
Preparation of Silicon Hydroxyapatite Nanopowders under Microwave-Assisted Hydrothermal Method


 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Microwave-Assisted Hydrothermal Synthesis
Ca10(PO43−)6−x(SiO44−)y(OH)2−x(s) + 10Na+ + xOH−(aq) + xNH4δ+NO3δ −(aq) + xH2O +yCH4δ+ + (y−x)[(Si-O-Si)3O−)](aq)
2.3. Characterization
3. Results and Discussion
3.1. Effect of Si4+ Saturation on the Hydrothermal Synthesis of Si-HAp
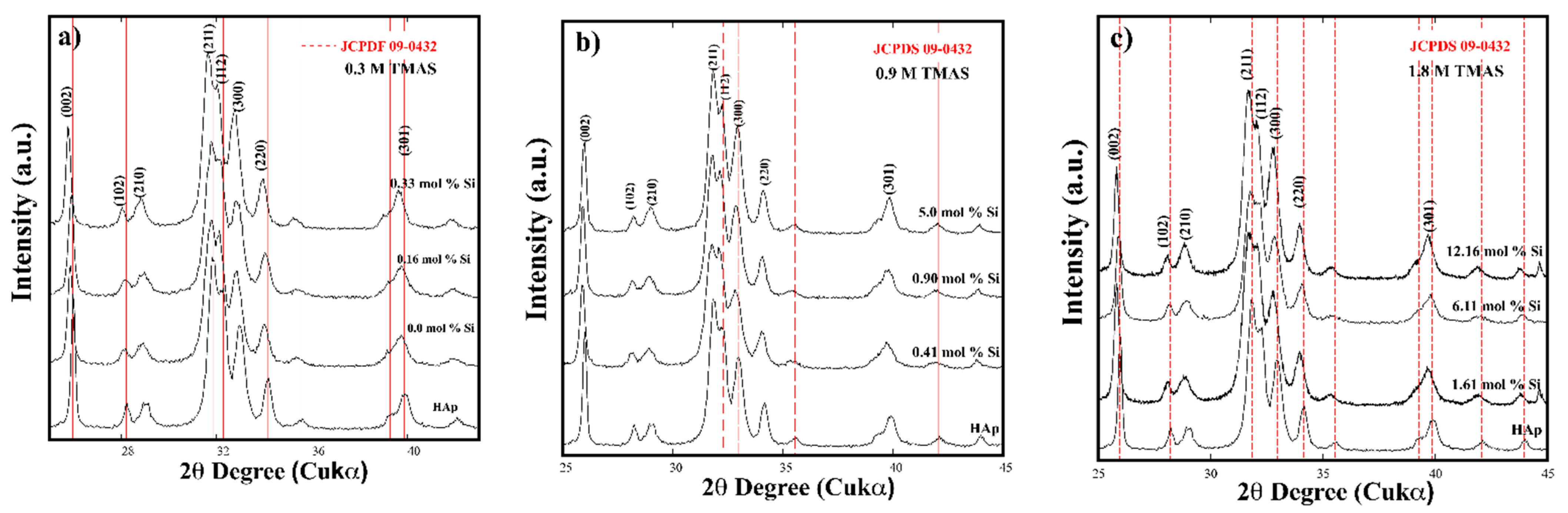
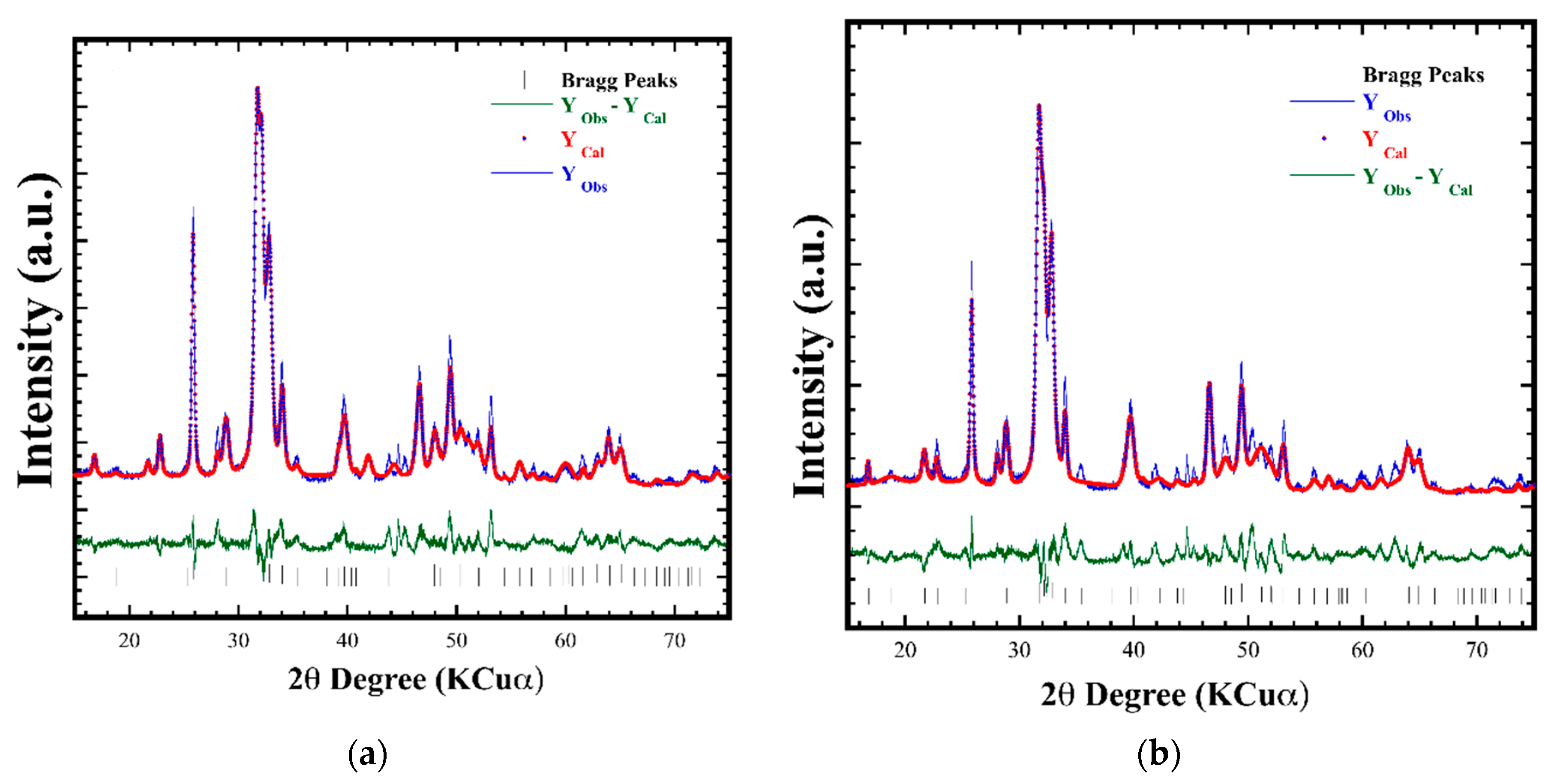
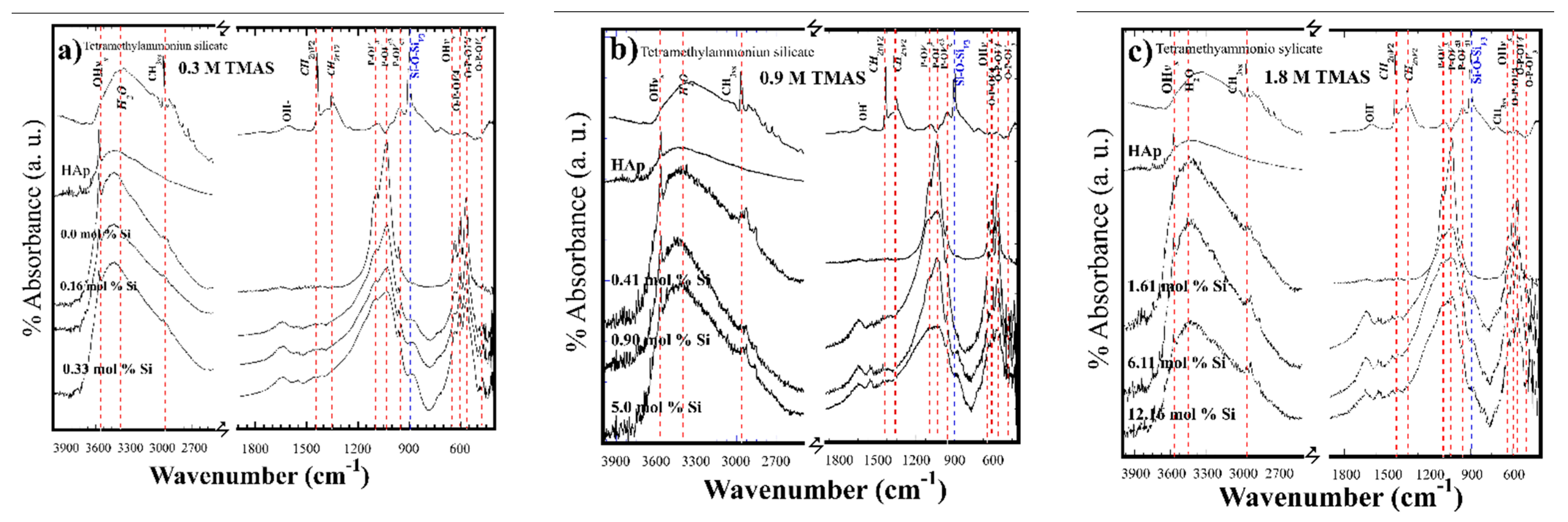
