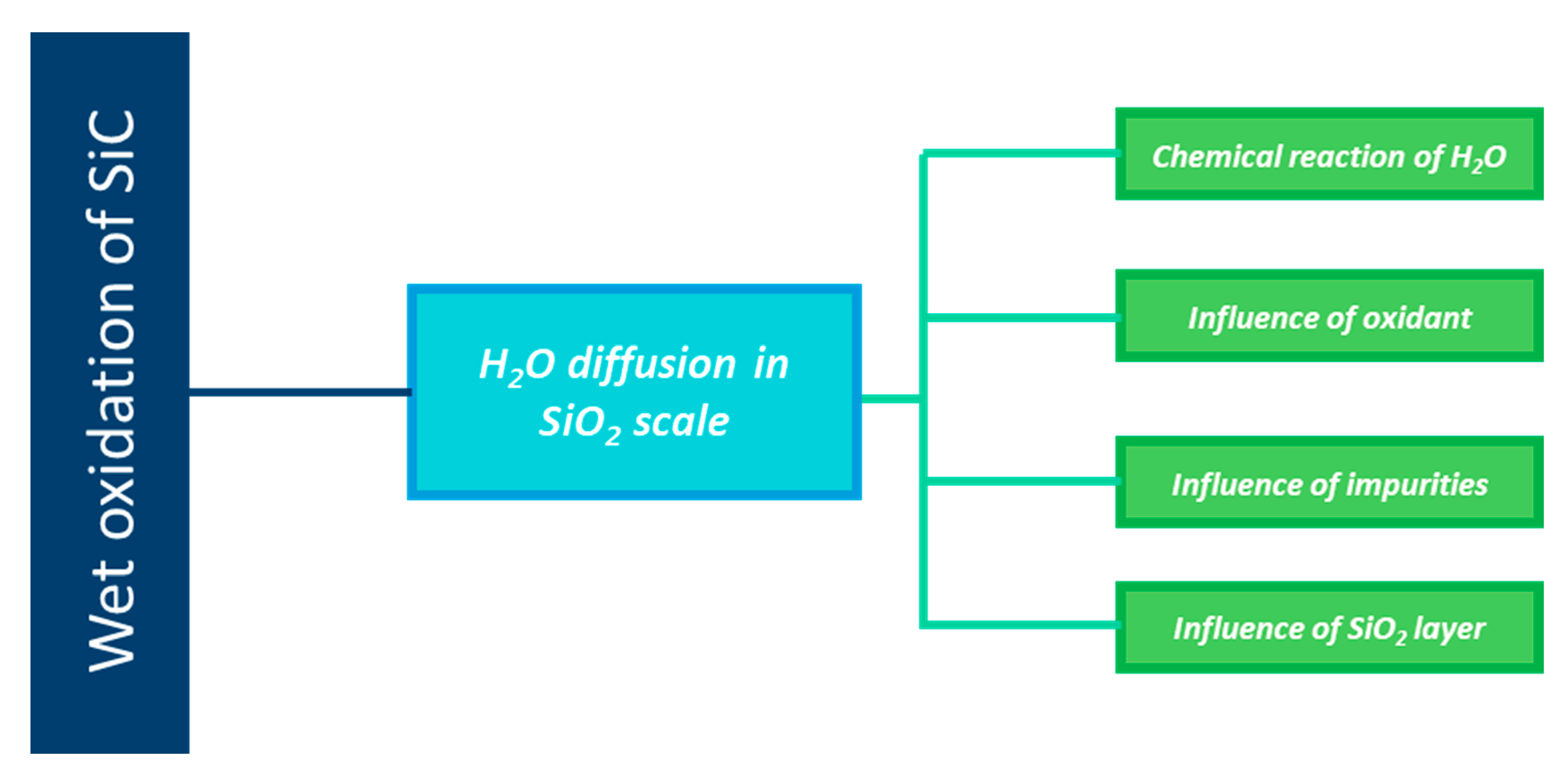
3.2.5. Particular Reaction of Water with the Oxide Layer
3.2.6. Oxidation Rate-Determining Step
3.2.7. Crystal Orientation Effect
3.3. Conclusions
- −
- At short oxidation time, a gas diffusion mechanism is dominant (parabolic regime) whereas at long times, a surface-reaction mechanism is dominant (linear regime),
- −
- For the gas diffusion mechanism, temperature plays an important role: at low temperatures, the oxygen diffusion is molecular whereas above 1350 °C, the diffusing species are ionic oxygen,
- −
- The oxidation behavior is complicated when the crystallization of amorphous silica takes part in the oxidation process. This reduces the oxidant transport and leads to the decrease of the oxidation rates,
- −
- Finally, the presence of impurities is not negligible and could be involved in all these studies. On one hand, it enhances the crystallization of the scale which leads to an increase of defects and, on the other hand, it creates high-permeable and viscous oxides. Both mechanisms result in a faster oxygen diffusion.
3.4. Dissolution-Reaction Model for Water through Oxide Scales
- ➔
- When C >> S, the diffusion process is not influenced by the reaction, so and
- ➔
- When S >> C, the effective diffusion coefficient decreases, as the reaction of molecular water occurs in the oxide layer, as described by the equilibrium Equation (31). Therefore, the effective diffusion coefficient takes the following form:
- −
- The open porosity of the network allows the permeation of molecular species and,
- −
- The Si-O-Si bridges network provides defects (as lattice vacancies) through which structural self-diffusion occurs with breaking and reforming of the bonds.
3.5. Conclusions
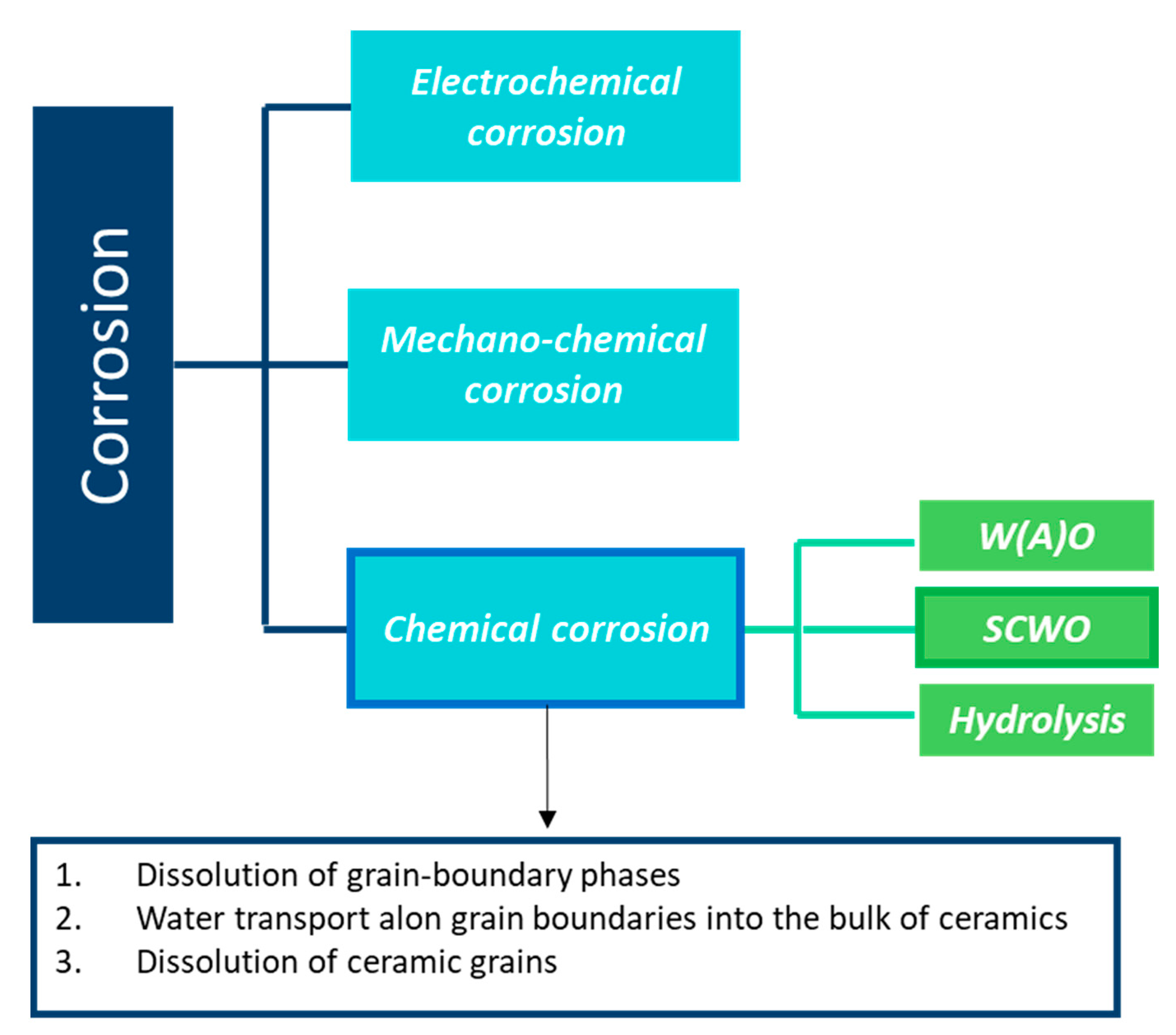
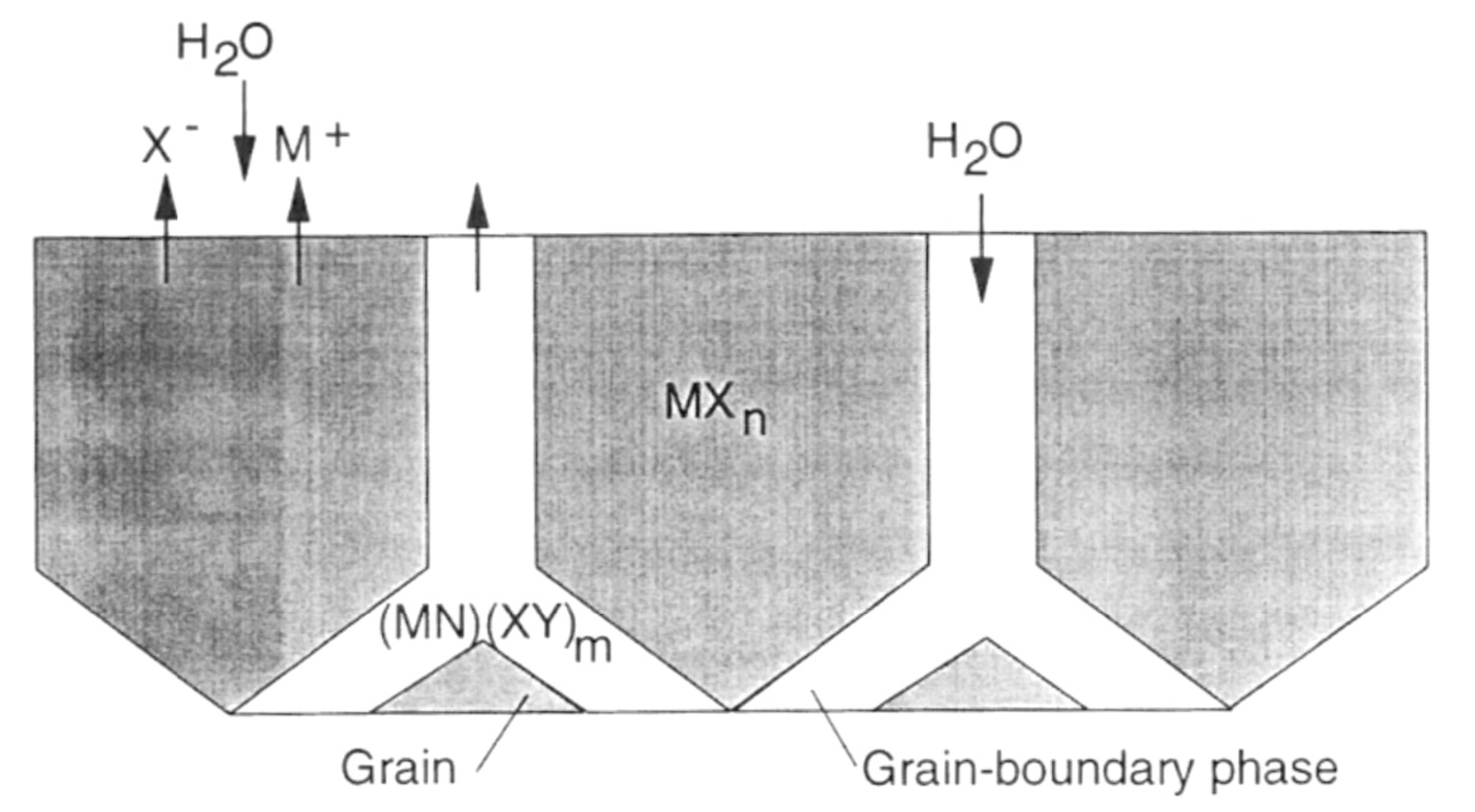
4. Hydrothermal Corrosion of Silicon Carbides Materials
- Reaction of grain-boundary phases,
- Water transport along grain boundaries into the bulk of ceramics,
- Reaction of ceramic grains.
4.1. Reaction Model for Chemical Corrosion
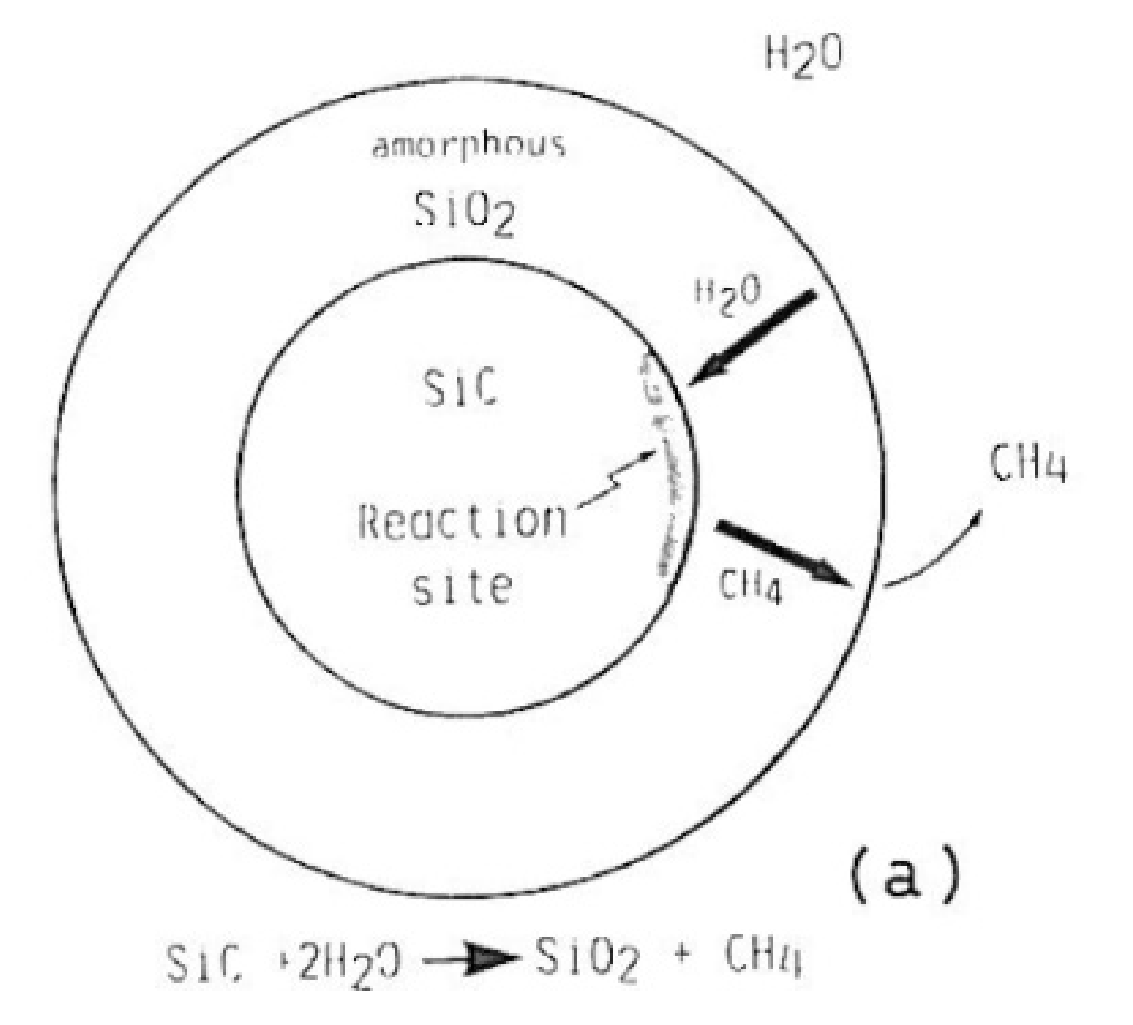
4.1.1. Yoshimura’s Model for Hydrothermal Oxidation of SiC in Supercritical Water
4.1.2. Hirayama’s Model for SiC Corrosion in Water Vapor
- −
- For a deoxygenated solution:SiC + 4H2O = H2SiO42− + 2H+ + CH4
- −
- For an oxygenated solution:SiC + 2O2 + 2H2O = H3SiO4− + CO32− + 4H+
4.1.3. Allongue’s Model for Dissolution of Silicon in Liquid Water
4.1.4. Conclusions
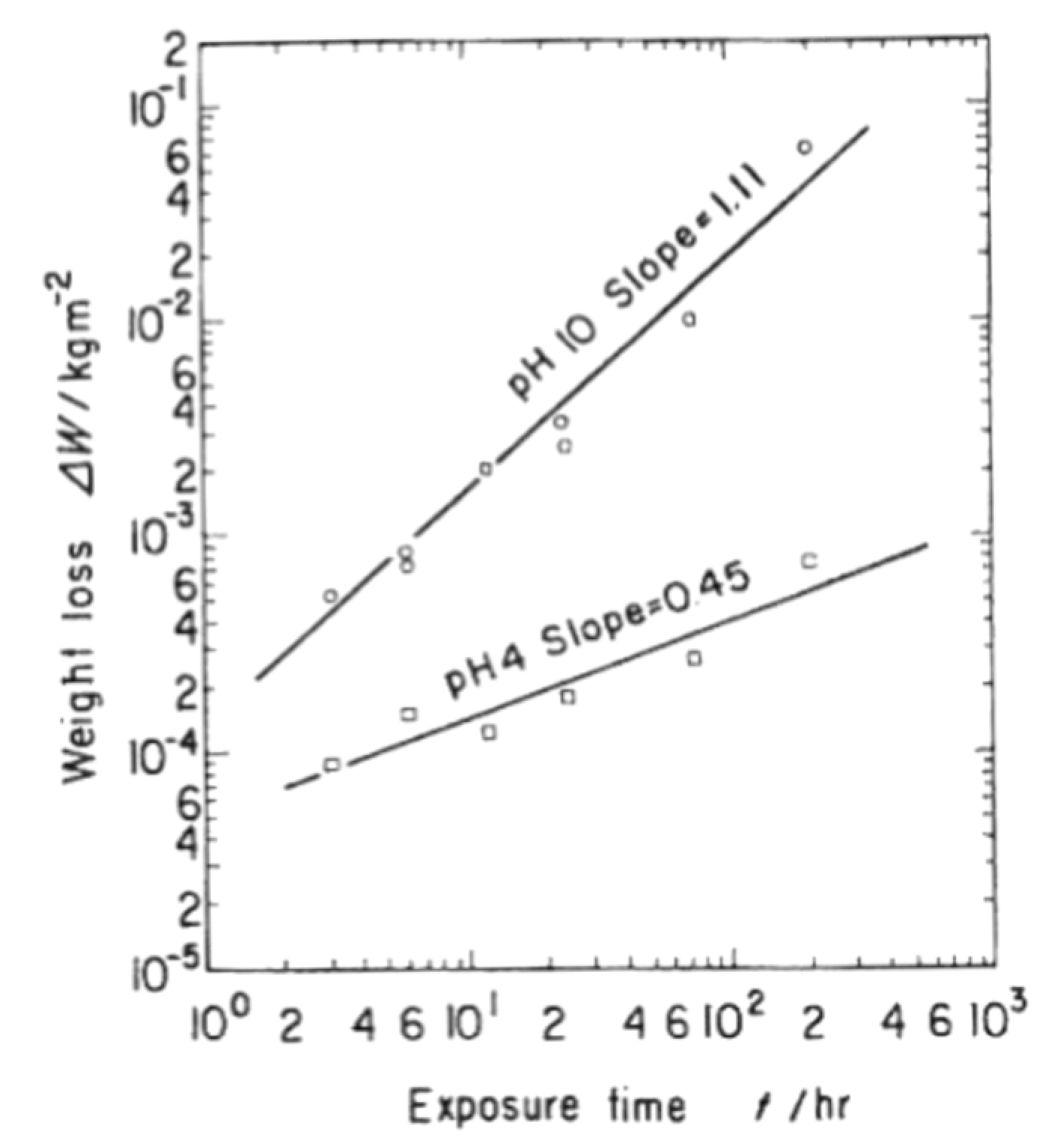
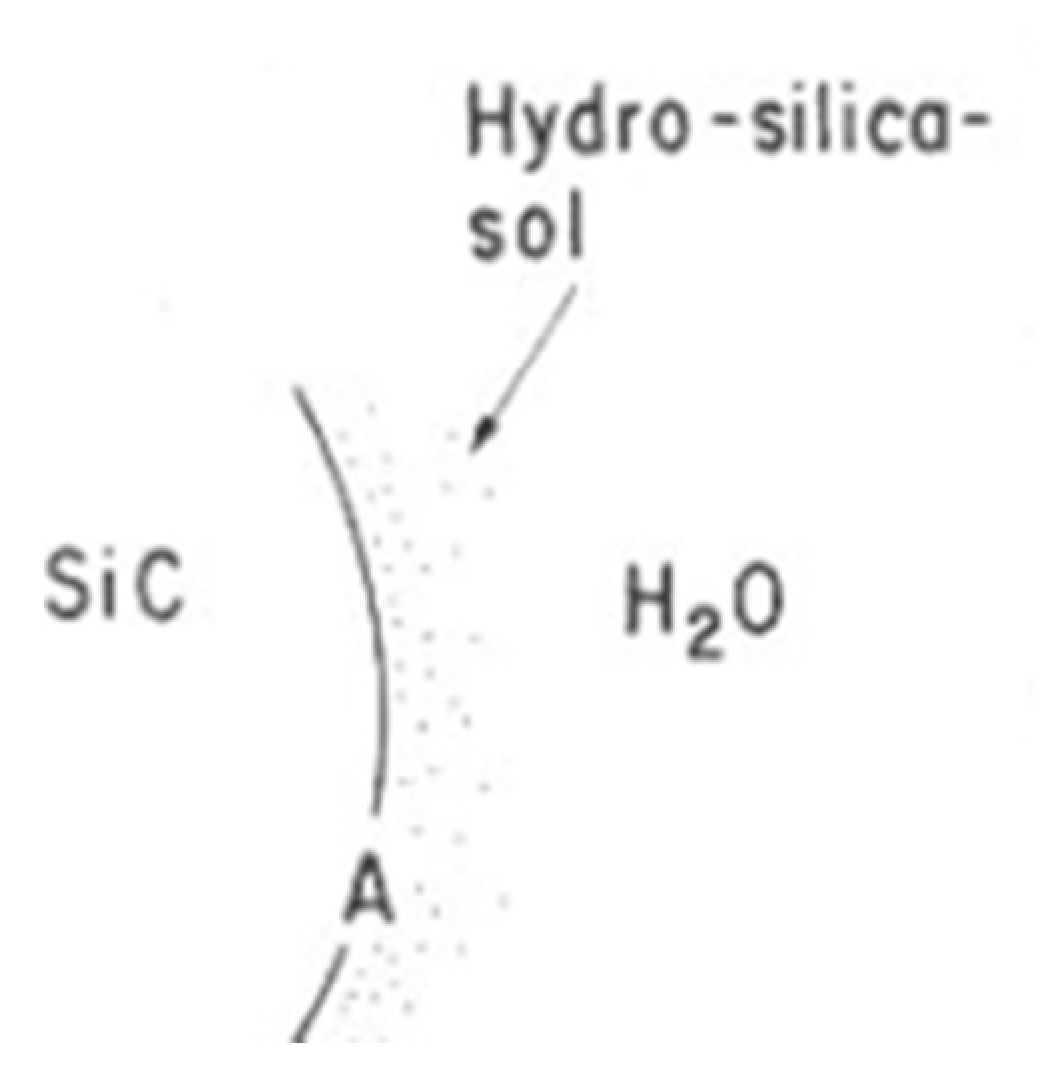
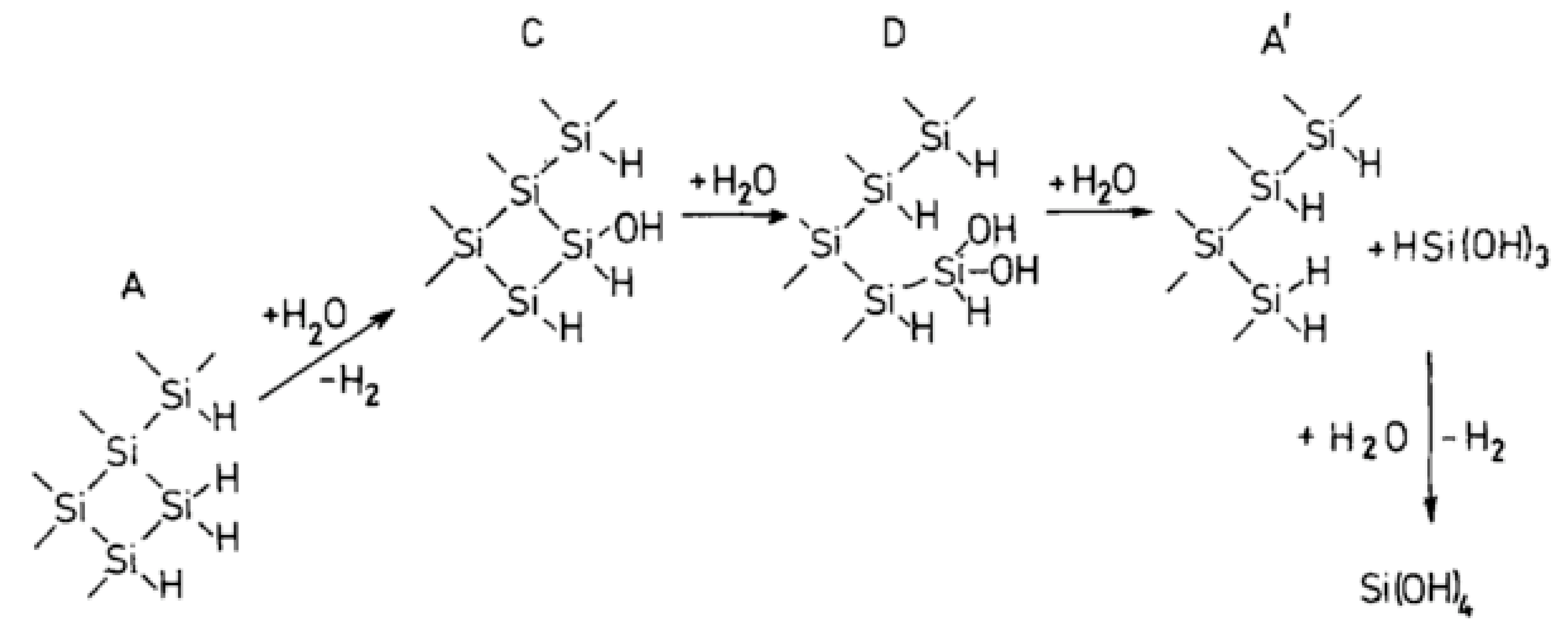
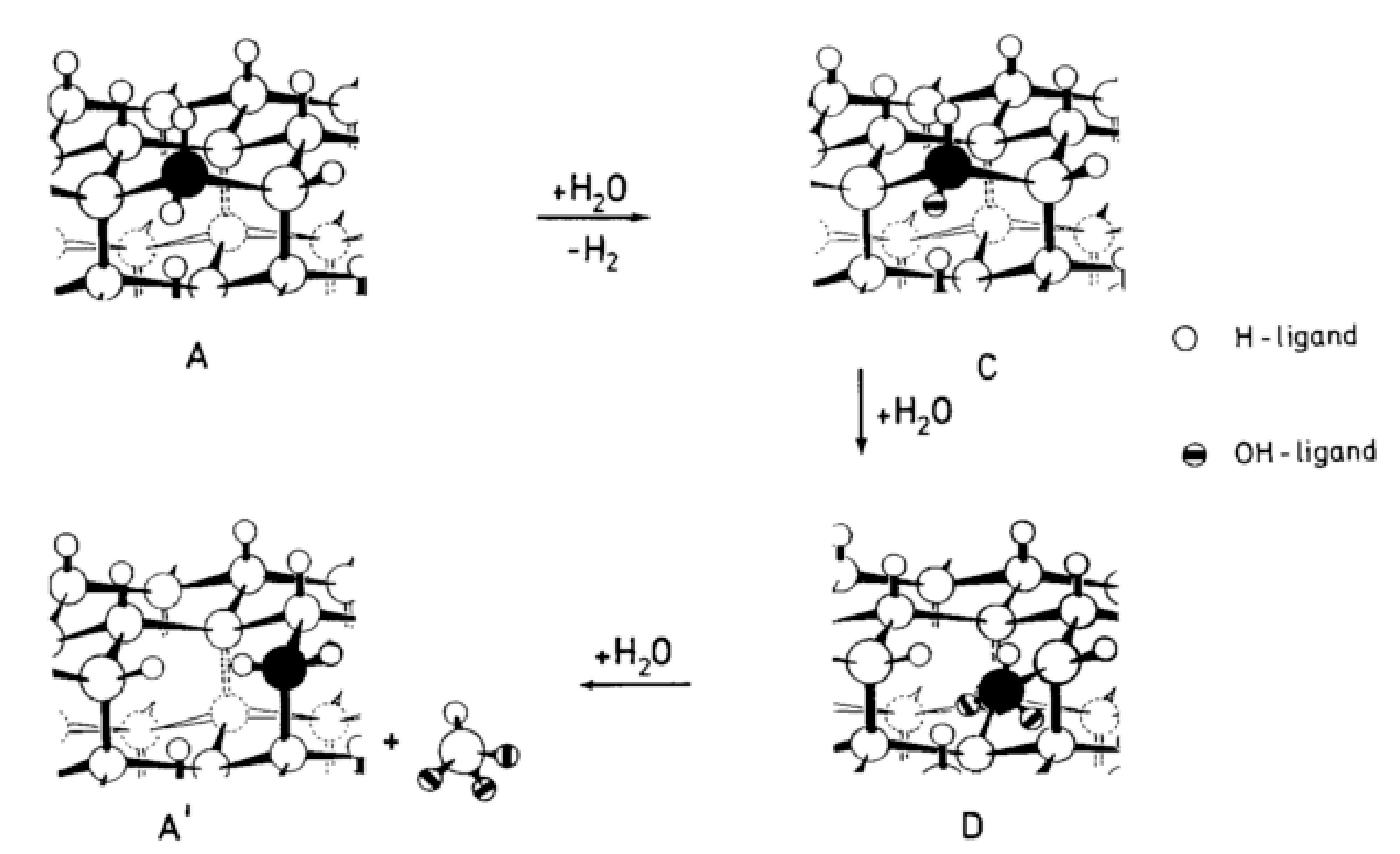
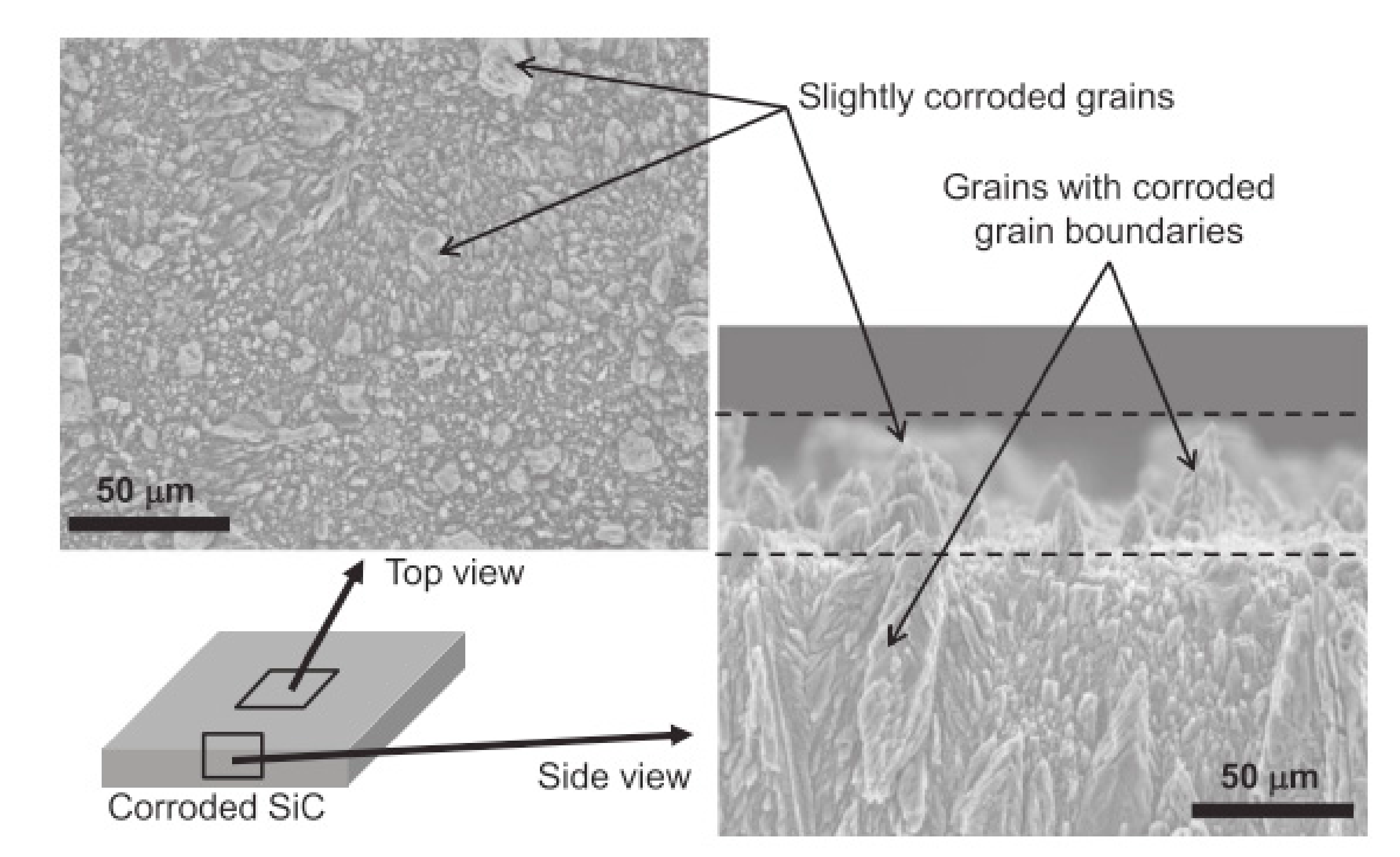
4.2. Hydrothermal Corrosion of the Surface
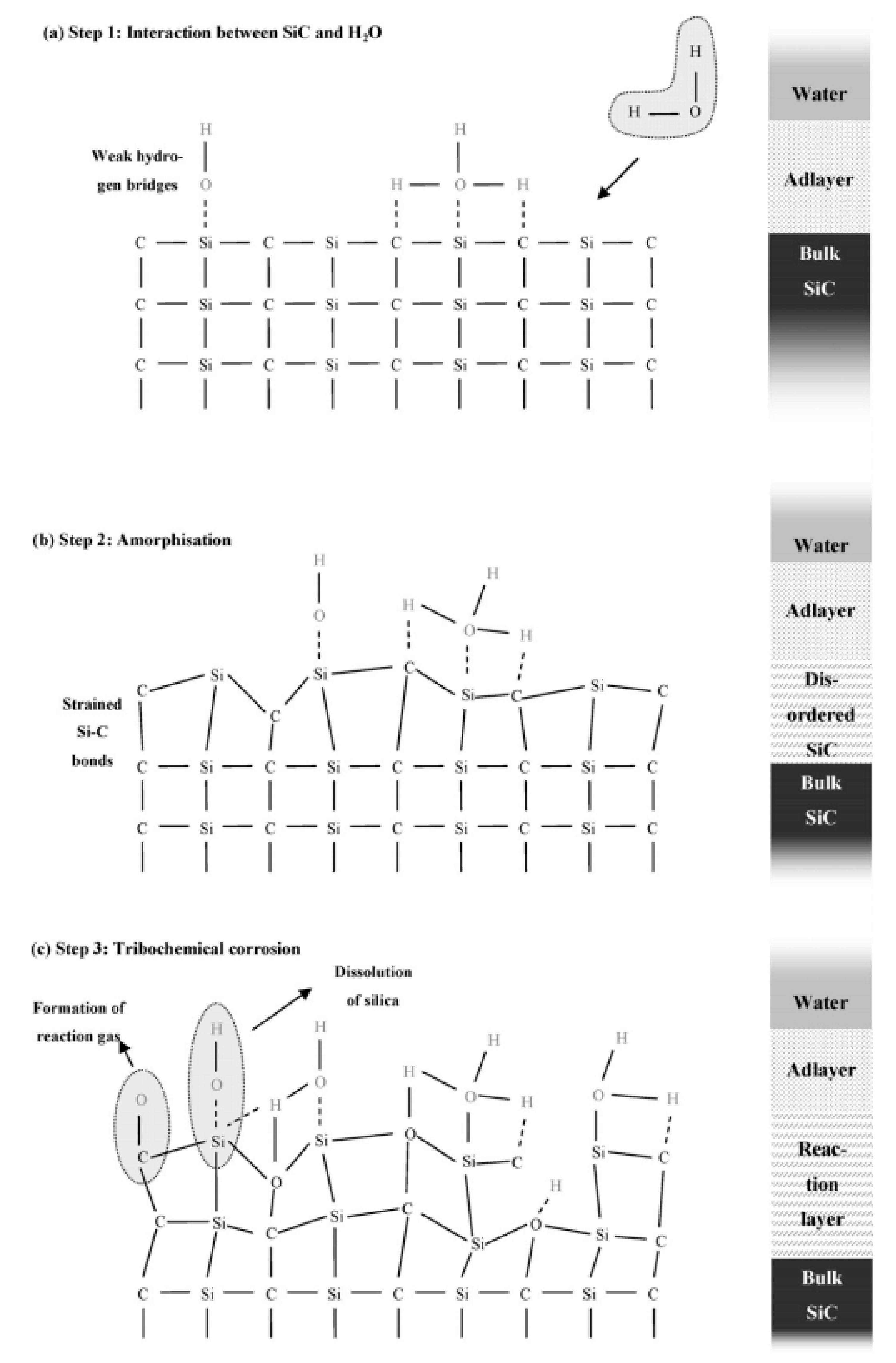
4.3. Reaction Model for Tribochemical Corrosion
- Interaction between SiC and water: The bulk reaction leads to the formation of OH groups and to the saturation of dangling bonds. Moreover, weak hydrogen bridges are created.