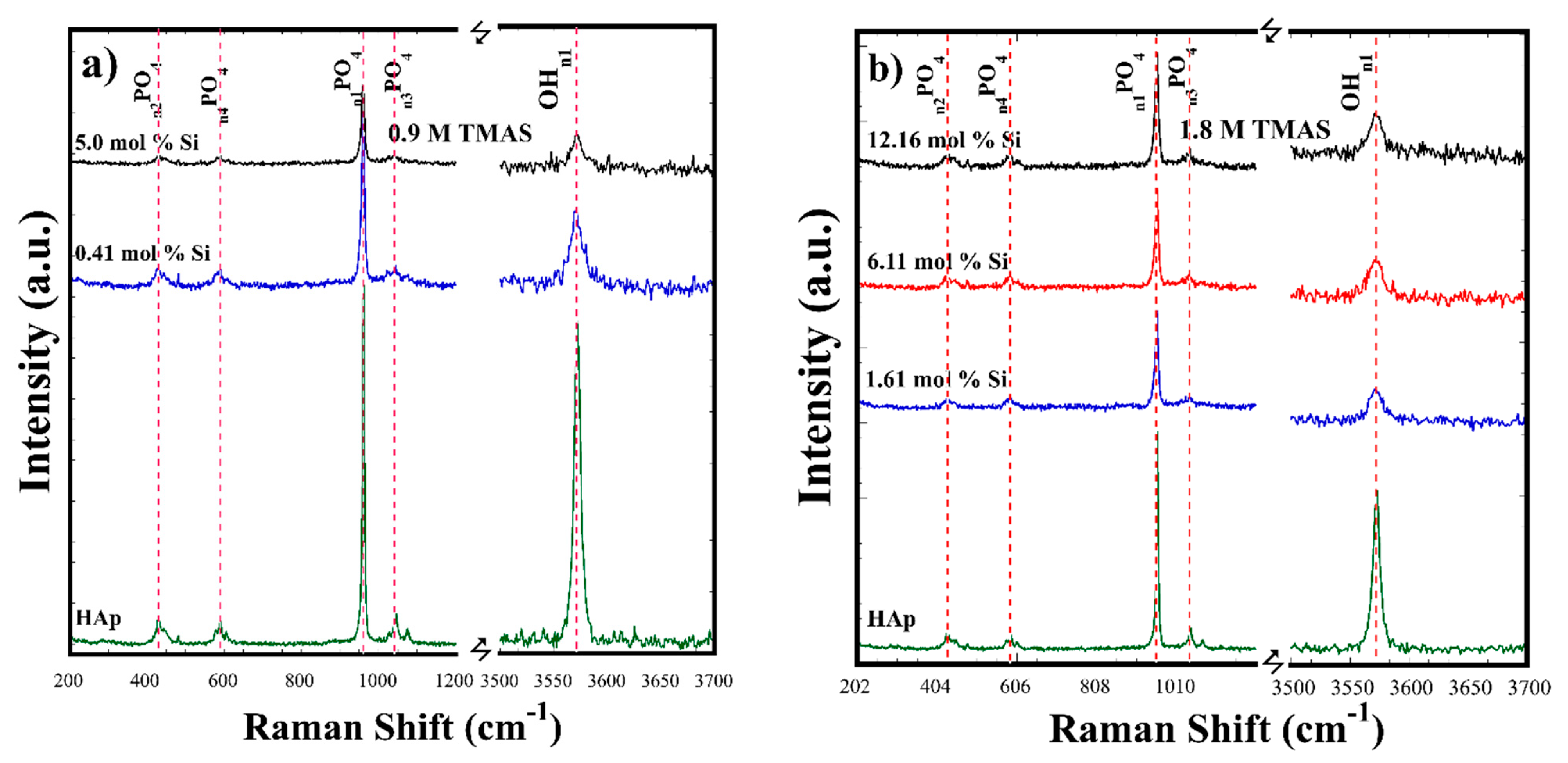
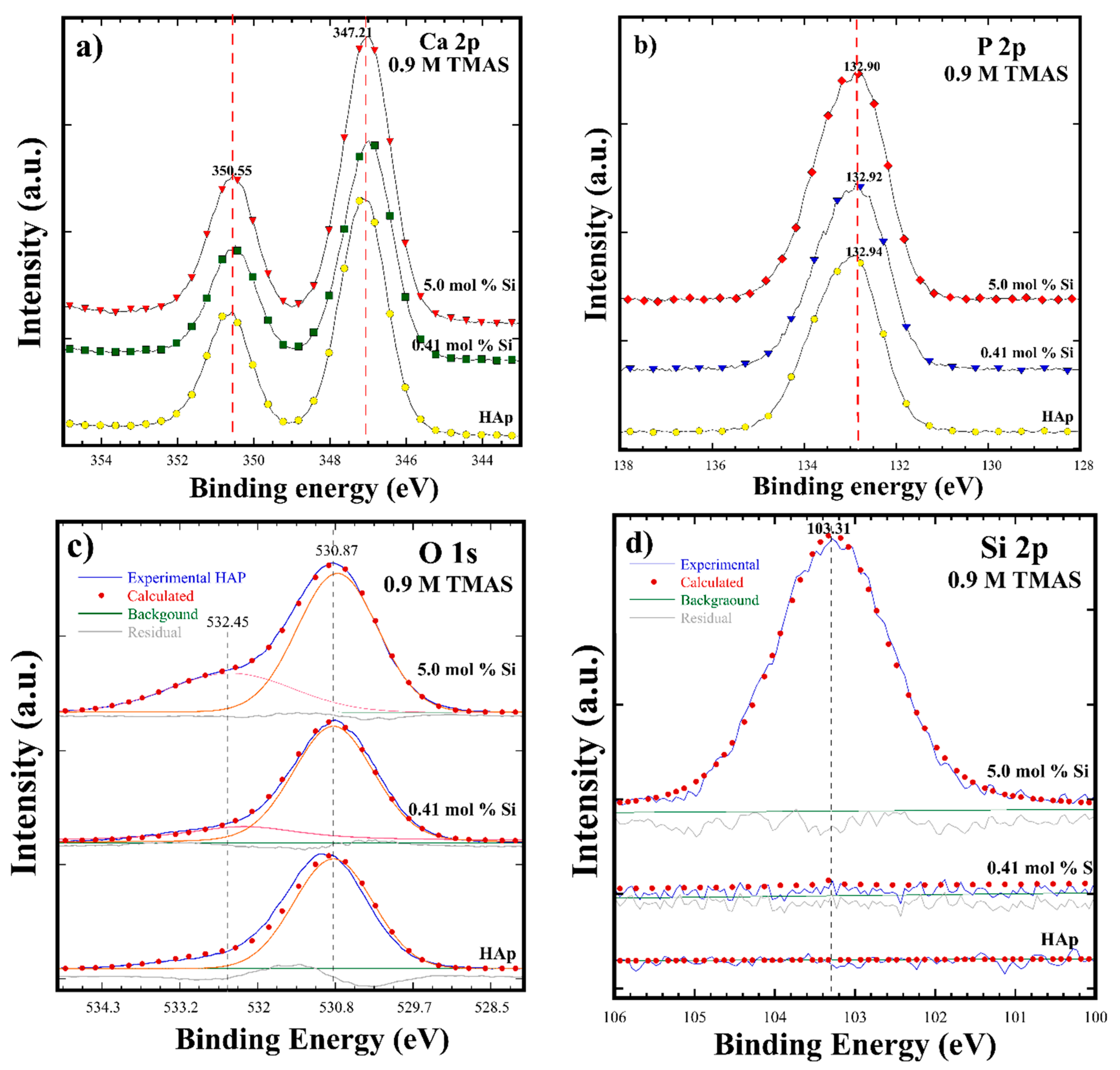
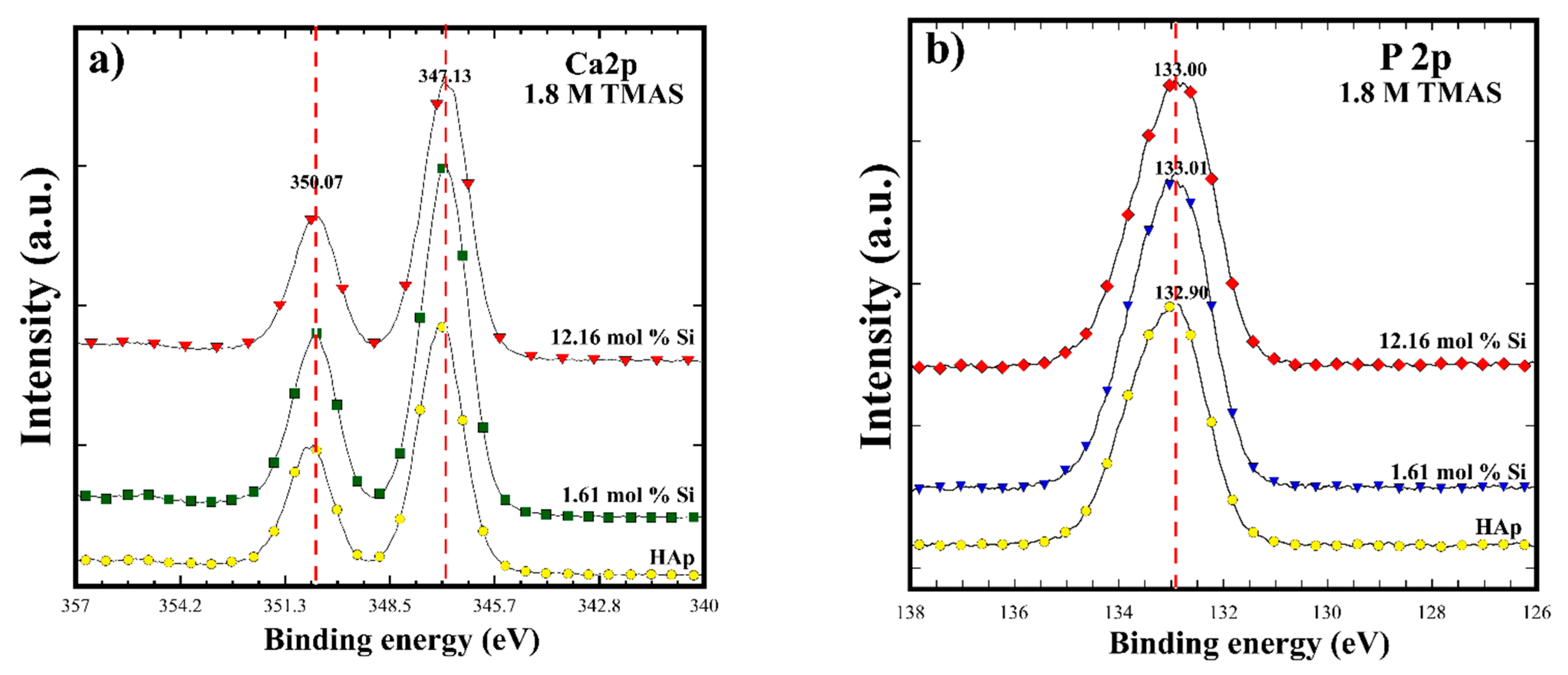
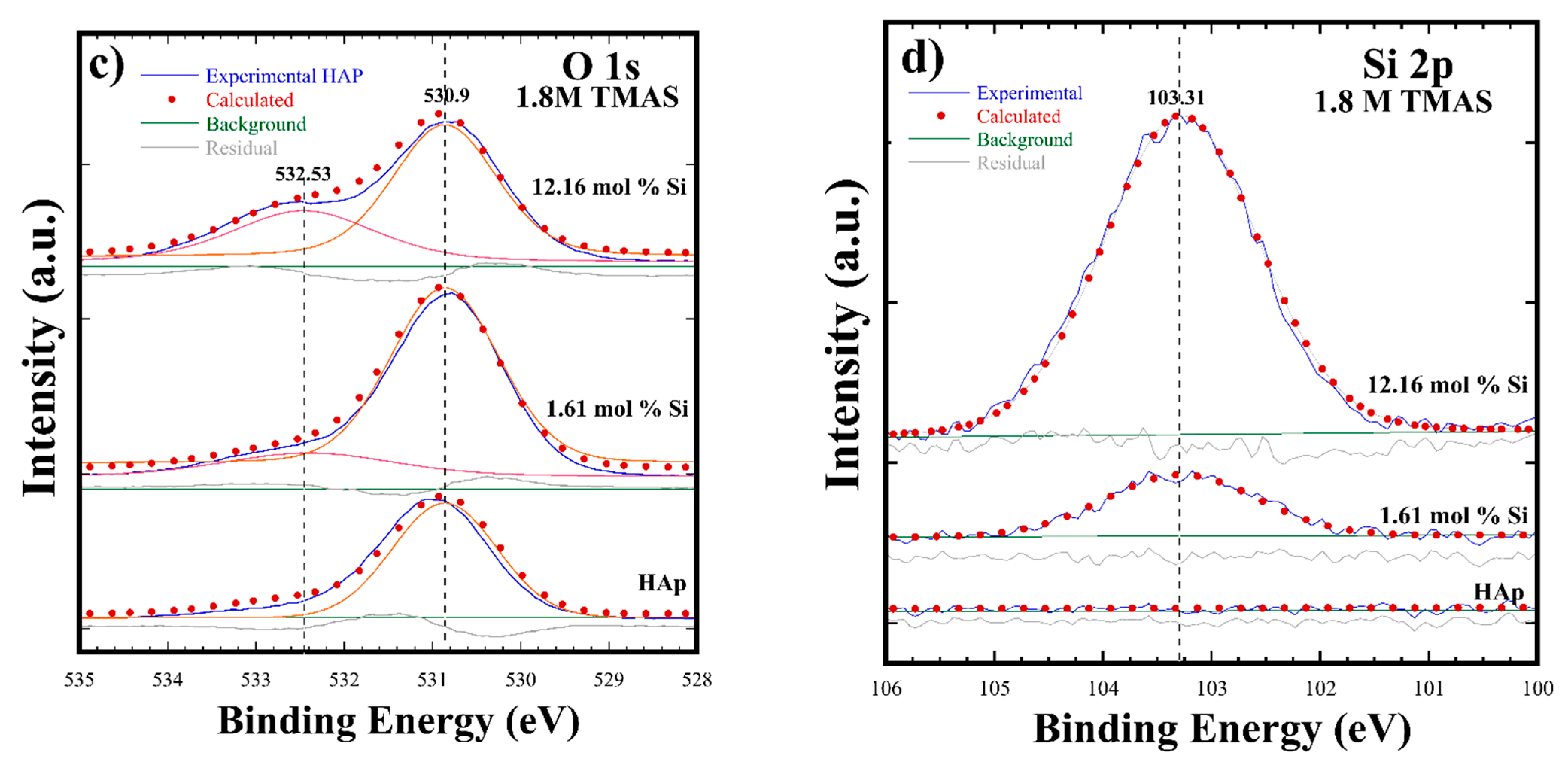
3.2. Crystalline Structural and Chemical Compositional Analyses of Si-HAp Powders
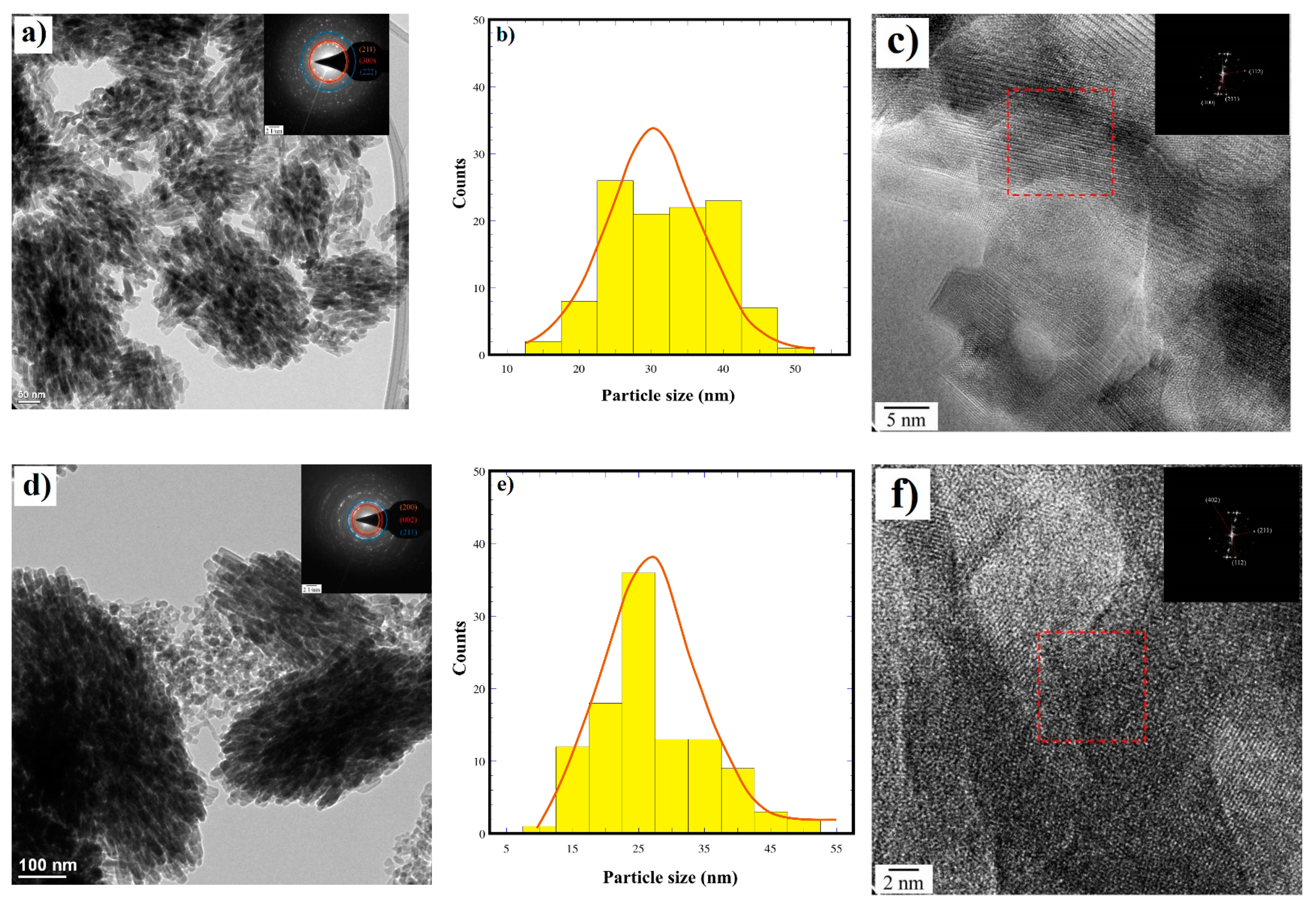