- Amorphization: Initially, mechanical stress causes the superficial amorphization of SiC. Therefore, disordered layers and strained Si-C bonds might form with higher susceptibility to be attacked by water. However, neither silica nor oxycarbidic phases were identified knowing that the detection limit of XRD and Raman spectroscopy is about 100 nm.
- Tribochemical corrosion: Simultaneously, silica dissolves in water, and, for low water-to-SiC ratios, it precipitates. Likewise, a cavitation-like wear phenomenon created by the release of gaseous compounds can cause the delamination of the layer.
4.4. Conclusions
5. Supercritical Water Corrosion of Silicon Carbide Materials
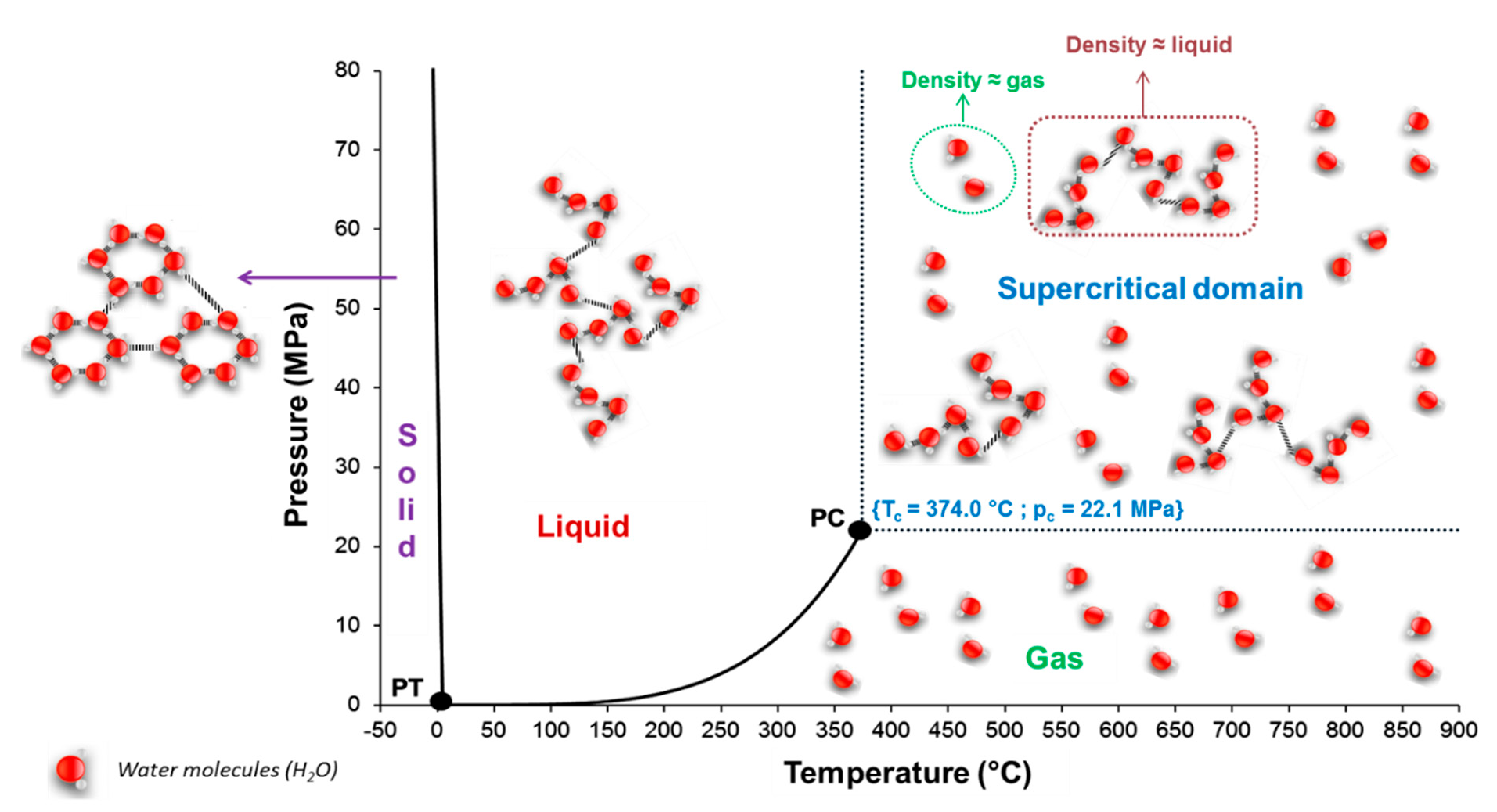
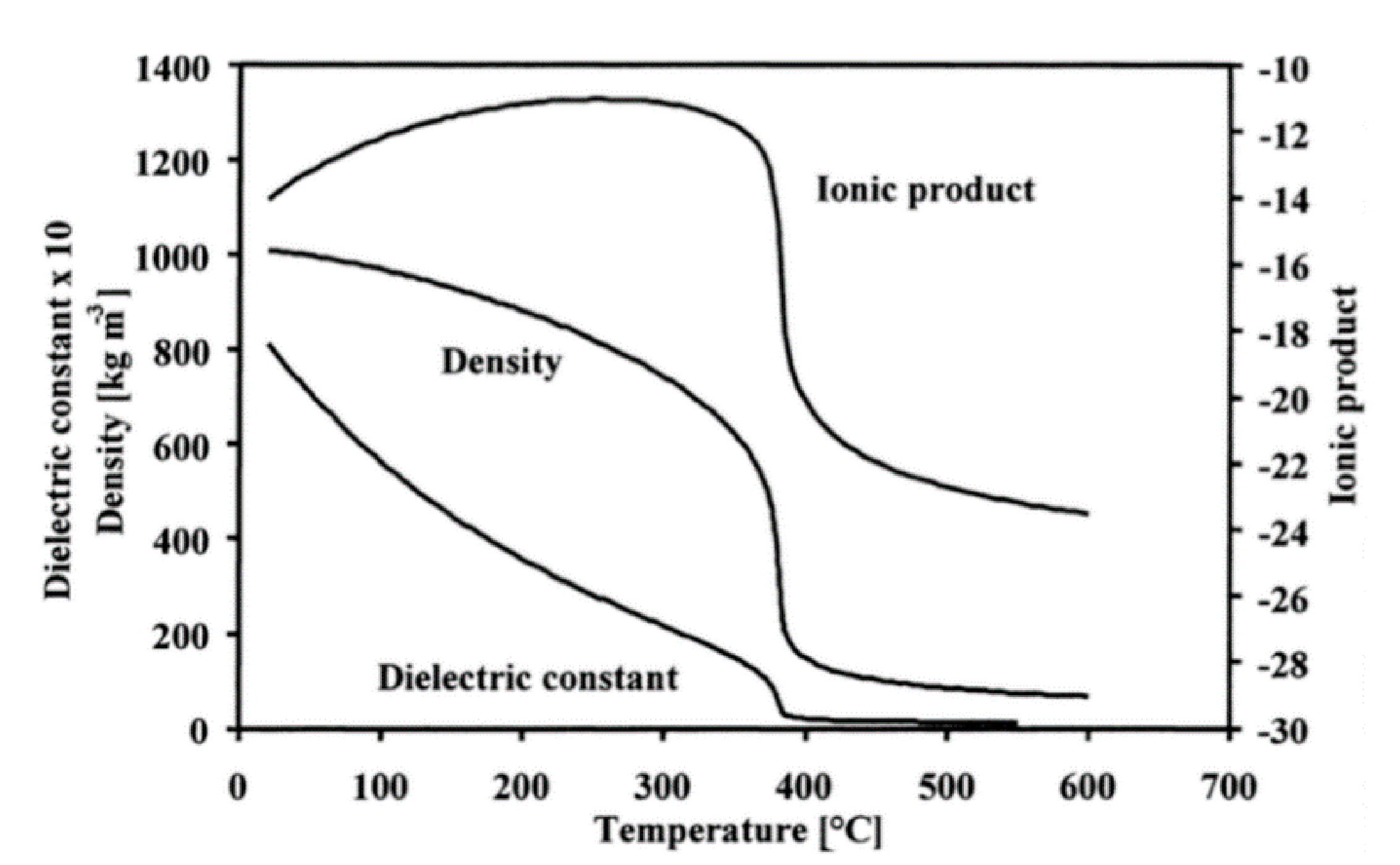
5.1. Supercritical Water Characteristics
5.2. Reaction Model of the SiC Hydrothermal Oxidation
- −
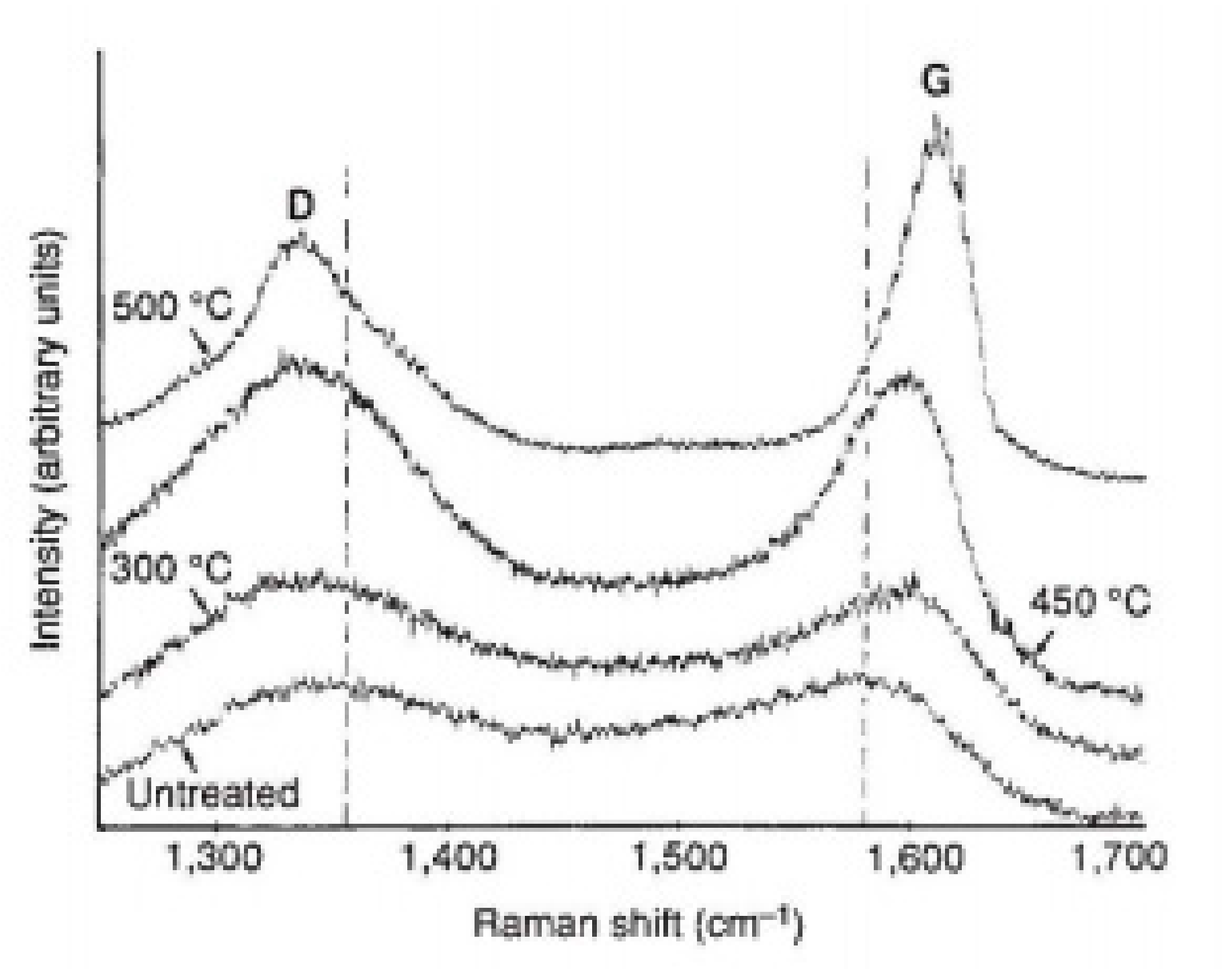
- The temperature and time of treatment can affect the composition of the carbon film from amorphous to graphitic carbon,
- −
- Above a certain temperature and reaction time, the yield of carbon reaches a maximum value,
- −
- Above a certain temperature, the carbon film is oxidized,
- −
- The influence of H2O:SiC is not well understood.
- At low H2O:SiC molar ratios (1:10), both carbon and silica were deposited,
- At intermediate H2O:SiC molar ratios (2:1), both carbon and silica were produced, but silica is dissolved in the water as follows (70):
- 3.
- At higher H2O:SiC molar ratios (10:1), neither carbon nor silica was identified on the surface of SiC (for a nanoscale detection limit) as the carbon reacts with water to form CO/CO2 and silica dissolves in water.
- ➔
- SCW can facilitate the transfer of carbon by-products (CO and CO2) because of its high diffusivity. Therefore, the transfer of carbon by-products is one of the key factors promoting SiC oxidation.
- ➔
- For NCW, the temperature is too low for the release of carbon by-products, so no SiC oxidation occurs.
5.3. Nanoporous Carbon Film Formation
- (1)
- The presence of a good substrate: by acting as a template, the cubic structure of β-SiC could allow the diamond growth,
- (2)
- The formation of hydrogen during hydrothermal treatment of SiC suggests that diamond was produced as hydrogen plays a role in the nucleation during diamond growth,
- (3)
- Tetrahedral carbon in SiC is believed to be transformed into diamond and not into graphite for energetical reason,
- (4)
- Preferential oxidation of sp2-bonded carbon by water seems to lead to the formation of carbon nuclei if the reaction (82) is replaced by the two following ones (85) and (86):
5.4. The Use of Silicon Carbide in the Nuclear Field
5.5. Conclusions
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Riedel, R. Handbook of Ceramic Hard Materials; Wiley-VCH: Hoboken, NJ, USA, 2000; ISBN 9783527299720. [Google Scholar]
- Yajima, S.; Hayashi, J.; Omori, M. Continuous silicon fibre of high tensile strength. Chem. Lett. 1975, 9, 931–934. [Google Scholar] [CrossRef]
- Roewer, G.; Herzog, U.; Trommer, K.; Muller, E.; Fruhauf, S. Silicon Carbide–A Survey of Synthetic Approaches, Properties and Applications. In Structure and Bonding; Jansen, M., Ed.; Springer: Berlin/Heidelberg, Germany, 2002; Volume 12, pp. 59–127. [Google Scholar]
- Gulbransen, E.A.; Jansson, S.A. The high-temperature oxidation, reduction, and volatilization reactions of silicon and silicon carbide. Oxid. Met. 1972, 4, 181–201. [Google Scholar] [CrossRef]
- Rosenqvist, T. Principles of Extractive Metallurgy; McGraw-Hill: New York, NY, USA, 1974. [Google Scholar]
- Hinze, J.W.; Tripp., W.C.; Graham, H.C. Mass Transport Phenomena in Ceramics; Cooper, A.R., Heuer, A.H., Eds.; Plenum Press: New York, NY, USA; London, UK,, 1975; p. 383. [Google Scholar]
- Jacobson, N.S. Corrosion of silicon-based ceramics in combustion environments. J. Am. Ceram. Soc. 1993, 76, 3–28. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, R.T. Oxidation effects on the mechanical properties of SiC fiber-reinforced reaction-bonded silicon nitride matrix composites. J. Am. Ceram. Soc. 1992, 75, 406–412. [Google Scholar] [CrossRef]
- Zheng, Z.; Tressler, R.E.; Spear, K.E. Oxidation of Single-Crystal Silicon Carbide. Part 1990, I. Experimental Studies. J. Electrochem. Soc. 1990, 137, 854–858. [Google Scholar] [CrossRef]
- Park, D.J.; Jung, Y.I.; Kim, H.G.; Park, J.Y.; Koo, Y.H. Oxidation behavior of silicon carbide at 1200 °C in both air and water-vapor-rich environments. Corros. Sci. 2014, 88, 416–422. [Google Scholar] [CrossRef]
- Deal, B.E.; Grove, A.S. General Relationship for the Thermal Oxidation of Silicon. J. Appl. Phys. 1965, 36, 3770–3778. [Google Scholar] [CrossRef] [Green Version]
- Harris, R.C.A. Oxidation of 6H- Silicon Carbide Platelets. J. Am. Ceram. Soc. 1975, 58, 7–9. [Google Scholar] [CrossRef]
- Massoud, H.Z. Thermal Oxidation of Silicon in Dry Oxygen Growth-rate Enchancement in the Thin Regime. I. Experimental Results. J. Electrochem. Soc. Solid State Sci. Technol. 1985, 132, 2685. [Google Scholar]
- Massoud, H.Z. Thermal Oxidation of Silicon in Dry Oxygen Growth-rate Enchancement in the Thin Regime. II. Physical Mechanisms. J. Electrochem. Soc. Solid State Sci. Technol. 1985, 132, 2693. [Google Scholar]
- Goto, D.; Hijikata, Y. Unified theory of silicon carbide oxidation based on the Si and C emission model. J. Phys. D Appl. Phys. 2016, 49, 225103. [Google Scholar] [CrossRef]
- Kageshima, H.; Shiraishi, K.; Uematsu, M. Universal theory of Si oxidation rate and importance of interfacial Si emission. Jpn. J. Appl. Phys. 1999, 38, L971. [Google Scholar] [CrossRef]
- Kouda, K.; Hijikaya, Y.; Yagi, S.; Yaguchi, H.; Yoshida, S. Oxygen partial pressure dependence of the SiC oxidation process studied by in-situ spectroscopic ellipsometry. J. Appl. Phys. 2012, 112, 24502. [Google Scholar] [CrossRef]
- Hijikata, Y. Macroscopic simulations of the SiC thermal oxidation process based on the Si and C emission model. Diam. Relat. Mater. 2019, 92, 253–258. [Google Scholar] [CrossRef]
- Hasunuma, R. Interfacial transition layer in thermally grown SiO2 film on 4H-SiC. In Proceedings of the IEEE International Conference on IC Design and Technology (ICICDT), Austin, TX, USA, 23–25 May 2017; pp. 1–4. [Google Scholar]
- Dartora, G.H.S.; Pitthan, E.; Stedile, F.C. Unraveling the mechanisms responsible for the interfacial region formation in 4H-SiC dry thermal oxidation. J. Appl. Phys. 2017, 122, 215301. [Google Scholar] [CrossRef]
- Akiyama, T.; Hori, S.; Nakamura, K.; Ito, T.; Kageshima, H.; Uematsu, M.; Shiraishi, K. Reaction mechanisms at 4H-SiC/SiO2 interface during wet SiC oxidation. Jpn. J. Appl. Phys. 2018, 57, 04FR08. [Google Scholar] [CrossRef]
- Gao, H.; Wang, H.; Niu, M.; Su, L.; Fan, X.; Wen, J.; Wei, Y. Oxidation simulation study of silicon carbide nanowires: A carbon-rich interface state. Appl. Surf. Sci. 2019, 493, 882–888. [Google Scholar] [CrossRef]
- Park, T.; Park, C.; Jung, J.; Yun, G.J. Investigation of silicon carbide oxidation mechanism using ReaxFF molecular dynamics simulation. J. Spacecr. Rocket. 2020, 57, 1328–1334. [Google Scholar] [CrossRef]
- Shimizu, T.; Akiyama, T.; Pradipto, A.-M.; Nakamura, K.; Ito, T.; Kageshima, H.; Uematsu, M.; Shiraishi, K. Ab initio calculations for the effect of wet oxidation condition on the reaction mechanism at 4H-SiC/SiO2 interface. Jpn. J. Appl. Phys. 2020, 59, SMMD01. [Google Scholar] [CrossRef]
- Ervin, G. Oxidation Behavior of Silicon Carbide. J. Am. Ceram. Soc. 1958, 41, 347–352. [Google Scholar] [CrossRef]
- Lu, W.J.; Steckl, A.J.; Chow, T.P.; Katz, W. Thermal oxidation of sputtered silicon carbide thin Films. J. Electrochem. Soc. Solid State Sci. Technol. 1984, 131, 1907–1914. [Google Scholar] [CrossRef]
- Ramberg, C.E.; Cruciani, G.; Spear, K.E.; Tressler, R.E. Passive-oxidation kinetics of high-purity silicon carbide from 800° to 1100 °C. J. Am. Ceram. Soc. 1996, 79, 2897–2911. [Google Scholar] [CrossRef]
- Singhal, S.C. Oxidation kinetics of hot-pressed silicon carbide. J. Mater. Sci. 1976, 11, 1246–1253. [Google Scholar] [CrossRef]
- Costello, J.A.; Tressler, R.E. Oxidation kinetics of hot-pressed and sintered alpha-SiC. J. Am. Ceram. Soc. 1981, 64, 327–331. [Google Scholar] [CrossRef]
- Costello, J.A.; Tressler, R.E. Oxidation kinetics of silicon carbide crystals and ceramics: I, In dry oxygen. J. Am. Ceram. Soc. 1986, 69, 674–681. [Google Scholar] [CrossRef]
- Narushima, T.; Goto, T.; Hirai, T. High-temperature passive oxidation of chemically vapor deposited silicon carbide. J. Am. Ceram. Soc. 1989, 72, 1386–1390. [Google Scholar] [CrossRef]
- Opila, E.J. Oxidation Kinetics of Chemically Vapor-Deposited Silicon Carbide in Wet Oxygen. J. Am. Ceram. Soc. 1994, 77, 730–736. [Google Scholar] [CrossRef]
- Ogbuji, L.U.J.T.; Opila, E.J. A comparison of the oxidation kinetics of SiC and Si3N. J. Electrochem. Soc. 1995, 142, 925–930. [Google Scholar] [CrossRef]
- Goto, T.; Homma, H.; Hirai, T. Effect of oxygen partial pressure on the high-temperature oxidation of CVD SiC. Corros. Sci. 2002, 44, 359–370. [Google Scholar] [CrossRef]
- Nakatogawa, T. Silicon carbide non-ohmic resistors. II. Oxidation rates of silicon carbide. J. Soc. Chem. Ind. Jpn. 1954, 57, 348–350. [Google Scholar]
- Adamsky, R.F. Oxidation of silicon carbide in the temperature range 1200 to 1500 °C. J. Phys. Chem. 1953, 63, 305–307. [Google Scholar] [CrossRef]
- Jorgensen, P.J.; Wadsworth, M.E.; Cutler, I.B. Oxidation of Silicon Carbide. J. Am. Ceram. Soc. 1959, 4, 613–616. [Google Scholar] [CrossRef]
- Jorgensen, P.J.; Wadsworth, M.E.; Cutler, I.B. Effects of Oxygen Partial Pressure on the Oxidation of Silicon Carbide. J. Mater. Chem. 1960, 43, 209–212. [Google Scholar] [CrossRef]
- Ainger, F.W. The formation and devitrification of oxides on silicon. J. Mater. Sci. 1966, 1, 1–13. [Google Scholar] [CrossRef]
- Pultz, W.W. Temperature and oxygen pressure dependence of silicon carbide oxidation. J. Phys. Chem. 1967, 71, 4556–4558. [Google Scholar] [CrossRef]
- Rosner, D.E.; Allendorf, D.H. High temperature kinetics of the oxidation and nitridation of pyrolytic silicon carbide in dissociated gases. J. Phys. Chem. 1970, 74, 1829–1839. [Google Scholar] [CrossRef]
- Antill, J.E.; Warburton, J.B. Oxidation of silicon and silicon carbide in gaseous atmospheres at 1000–1300 °C. In Proceedings of the AGARD Conference Proceedings, Paris, France, 1 January 1970. [Google Scholar]
- Norton, F.J. Permeation of gaseous oxygen through vitreous silica. Nature 1961, 191, 701. [Google Scholar] [CrossRef]
- Motzfeldt, K. On the rate of oxidation of silicon and of silicon carbide in oxygen, and correlation with the permability of silica glass. Acta Chem. Scand. 1964, 18, 1596–1606. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, P.J.; Wadsworth, M.E.; Cutler, I.B. Effects of water vapour on oxidation of silicon carbide. J. Am. Ceram. Soc. 1960, 44, 258–261. [Google Scholar] [CrossRef]
- Presser, V.; Loges, A.; Hemberger, Y.; Nickel, K.G. Microstructural evolution of silica on single-crystal silicon carbide. Part I: Devitrification and oxidation rates. J. Am. Ceram. Soc. 2009, 92, 724–731. [Google Scholar] [CrossRef]
- Wagstaff, F.E. Crystallization kinetics of internally nucleated vitreous silica. J. Mater. Chem. 1968, 51, 449–453. [Google Scholar] [CrossRef]
- Wei, W.-C.; Halloran, J.W. Phase transformation of diphasic aluminosilicate gels. J. Am. Ceram. Soc. 1988, 71, 166–172. [Google Scholar] [CrossRef]
- Lewis, E.A.; Irene, E.A. The Effect of Surface Orientation on Silicon Oxidation Kinetics. J. Electrochem. Soc. 1987, 134, 2332–2339. [Google Scholar] [CrossRef]
- Suzuki, A.; Ashida, H.; Furui, N.; Mameno, K.; Matsunami, H. Thermal oxidation of SiC and elelectric properties of Al-SiO2-SiC MOS structure. Jpn. J. Appl. Phys. Jpn. 1982, 21, 579–585. [Google Scholar] [CrossRef]
- Onneby, C.; Pantano, C.G. Silicon oxycarbide formation on SiC surfaces and at the SiC/SiO2 interface. J. Vac. Sci. Technol. A Vac. Surf. Film. 1997, 15, 1597–1602. [Google Scholar] [CrossRef]
- Vickridge, I.; Ganem, J.; Hoshino, Y.; Trimaille, I. Growth of SiO2 on SiC by dry thermal oxidation: Mechanisms. J. Phys. D Appl. Phys. 2007, 40, 6254. [Google Scholar] [CrossRef]
- Gavrikov, A.; Knizhnik, A.; Safonov, A.; Scherbinin, A.; Bagatur’yants, A.; Potapkin, B.; Chatterjee, A.; Matocha, K. First-principles-based investigation of kinetic mechanism of SiC 0001 dry oxidation including defect generation and passivation. J. Appl. Phys. 2008, 104, 093508. [Google Scholar] [CrossRef]
- Chang, K.C.; Nuhfer, N.T.; Porter, L.M.; Wahab, Q. High-carbon concentrations at the silicon dioxide-silicon carbide interface identified by electron energy loss spectroscopy. Appl. Phys. Lett. 2000, 77, 2186–2188. [Google Scholar] [CrossRef]
- Suzuki, H. A study of the oxidation of pure silicon carbide powders. Yogyo Kyokaishi 1966, 65, 88. [Google Scholar] [CrossRef] [Green Version]
- Chaudhry, M.I. A study of native oxides of B-SiC using Auger electron spectroscopy. J. Mater. Res. 1989, 4, 404–407. [Google Scholar] [CrossRef]
- Fitzer, E.; Ebi, R. Kinetic Studies on the Oxidation of Silicon Carbide; Marshall, R.C., Faust, J.W., Jr., Ryan, C.E., Eds.; University of South Carolina Press: Colombia, SC, USA, 1974; pp. 320–328. [Google Scholar]
- Singhal, S.C. Effect of water vapor on the oxidation of hot-pressed silicon nitride and silicon carbide. J. Am. Ceram. Soc. 1976, 59, 81–82. [Google Scholar] [CrossRef]
- Tedmon, C.S. The effect of oxide volatilization on the oxidation kinetics of Cr and Fe-Cr alloys. J. Electrochem. Soc. 1966, 113, 766. [Google Scholar] [CrossRef]
- Irene, E.; Ghez, R. Silicon oxidation studies: The role of H2O. J. Electrochem. Soc. Solid State Sci. Technol. 1977, 124, 1757–1761. [Google Scholar]
- Opila, E.J.; Hann, R.E. Paralinear oxidation of CVD SiC in water vapor. J. Am. Ceram. Soc. 1997, 80, 197–205. [Google Scholar] [CrossRef]
- Opila, E.J.; Fox, D.S.; Jacobson, N. Mass spectrometric identification of Si-O-H(g) species from the reaction of silica with water vapor at atmospheric pressure. J. Am. Ceram. Soc. 1997, 80, 1009–1012. [Google Scholar] [CrossRef]
- Opila, E.J.; Robinson, R.C. The oxidation rate of SiC in high pressure water vapor environments. In High-temperature Corrosion Materials Chemistry; McNallan, M.J., Opila, E.J., Maruyama, T., Narita, T., Eds.; The Electrochemical Soc.: Pennington, NJ, USA, 1999; pp. 398–406. [Google Scholar]
- Opila, E.J. Oxidation and volatilization of silica formers in water vapor. J. Mater. Chem. 2003, 86, 1238–1248. [Google Scholar] [CrossRef] [Green Version]
- Opila, E.J.; Robinson, R.C.; Cuy, M.D. High temperature corrosion of silicon carbide and silicon nitride in water vapor. In Advances in Science and Technology; Vincenzini, P., Ed.; NASA Glenn Researcher Center: Cleveland, OH, USA, 2003; pp. 243–254. [Google Scholar]
- Opila, E.J. Variation of the oxidation rate of silicon carbide with water-vapor pressure. J. Am. Ceram. Soc. 1999, 82, 625–636. [Google Scholar] [CrossRef]
- Tortorelli, P.F.; More, K.L. Effects of high water-vapor pressure on oxidation of silicon carbide at 1200 °C. J. Am. Chem. Soc. 2003, 86, 1249–1255. [Google Scholar] [CrossRef]
- Haycock, E.W. Transitions from Parabolic to Linear Kinetics in Scaling of Metals. J. Electrochem. Soc. 1959, 106, 771–775. [Google Scholar] [CrossRef]
- Palmour, J.W.; Kim, H.J.; Davis, R.F. WET and Dry Oxidation of Single Crystal β-SiC: Kinetics and Interface Characteristics. MRS Online Proc. Libr. 1985, 54, 553–559. [Google Scholar] [CrossRef]
- Cappelen, H.; Johansen, K.H.; Motzfeldt, K. Oxidation of silicon carbide in oxygen and in water vapour at 1500 °C. Acta Chem. Scand. 1981, 35, 247–254. [Google Scholar] [CrossRef] [Green Version]
- Fung, C.D.; Kopanski, J.J. Thermal oxidation of 3C silicon carbide single-crystal layers on silicon. Appl. Phys. Lett. 1984, 45, 757–759. [Google Scholar] [CrossRef]
- Narushima, T.; Goto, T.; Iguchi, Y.; Hirai, T. High-temperature oxidation of chemically vapor-deposited silicon carbide in wet oxygen at 1823 to 1923 K. J. Am. Ceram. Soc. 1990, 73, 1580–1584. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond; Ithaca, N., Ed.; Cornell University Press: Ithaca, NY, USA, 1960. [Google Scholar]
- Maeda, M.; Nakamura, K.; Ohkubo, T. Oxidation of silicon carbide in a wet atmosphere. J. Mater. Sci. 1988, 23, 3933–3938. [Google Scholar] [CrossRef]
- Sosman, R.B. The properties of silica: An introduction to the properties of substances in the solid non conducting state. Chem. Cat. Co. 1927, 37, 856. [Google Scholar]
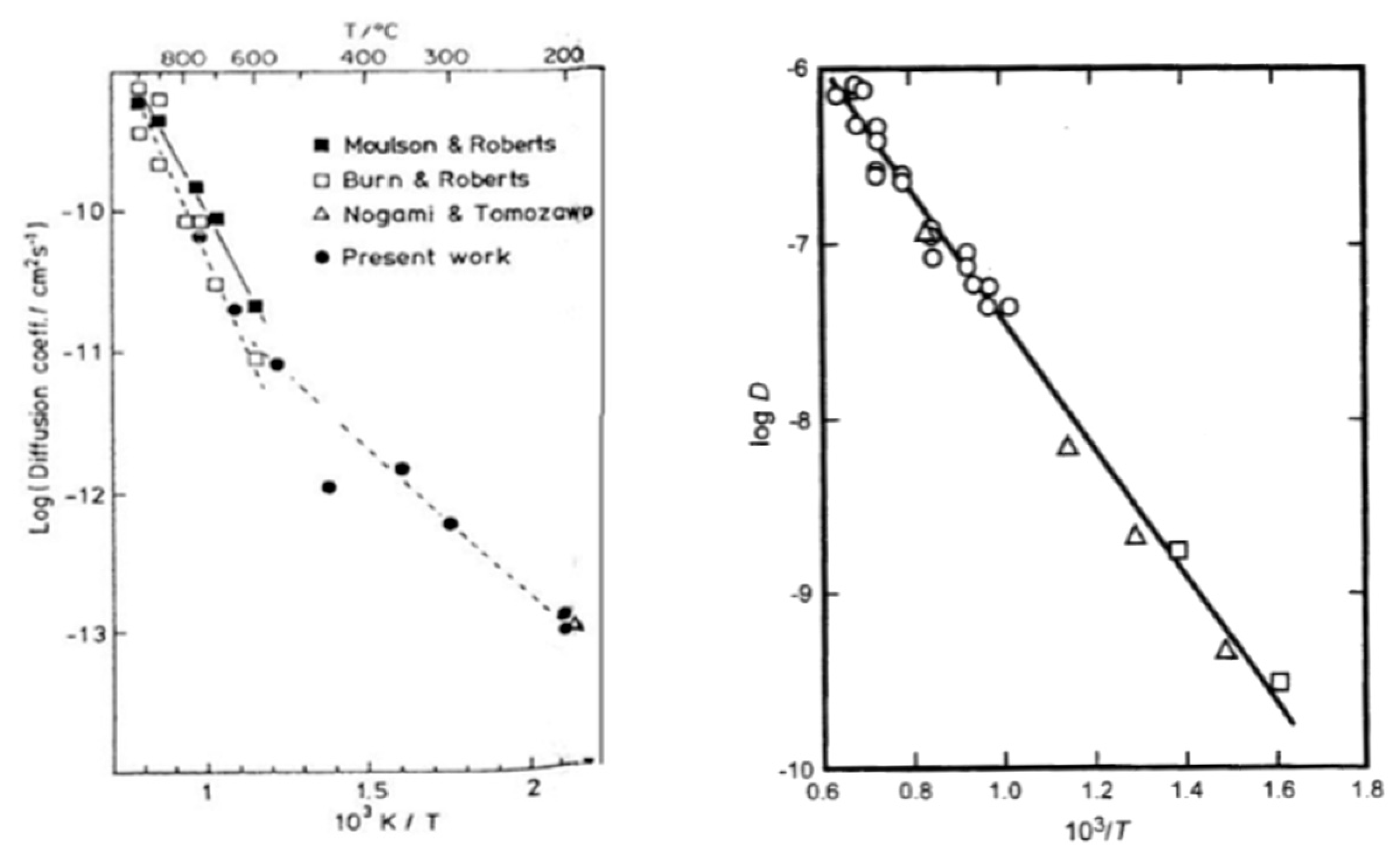
- Lamkin, M.A.; Riley, F.L. Oxygen mobility in silicon dioxide and silicate glasses: A review. J. Eur. Ceram. Soc. 1992, 10, 347–367. [Google Scholar] [CrossRef]
- Wagner, C. Passivity during the Oxidation of Silicon at Elevated Temperatures. J. Appl. Phys. 1985, 29, 1295–1297. [Google Scholar] [CrossRef]
- Moulson, A.J.; Roberts, J.P. Water in silica glass. Trans. Faraday Soc. 1961, 57, 1208–1216. [Google Scholar] [CrossRef]
- Hetherington, G.; Jack, K.H. Water in Vitreous Silica. J. Phys. Chem. Glasses 1962, 3, 129–133. [Google Scholar]
- Brückner, J. Properties and structure of vitreous silica. J. Non Cryst. Solids 1971, 5, 177. [Google Scholar] [CrossRef]
- Doremus, R.H. The diffusion of water in fused silica. In Reactivity of Solids; Mitchell, J.W., Devries, R.C., Eds.; Wiley: New York, NY, USA, 1969; pp. 667–673. [Google Scholar]
- Doremus, R.H. Diffusion of water in crystalline; glass oxides: Diffusion-reaction-model. J. Mater. Res. 1999, 14, 3754–3758. [Google Scholar] [CrossRef]
- Shewmon, P.G. Diffusion in Solids; McGraw-Hill Book Company: New York, NY, USA, 1963. [Google Scholar]
- Roberts, G.J.; Roberts, J.P. An oxygen tracer investigation of the diffusion of ‘water’ in silica glass. Phys. Chem. Glasses 1966, 7, 82–89. [Google Scholar]
- Wakabayashi, H.; Tomozawa, M. Diffusion of Water into Silica Glass at Low Temperature. J. Am. Ceram. Soc. 1989, 72, 1850–1855. [Google Scholar] [CrossRef]
- Fortier, S.M.; Giletti, B.J. An empirical model for predicting diffusion coefficients in silicate minerals. Science 1989, 245, 1481–1484. [Google Scholar] [CrossRef] [PubMed]
- Bremen, W.; Naoumidis, A.; Nickel, H. Oxidationsverhalten des pyrolytisch abgeschiedenen B-SiC unter einer atmosphäre aus CO-CO2-gasgemischen. J. Nucl. Mater. 1977, 71, 56–64. [Google Scholar] [CrossRef]
- Schachtner, R.; Sockel, H. Study of Oxygen Diffusion in Quartz by Activation Analysis. In Reactivity of Solids; Wood, J., Lindqvist, O., Helgesson, C., Vannerberg, N.-G., Eds.; Springer: Berlin/Heidelberg, Germany, 1977; pp. 605–609. [Google Scholar]
- Giletti, B.J.; Semet, M.P.; Yund, R.A. Studies in diffusion: III. Oxidation in feldspars: An ion microprobe determination. Geochem. Cosmochim. Acta 1978, 42, 45–57. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Yoshimura, M. Water effects on corrosion behavior of SiC ceramics. MRS Bull. 1994, XIX, 39–45. [Google Scholar] [CrossRef]
- Kim, W.-J.; Hwang, H.S.; Park, J.Y.; Ryu, W.-S. Corrosion behaviors of sintered; chemically vapor deposited silicon carbide ceramics in water at 360 °C. J. Mater. Sci. Lett. 2003, 22, 581–584. [Google Scholar] [CrossRef]
- Yoshimura, M.; Kase, J.; Somiya, S. Oxidation of SiC powder by high-temperature, high-pressure H2O. J. Mater. Res. 1986, 1, 100–103. [Google Scholar] [CrossRef]
- Hirayama, H.; Kawakubo, T.; Goto, A. Corrosion behavior of silicon carbide in 290 °C water. J. Am. Ceram. Soc. 1989, 72, 2049–2053. [Google Scholar] [CrossRef]
- Allongue, P.; Brune, H.; Gerischer, H. In situ STM observations of the etching of n-Si (111) in NaOH solutions. Surf. Sci. 1992, 275, 414–423. [Google Scholar] [CrossRef] [Green Version]
- Allongue, P.; Costa-Kieling, V.; Gerischer, H. Etching of Silicon in NaOH Solutions. Part II. Electrochemical studies of n-Si (111) and (100) and mechanism of the dissolution. J. Electrochem. Soc. 1993, 140, 1018–1026. [Google Scholar] [CrossRef]
- Smialek, J.L.; Jacobson, N.S. Mechanism of Strength Degradation for Hot Corrosion of alpha-SiC. J. Am. Ceram. Soc. 1986, 69, 741–752. [Google Scholar] [CrossRef]
- Henager, C.H.; Schemer-Kohrn, A.L.; Pitman, S.G.; Senor, D.J.; Geelhood, K.J.; Painter, C.L. Pitting corrosion in CVD SiC at 300 °C in deoxygenated high-purity water. J. Nucl. Mater. 2008, 378, 9–16. [Google Scholar] [CrossRef]
- Barringer, E.; Faiztompkins, Z.; Feinroth, H.; Allen, T.; Lance, M.; Meyer, H.; Gog Lara-Curzio, E. Corrosion of CVD Silicon Carbide in 500 °C Supercritical Water. J. Am. Ceram. Soc. 2007, 90, 315–318. [Google Scholar] [CrossRef]
- Kim, W.-J.; Hwang, H.S.; Park, J.Y. Corrosion behavior of reaction-bonded silicon carbide ceramics in high-temperature water. J. Mater. Sci. Lett. 2002, 21, 733–735. [Google Scholar] [CrossRef]
- Park, J.-Y.; Kim, I.-H.; Jung, Y.-I.; Kim, H.-G.; Park, D.-J.; Kim, W.-J. Long-term corrosion behavior of CVD SiC in 360 °C water and 400 °C steam. J. Nucl. Mater. 2013, 443, 603–607. [Google Scholar] [CrossRef]
- Tan, L.; Allen, T.R.; Barringer, E. Effect of microstructure on the corrosion of CVD-SiC exposed to supercritical water. J. Nucl. Mater. 2009, 394, 95–101. [Google Scholar] [CrossRef]
- Quinn, T. Oxidation wear modeling: I. Wear 1992, 153, 179–200. [Google Scholar] [CrossRef]
- Quinn, T. Oxidational wear modelling: Part II. The general theory of oxidational wear. Wear 1994, 175, 199–208. [Google Scholar] [CrossRef]
- Boch, P.; Platon, F.; Kapelski, G. Tribological and interfacial phenomena in Al2O3/SiC and SiC/SiC couples at high temperature. J. Eur. Ceram. Soc. 1989, 5, 223–228. [Google Scholar] [CrossRef]
- Sasaki, S. The effect of the surrounding atmosphere on the friction and wear of alumina, zirconia, silicon-carbide and silicon-nitride. Wear 1989, 134, 185–200. [Google Scholar] [CrossRef]
- Zum Gahr, K.-H.; Blattner, R.; Hwang, D.-H.; Pöhlmann, K. Micro- and macro-tribological properties of SiC ceramics in sliding contact. Wear 2001, 250, 299–310. [Google Scholar] [CrossRef]
- Presser, V.; Krummhauer, O.; Nickel, K.; Kailer, A.; Berthold, C.; Raisch, C. Tribological and hydrothermal behaviour of silicon carbide under water lubrication. Wear 2009, 266, 771–781. [Google Scholar] [CrossRef]
- Loppinet-Serani, A.; Aymonier, C.; Cansell, F. Supercritical water for environmental technologies. J. Chem. Technol. Biotechnol. 2010, 85, 583–589. [Google Scholar] [CrossRef]
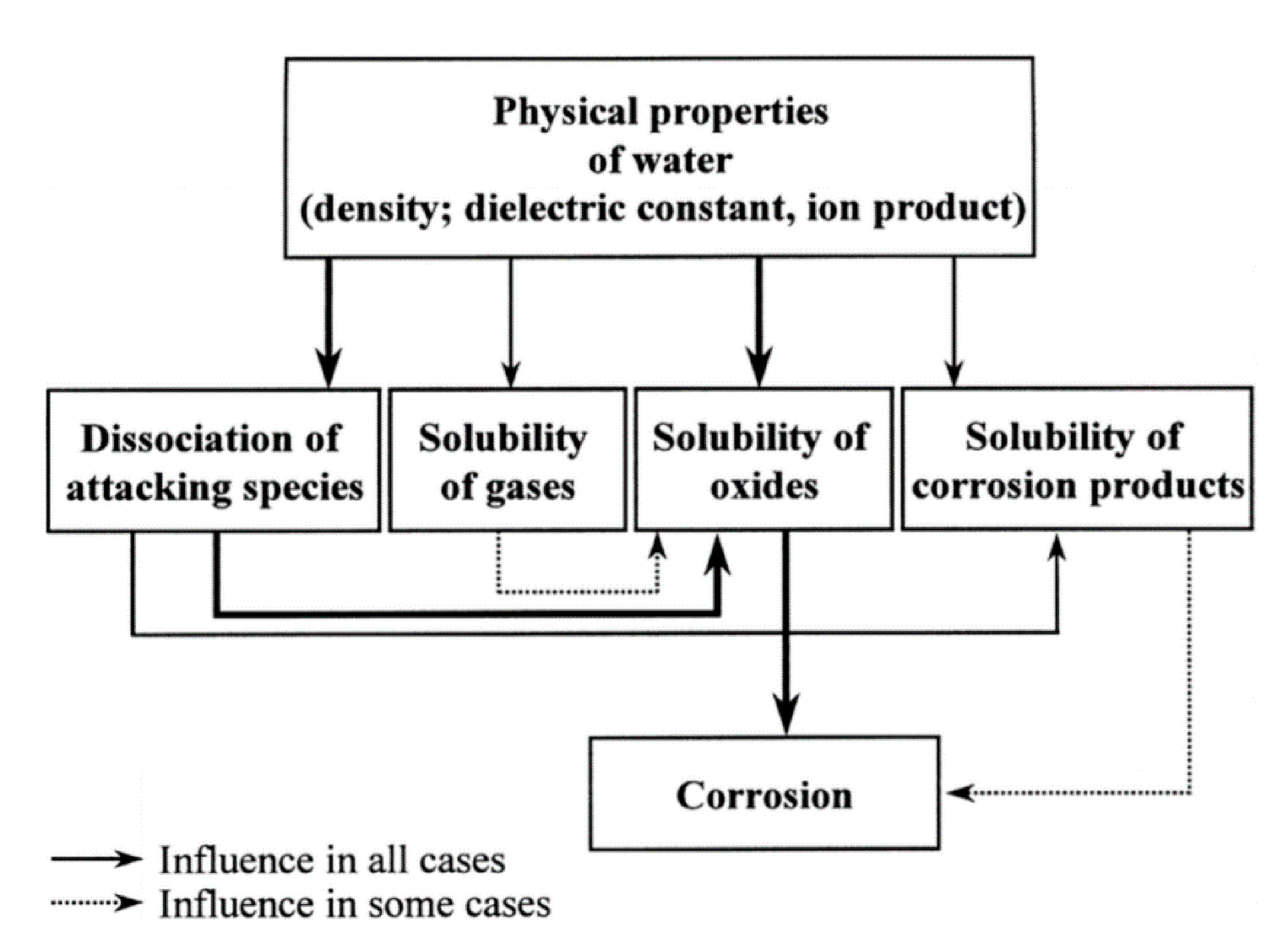
- Kritzer, P.; Boukis, N.; Dinjus, E. Factors controlling corrosion in high-temperature aqueous solutions: A contribution to the dissociation and solubility data influencing corrosion processes. J. Supercrit. Fluids 1999, 15, 205–227. [Google Scholar] [CrossRef]
- Duverger-Nédellec, E.; Voisin, T.; Erriguible, A.; Aymonier, C. Unveiling the complexity of salt(s) in water under transcritical conditions. J. Supercrit. Fluids 2020, 165, 104977. [Google Scholar] [CrossRef]
- Bröll, D.; Kaul, C.; Krämer, A.; Krammer, P.; Richter, T.; Jung, M.; Vogel, H.; Zehner, P. Chemistry in Supercritical Water. Angew. Chem. Int. Ed. 1999, 38, 2998–3014. [Google Scholar] [CrossRef]
- Aymonier, C.; Philippot, G.; Erriguible, A.; Marre, S. Playing with chemistry in supercritical solvents and the associated technologies for advanced materials by design. J. Supercrit. Fluids 2018, 134, 184–196. [Google Scholar] [CrossRef]
- Loppinet-Serani, A.; Aymonier, C.; Cansell, F. Current and foreseeable applications of supercritical water for energy and the environment. ChemSusChem 2008, 1, 486–503. [Google Scholar] [CrossRef]
- Morin, C.; Loppinet-Serani, A.; Cansell, F.; Aymonier, C. Near- and supercritical solvolysis of carbon fibre reinforced polymers (CFRPs) for recycling carbon fibers as a valuable resource: State of the art. J. Supercrit. Fluids 2012, 66, 232–240. [Google Scholar] [CrossRef] [Green Version]
- Adachi, Y.; Lu, B.C.-Y. Supercritical fluid extraction with carbon dioxide and ethylene. Fluid Phase Equilibria 1983, 14, 147–156. [Google Scholar] [CrossRef]
- Jessop, P.G.; Ikariya, T.; Noyori, R. Homogeneous catalysis in supercritical fluid. Chem. Rev. 1999, 99, 475–493. [Google Scholar] [CrossRef] [PubMed]
- Savage, P.E. A perspective on catalysis in sub and supercritical water. J. Supercrit. Fluids 2009, 47, 407–414. [Google Scholar] [CrossRef]
- Kazarian, S. Polymer processing with supercritical fluids. Polym. Sci. Ser. C 2000, 42, 78–101. [Google Scholar]
- Aymonier, C.; Loppinet-Serani, A.; Reveron, H.; Garrabos, Y.; Cansell, F. Review of supercritical fluids in inorganic materials science. J. Supercrit. Fluids 2006, 38, 242–251. [Google Scholar] [CrossRef]
- Adschiri, T.; Kanazawa, K.; Arai, K. Rapid and continuous hydrothermal crystallization of metal oxide particles in supercritical water. J. Am. Chem. Soc. 1992, 75, 1019–1022. [Google Scholar] [CrossRef]
- Cansell, F.; Aymonier, C.; Loppinet-Serani, A. Review on materials science and supercritical fluids. Curr. Opin. Solid State Mater. Sci. 2003, 7, 331–340. [Google Scholar] [CrossRef]
- Marre, S.; Jensen, K.F. Synthesis of micro and nanostructures in microfluidic systems. Chem. Soc. Rev. 2010, 39, 1183–1202. [Google Scholar] [CrossRef]
- Byrappa, K.; Adschiri, T. Hydrothermal technology for nanotechnology. Progress Cryst. Growth Charact. Mater. 2007, 53, 117–166. [Google Scholar] [CrossRef] [Green Version]
- Campardelli, R.; Baldino, L.; Reverchon, E. Supercritical fluids applications in nanomedicine. J. Supercrit. Fluids 2015, 101, 193–214. [Google Scholar] [CrossRef]
- Hirano, S.-I.; Nakamura, K.; Sōmiya, S. Graphitization of carbon in presence of calcium compounds under hydrothermal condition by use of high gas pressure apparatus. In Hydrothermal Reactions for Materials Science and Engineering; Sōmiya, S., Ed.; Springer: Dordrecht, The Netherlands, 1989; pp. 331–336. [Google Scholar]
- Gogotsi, Y.; Yoshimura, M. Formation of carbon films on carbides under hydrothermal conditions. Nature 1994, 367, 630–638. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Yoshimura, M.; Kakihanna, M.; Kanno, Y.; Shibuya, M. Hydrothermal synthesis of carbon films on SiC fibers and particles. Ceram. Trans. 1995, 51, 243–247. [Google Scholar]
- Jacobson, N.; Gogotsi, Y.; Yoshimura, M. Thermodynamic and experimental study of carbon formation on carbides under hydrothermal conditions. J. Mater. Chem. 1995, 5, 595–601. [Google Scholar] [CrossRef]
- Ito, S.; Tomozawa, M. Stress corrosion of silica glass. J. Am. Ceram. Soc. 1981, 64, C160. [Google Scholar] [CrossRef]
- Kase, J. Master’s Thesis. Tokyo Institute of Technology, Yokohama, Japan, 1987; pp. 189–190. [Google Scholar]
- Gogotsi, Y.; Yoshimura, M. Oxidation and properties degradation of SiC fibres below 850 °C. J. Mater. Sci. Lett. 1994, 13, 680–683. [Google Scholar] [CrossRef]
- Gogotsi, Y.G.; Yoshimura, M. Low-temperature oxidation, hydrothermal corrosion, and their effects on properties of SiC (tyranno) fibers. J. Am. Ceram. Soc. 1995, 78, 1439–1450. [Google Scholar] [CrossRef]
- Li, J.; Eveno, P.; Huntz, A.M. Oxidation of silicon carbide. Werkst. Korros. 1987, 41, 716–725. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Yoshimura, M. Degradation of SiC-based fibres in high-temperature, high pressure. J. Mater. Sci. Lett. 1994, 13, 395–399. [Google Scholar] [CrossRef]
- Kraft, T.; Nickel, K.G.; Gogotsi, Y. Hydrothermal degradation of chemical vapour deposited SiC fibres. J. Mater. Sci. 1998, 33, 4357–4364. [Google Scholar] [CrossRef]
- Futatsuki, T.; Oe, T.; Aoki, H.; Kimura, C.; Komatsu, N.; Sugino, T. Low-temperature oxidation of SiC surfaces by supercritical water oxidation. Appl. Surf. Sci. 2010, 256, 6512–6517. [Google Scholar] [CrossRef]
- Yoko, A.; Oshima, Y. Recovery of silicon from silicon sludge using supercritical water. J. Supercrit. Fluids 2013, 75, 1–5. [Google Scholar] [CrossRef]
- Le Coustumer, P.; Monthioux, M.; Oberlin, A. Understanding Nicalon Fibre. J. Eur. Ceram. Soc. 1993, 11, 95–103. [Google Scholar] [CrossRef]
- Yajima, S.; Iwai, T.; Yamamura, T.; Okamura, K.; Hasegawa, Y. Synthesis of a polytitanocarbosilane and its conversion into inorganic compounds. J. Mater. Res. 1981, 16, 1349–1355. [Google Scholar] [CrossRef]
- Gogotsi, Y. Nanostructure Carbon Coatings. NATO Advanced Research Workshop on Nanostructured Films and Coating. Ser. 3 High Technol. 2000, 78, 25–40. [Google Scholar]
- Gogotsi, Y.; Kofstad, P.; Yoshimura, M.; Nickel, K.G. Formation of sp3-bonded carbon upon hydrothermal treatment of SiC. Diam. Relat. Mater. 1996, 5, 151–162. [Google Scholar] [CrossRef]
- Gogotsi, Y.; Nickel, K.G.; Bahloul-Hourlier, D.; Merle-Mejean, T.; Khomenko, G.E.; Skjerlie, K.P. Structure of carbon produced hydrothermal treatment of β-SiC powder. J. Mater. Chem. 1996, 6, 595–604. [Google Scholar] [CrossRef]
- Wang, W.; Wang, T.; Chen, B. Primary study on the irradiation effects of high energy C+ and H+ on diamond-like carbon films. J. Appl. Phys. 1992, 72, 69–72. [Google Scholar] [CrossRef]
- Badzian, A.; Badzian, T. Diamond homoepitaxy by Chemical Vapor Deposition. Diam. Relat. Mater. 1993, 2, 147–157. [Google Scholar] [CrossRef]
- Prawer, S.; Nugent, K.W.; Jamieson, D.N. The Raman Spectrum of Amorphous Diamond. Diam. Relat. Mater. 1998, 7, 106–110. [Google Scholar] [CrossRef]
- Peng, T.; Lv, H.; He, D.; Pan, M.; Mu, S. Direct transformation of amorphous silicon carbide into graphene under low temperature and ambient pressure. Sci. Rep. 2013, 3, 1148. [Google Scholar] [CrossRef] [PubMed]
- DeVries, R.C. Hydrothermal carbon: A review from carbon in Herkimer diamonds to that in real diamonds. Adv. Ceram. 1990, 3, 181–205. [Google Scholar]
- Rumble, D., III; Hoering, T.C. Carbon isotope geochemistry of graphite vein deposits from New Hampshire, U.S.A. Geochim. Cosmochim. Acta 1986, 50, 1239–1247. [Google Scholar] [CrossRef]
- Pint, B.A.; Terrani, K.A.; Brady, M.P.; Cheng, T.; Keiser, J.R. High temperature oxidation of fuel cladding candidate materials in steam-hydrogen environments. J. Nucl. Mater. 2013, 440, 420–427. [Google Scholar] [CrossRef]
- Seibert, R.L.; Jolly, B.C.; Balooch, M.; Schappel, D.P.; Terrani, K.A. Production and characterization of TRISO fuel particles with multilayered SiC. J. Nucl. Mater. 2019, 515, 215–226. [Google Scholar] [CrossRef]
- Terrani, K.A.; Yang, Y.; Kim, Y.-J.; Rebak, R.; Meyer, H.M., III; Gerczak, T.J. Hydrothermal corrosion of SiC in LWR coolant environments in the absence of irradiation. J. Nucl. Mater. 2015, 465, 488–498. [Google Scholar] [CrossRef] [Green Version]
- Kondo, S.; Lee, M.; Hinoki, T.; Hyodo, Y.; Kano, F. Effect of irradiation damage on hydrothermal corrosion of SiC. J. Nucl. Mater. 2015, 464, 36–42. [Google Scholar] [CrossRef]
- Kim, D.; Lee, H.-G.; Park, J.Y.; Park, J.-Y.; Kim, W.-J. Effect of dissolved hydrogen on the corrosion behavior of chemically vapor deposited SiC in a simulated pressurized water reactor environment. Corros. Sci. 2015, 98, 304–309. [Google Scholar] [CrossRef]
- Doyle, P.J.; Zinkle, S.; Raiman, S.S. Hydrothermal corrosion behavior of CVD SiC in high temperature water. J. Nucl. Mater. 2020, 539, 152241. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, D.; Lee, H.-G.; Park, J.Y.; Kim, W.-J. Factors affecting the hydrothermal corrosion behavior of chemically vapor deposited silicon carbides. J. Nucl. Mater. 2019, 518, 35–356. [Google Scholar] [CrossRef]
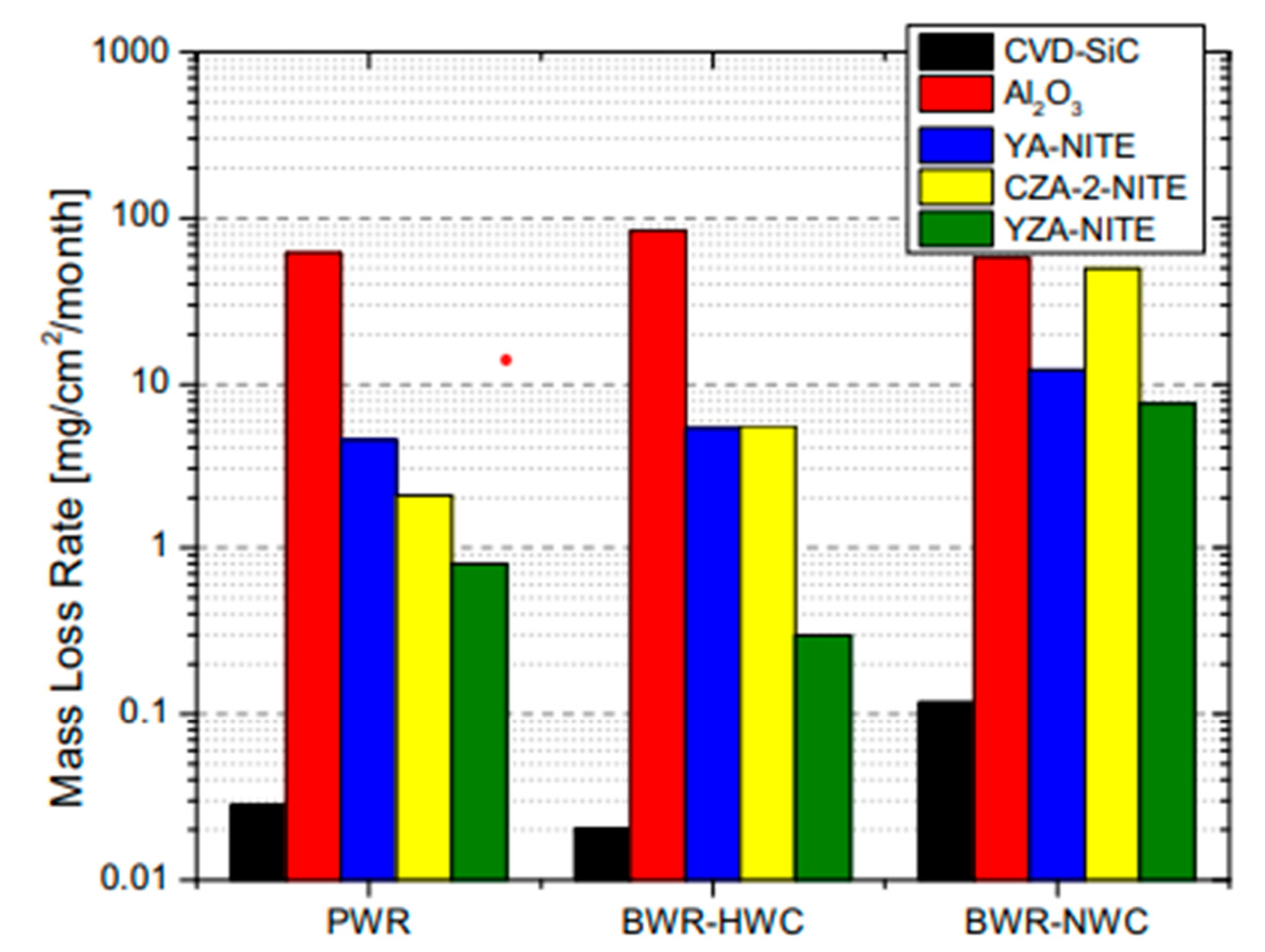
- Parish, C.M.; Terrani, K.A.; Kim, Y.-J.; Koyanagi, T.; Katoh, Y. Microstructure and hydrothermal corrosion behavior of NITE-SiC with various sintering additives in LWR coolant environments. J. Eur. Ceram. Soc. 2017, 37, 1261–1279. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Liu, B.; Liu, R.; Zhao, X.; Ni, N.; Xiao, P. Comparison of hydrothermal corrosion behavior of SiC with Al2O3 and Al2O3 + Y2O3 sintering additives. J. Am. Chem. Soc. 2020, 103, 2024–2034. [Google Scholar]
- Lobach, K.V.; Sayenko, S.Y.; Shkuropatenko, V.A.; Voyevodin, V.M.; Zykova, H.V.; Zuyok, V.A.; Bykov, A.O.; Tovazhnyans’kyy, L.L.; Chunyaev, O.M. Corrosion resistance of ceramics based on SiC under hydrothermal conditions. Mater. Sci. 2020, 55, 672–682. [Google Scholar] [CrossRef]
- Suyama, S.; Ukai, M.; Akimoto, M.; Nishimura, T.; Tajima, S. Hydrothermal corrosion behaviors of constituent materials of SiC/SiC composites for LWR applications. Ceramics 2019, 2, 47. [Google Scholar] [CrossRef] [Green Version]
- Henry, L.; Biscay, N.; Huguet, C.; Loison, S.; Aymonier, C. A water-based process for the surface functionalization of ceramic fibres. Green Chem. 2020, 22, 8308–8315. [Google Scholar] [CrossRef]

















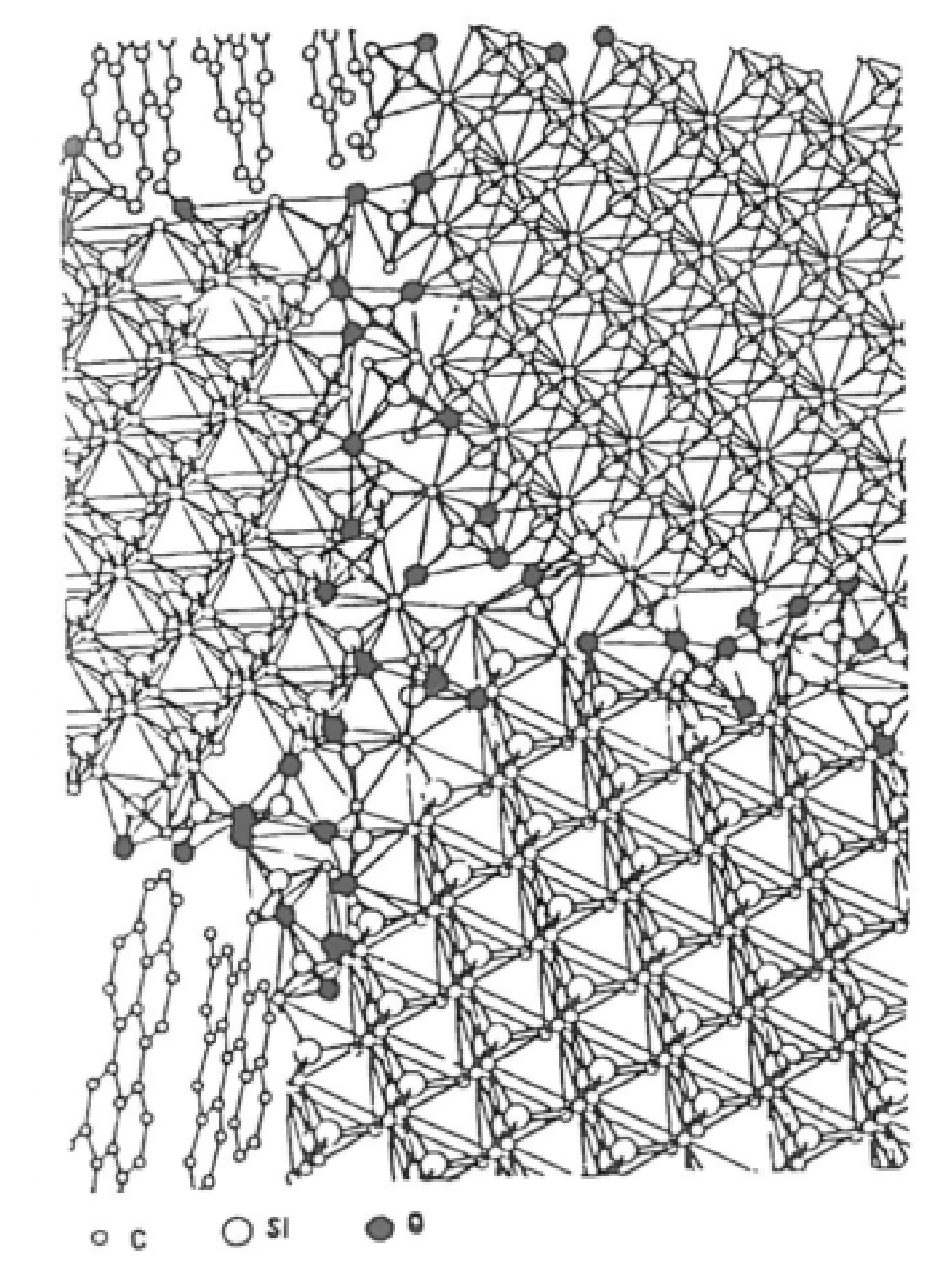
 ) mullite. Reprinted with permission from Reference [74]. Copyright 1988 Springer Nature.
) mullite. Reprinted with permission from Reference [74]. Copyright 1988 Springer Nature.
) mullite. Reprinted with permission from Reference [74]. Copyright 1988 Springer Nature.
) mullite. Reprinted with permission from Reference [74]. Copyright 1988 Springer Nature.



























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
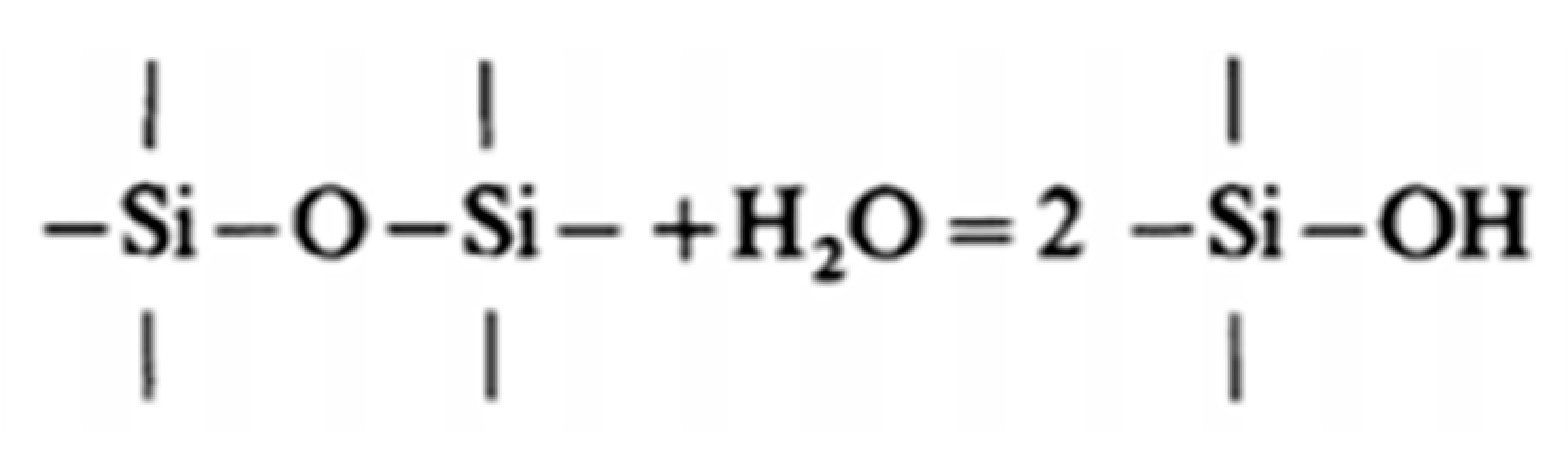{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Types of SiC | T (°C) | Kinetics | Activation Energy for Linear Oxidation (kJ/mol) |
|---|---|---|---|
| Powder SiC (green) short heating and cooling cycles | 1100 | Linear law | No data [25] |
| Si slices (111) | 900–1200 | Linear parabolic | 193 [11] |
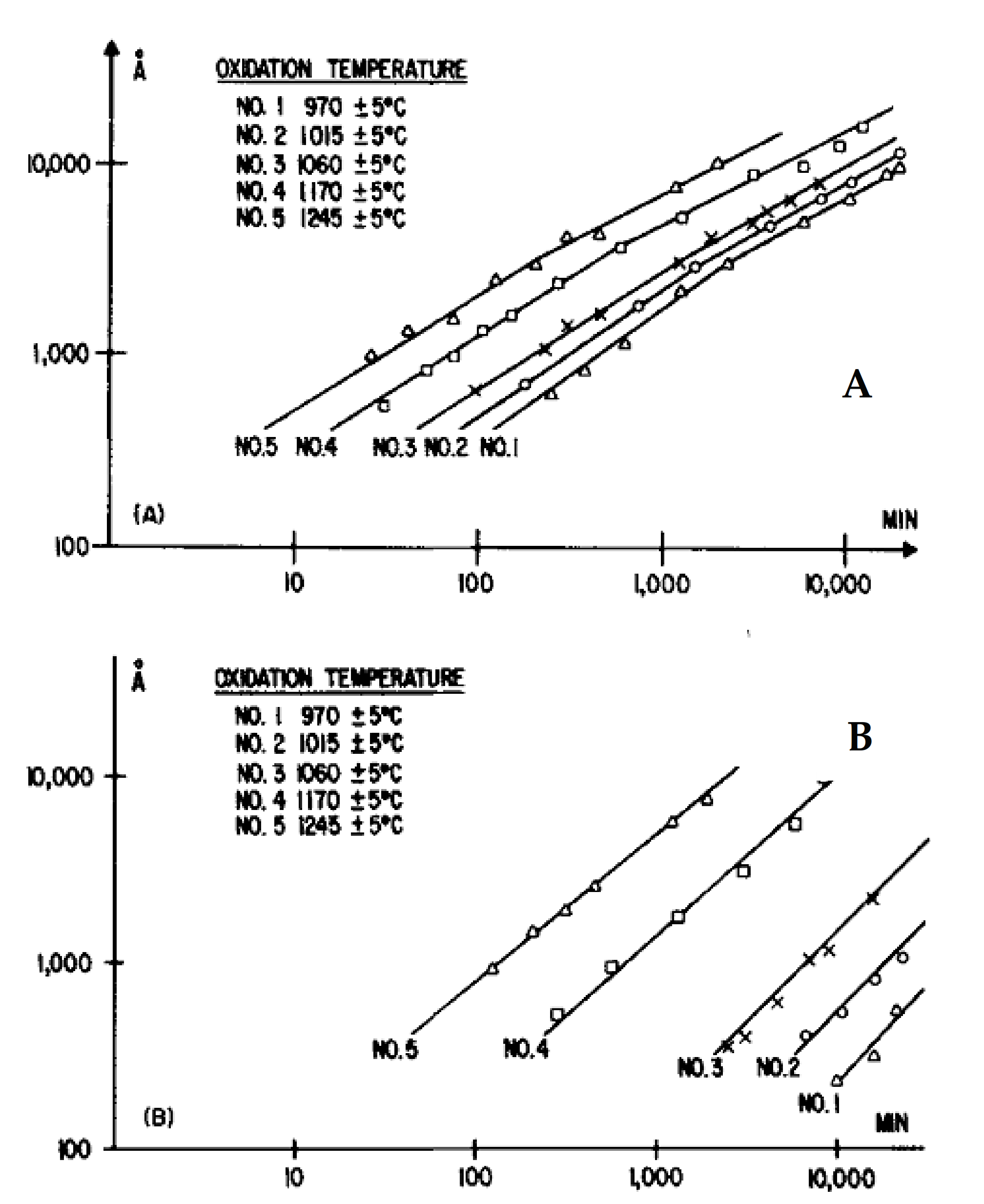
| Single crystals SiC (thin oxide face) | 970–1245 | Linear parabolic | 355 [12] |
| RF-Sputtered thin α-SiC films (C-face) | 950–1100 | Linear law | 155–200 [26] |
| Single crystal Si | 800–1100 | Linear parabolic | 155 [27] |
| Single crystal SiC (fast oxidation face) | 800–1100 | Linear parabolic | 159 [27] |
| Single crystal SiC (slow oxidation face) | 800–1100 | Linear parabolic | 330 [27] |
| CVD-SiC thick films (fast oxidation face) | 800–1100 | Linear parabolic | 170 [27] |
| CVD-SiC thick films (slow oxidation face) | 800–1100 | Linear parabolic | 334 [27] |
| Single crystals SiC (thick oxide face) | 970–1245 | Linear parabolic | 196 [12] |
| HfB2 + 20 v/o SiC composite | 1350–1550 | Parabolic law | 452 [6] |
| Hot-pressed SiC | 1200–1400 | Parabolic law | 481 [28] |
| Hot-pressed SiC | 1200–1500 | Parabolic law | 134–389 [29] |
| Sintered α-SiC | 1200–1500 | Parabolic law | 155–498 [29] |
| Single-crystals Si | 1200–1400 | Linear parabolic | 120 [30] |
| Single-crystal SiC (green) (fast-grow face) | 1200–1400 | Linear parabolic | 121–297 [30] |
| Single-crystal SiC (green) (slow-grow face) | 1200–1400 | Linear parabolic | 339 [30] |
| Controlled nucleation thermally deposited SiC | 1200–1400 | Linear parabolic | 142–293 [30] |
| Sintered α-SiC | 1200–1400 | Linear parabolic | 217–289 [30] |
| Hot-pressed SiC | 1200–1400 | Linear parabolic | 221 [30] |
| CVD-SiC | 1550–1675 | Linear parabolic | 345 (amorphous silica) and 387 (cristobalite) [31] |
| Single crystal SiC (green) (C face) | 1200–1350 | Parabolic law | 120 [9] |
| Single crystal SiC (green) (C face) | 1350–1500 | Parabolic law | 260 [9] |
| Single crystal SiC (green) (Si face) | 1350–1500 | Parabolic law | 223–298 [9] |
| CVD-SiC | 1200–1400 | Linear parabolic | 142 [32] |
| Single crystal Si | 800–1100 | Linear parabolic | 96 [27] |
| Single crystal SiC (fast oxidation face) | 800–1100 | Linear parabolic | 99 [27] |
| Single crystal SiC (slow oxidation face) | 800–1100 | Linear parabolic | 292 [27] |
| CVD-SiC thick films (fast oxidation face) | 800–1100 | Linear parabolic | 94 [27] |
| CVD-SiC thick films (slow oxidation face) | 800–1100 | Linear parabolic | 285 [27] |
| CVD-SiC | 1200–1500 | Linear parabolic | 118 [33] |
| CVD-SiC | 1397–1737 | Linear parabolic | 210 [34] |
| Types of SiC | T (°C) | Kinetics | Activation energy for parabolic oxidation (kJ/mol) |
| Powder SiC (black) | 1000–1200 | Parabolic law | 209 [35] |
| Powder SiC (green) Oxidation time <30 min | 1000–1200 | Parabolic law | 117 [35] |
| Powder SiC (green) Oxidation time >60 min | 1000–1200 | Parabolic law | 263 [35] |
| Powder SiC (green) short time oxidation | 1100–1300 | Parabolic law | 209 [25] |
| Single-crystals SiC (green) | 1200–1500 | Parabolic law | 276 [36] |
| High purity SiC | 900–1200 | Parabolic law | 85 (amorphous silica) and 65 (cristobalite) [37] |
| High purity SiC | 1380–1556 | Parabolic law | 190 [38] |
| Si slices (111) | 900–1200 | Linear parabolic | 119 [11] |
| Si slices (111) | 1000–1200 | Parabolic law | 125 [39] |
| Powder SiC | 1200–1500 | Parabolic law | 632 [40] |
| Polycrystalline CVD SiC | 1477–1627 | Linear parabolic | 1130 [41] |
| Self-bonded SiC (50/50 α/β) | 1000–1300 | Parabolic law | No data [42] |
| Types of SiC | Oxidant Species | T (°C) | Kinetics | Activation Energy for Linear Oxidation (kJ/mol) |
|---|---|---|---|---|
| Si slices (111) | Wet O2 | 900–1200 | Linear parabolic | 190 [11] |
| Powder SiC | Wet air | 1200–1400 | Linear law | 146 [57] |
| Powder SiC | Wet O2, Ar, N2 | 1500 | Linear law | No data [70] |
| Single-crystal α-SiC (Si + C faces) | 84% vol H2O | 850–1050 | Linear parabolic | 109 [51] |
| Single-crystal α-SiC (C face) | 84% vol H2O | 850–1050 | Linear parabolic | 200 [51] |
| Single-crystal β-SiC (C face) | 98 °C water + O2 | 1000–1200 | Linear parabolic | 251 [69] |
| Single-crystal β-SiC (C face) | 98 °C water + Ar | 1000–1200 | Linear parabolic | 280 [69] |
| CVD-SiC | 84% vol H2O | 1000–1250 | Linear parabolic | 309 [71] |
| RF-Sputtered thin α-SiC films | 84% vol H2O | 950–1100 | Linear law | 205–218 [25] |
| CVD-SiC | 10% H2O in O2 | 1550–1650 | Linear parabolic | 428 [72] |
| Powder-SiC | 50%H2O/50%O2 | 1200–1400 | Parabolic law | No data [61] |
| Sintered α-SiC | 12.3% H2O, 2.1% O2, 11.0% CO2, 71.8% N2 50% H2O/50% O2 | 1316 | Paralinear | No data [63] |
| CVD-SiC | 1100–1400 | Paralinear | No data [65] | |
| Sintered α-SiC | 50%H2O/50%O2 | 1100–1400 | Paralinear | No data [65] |
| Fused quartz | 50%H2O/50%O2 | 1100–1400 | Paralinear | No data [65] |
| Types of SiC | Oxidants | T (°C) | Kinetics | Activation Energy for Parabolic Oxidation (kJ/mol) |
|---|---|---|---|---|
| High purity SiC | H2O/Ar | 1218–1514 | Parabolic law | 102 [46] |
| Si slices (111) | Wet O2 | 900–1200 | Linear parabolic | 68 [11] |
| Si slices (111) | 90 °C water + O2 | 1000–1200 | Parabolic law | 85 [40] |
| Si slices (111) | Steam | 1000–1200 | Parabolic law | 102 [40] |
| SiC (50/50 of α/β) | Wet O2 | 1000–1300 | Parabolic law | No data [43] |
| Hot pressed SiC | 3% H2O in O2 | 1200–1400 | Parabolic law | 527 [58] |
| Single crystals Si (100) | H2O/O2 (1 to 2000 ppm) | 780–980 | Linear parabolic | No data [62] |
| Single crystals Si (100) | H2O/N2 (1 to 2000 ppm) | 780–980 | Linear parabolic | No data [62] |
| Single-crystal β-SiC (C face) | 98 °C water + O2 | 1000–1200 | Linear parabolic | 531 [69] |
| Single-crystal β-SiC (C face) | 98 °C water + Ar | 1000–1200 | Linear parabolic | 656 [69] |
| CVD-SiC | 84% vol H2O | 1000–1250 | Linear parabolic | 209 [71] |
| Pressureless-sintered α-SiC | H2O in Air (10 to 40% vol) | 1300 | Parabolic law | No data [73] |
| CVD-SiC | 10% H2O in O2 | 1550–1650 | Linear parabolic | 397 [74] |
| CVD-SiC in fused quartz tubes | 10% H2O in O2 | 1200–1400 | Linear parabolic | 41 [32] |
| CVD-SiC in high purity Al2O3 tubes | 10% H2O in O2 | 1200–1400 | Linear parabolic | 249 [32] |
| CVD-SiC | H2O/O2 (10 to 90% vol) | 1200–1400 | Parabolic | 28-156 [66] |
| CVD-SiC | H2O/Ar | 1200–1400 | Parabolic | No data [66] |
| Si, Sintered α-SiC, CVD-SiC | Air + 15% vol H2O | 1200 | Paralinear (adapted from the model of Haycock) | No data [67] |
| Water Vapor Defect Species | Defect Formation Reaction | Mass Action Expression | Electro-Neutrality Expression | Water Vapor Partial Pressure Dependence | Power Low Exponent for Water |
|---|---|---|---|---|---|
| none | 1 | ||||
| no | none | 2 | |||
| 1/2 | |||||
| 1/3 | |||||
| 1/3 |
| Material/Silicon Bonding | Activation Energy in the Parabolic Regime Which Is Limited by Diffusion of Oxidant Species through Silica | Linear Regime Limited by the Interface Reaction | |||
|---|---|---|---|---|---|
| Conditions (°C) | Oxygen Permeation (kJ/mol) | Conditions (°C) | Water Permeation (kJ/mol) | Breaking Energy (kJ/mol) | |
| Fused silica | 950–1100 | 113 [44] | 300–1100 | 70 [81] | |
| Cristobalite | 1000–1400 on SiC substrate | 430 [87] | / | ||
| Tridymite | 1070–1280 | 195 [88] | / | ||
| β-Quartz | 870–1180 | 195 [88] | 600–800 + 100 MPA H2O | 142 (//to c) [89] | |
| Si-Si | 177 [73] | ||||
| Si-C | 290 [73] | ||||
| Si-O or Si-OH | 377 [81] | ||||
| “Normal Water” | “Subcritical Water” | “Supercritical Water” | Superheated Steam | ||
|---|---|---|---|---|---|
| T [°C] | 25 | 250 | 400 | 400 | 400 |
| P[MPa] | 0.1 | 5 | 25 | 50 | 0.1 |
| ρ[g·cm−3] | 0.997 | 0.80 | 0.17 | 0.58 | 0.0003 |
| ε | 78.5 | 27.1 | 5.9 | 10.5 | 1 |
| pKw | 14.0 | 11.2 | 19.4 | 11.9 | / |
| Cp[kJ.kg−1·K−1] η[mPa.s] | 4.22 0.89 | 4.86 0.11 | 13 0.03 | 6.8 0.07 | 2.1 0.02 |
| λ[mW·m−1·K−1] | 608 | 620 | 160 | 438 | 55 |
| P = 10–100 MPa | Low (H2O:SiC) Molar Ratios (1: 10) | Intermediates (H2O:SiC) Molar Ratios (2: 1) | High (H2O:SiC) Molar Ratios (10: 1) |
|---|---|---|---|
| Observations | Deposition of carbon and silica, according to Yoshimura | Formation of a carbon layer and dissolution of silica, according to Gogotsi | Oxidation of carbon and dissolution of silica, according to Hirayama |
| 300 °C | No reactions | SiCxOy + nH2O → SiO2 +xC + nH2 SiO2 + H2O → SiO32− + 2 H+ | SiC + 4 H2O → Si(OH)4 + CH4 SiCxOy + 4 H2O → Si(OH)4 + xCH4 |
| 400 °C | |||
| 500 °C | SiC + 2 H2O → SiO2 + C + 2 H2 SiC + 2 H2O → SiO2 + CH4 O2 + CH4 → CO2 + 2 H2O | SiC + 2 H2O → SiO2 + C + 2 H2 SiCxOy + nH2O → SiO2 +xC + nH2 SiO2 + H2O → SiO32− + 2 H+ | SiC + 2 H2O → SiO2 + C + 2 H2 SiCxOy + nH2O → SiO2 +xC + nH2 SiO2 + H2O → SiO32− + 2 H+ |
| 600 °C | SiC + 2 H2O → SiO2 + C + 2 H2 SiC + 3 H2O → SiO2 + CO + 3 H2 SiC + 4 H2O → SiO2 + CO2 + 4 H2 | C + H2O → CO + H2 2C + 2H2O → CO2 + CH4 3C + 2H2O → 2CO + CH4 | C + H2O → CO + H2 2C + 2H2O → CO2 + CH4 3C + 2H2O → 2CO + CH4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Biscay, N.; Henry, L.; Adschiri, T.; Yoshimura, M.; Aymonier, C. Behavior of Silicon Carbide Materials under Dry to Hydrothermal Conditions. Nanomaterials 2021, 11, 1351. https://doi.org/10.3390/nano11051351
Biscay N, Henry L, Adschiri T, Yoshimura M, Aymonier C. Behavior of Silicon Carbide Materials under Dry to Hydrothermal Conditions. Nanomaterials. 2021; 11(5):1351. https://doi.org/10.3390/nano11051351
Chicago/Turabian StyleBiscay, Nicolas, Lucile Henry, Tadafumi Adschiri, Masahiro Yoshimura, and Cyril Aymonier. 2021. "Behavior of Silicon Carbide Materials under Dry to Hydrothermal Conditions" Nanomaterials 11, no. 5: 1351. https://doi.org/10.3390/nano11051351
APA StyleBiscay, N., Henry, L., Adschiri, T., Yoshimura, M., & Aymonier, C. (2021). Behavior of Silicon Carbide Materials under Dry to Hydrothermal Conditions. Nanomaterials, 11(5), 1351. https://doi.org/10.3390/nano11051351