3.3. Morphological Aspects of the Partially Substituted Si-HAp Particles Prepared Hydrothermally
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kongjun, Z.; Yanagisawa, K.; Shimanouchi, R.; Onda, A.; Kajiyoshi, K.; Qiu, J. Synthesis and crystallographic study of Pb-Sr hydroxyapatite solid solutions by high temperature mixing method under hydrothermal conditions. J. Ceram. Soc. Jpn. 2007, 115, 873–876. [Google Scholar] [CrossRef] [Green Version]
- Carlisle, E.M. Silicon: A possible factor in bone calcification. Science 1970, 167, 279–280. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Lee, K.S.; Chang, J.S.; Cho, W.S.; Kim, Y.; Kim, S.R.; Kim, Y.T. Biocompatibility of Si-substituted hydroxyapatite. J. Key Eng. Mater. 2004, 254–256, 135–138. [Google Scholar] [CrossRef]
- Gibson, I.R.; Huang, J.; Best, S.M.; Bonfield, W. Enhanced in vitro cell activity and surface apatite layer formation on novel silicon-substituted hydroxyapatites. World Sci. 2009, 12, 191–194. [Google Scholar] [CrossRef]
- Jamil, M.; Elouatli, B.; Khallok, H.; Jamil, M.; Elouahli, A.; Gourri, E.; Ezzahmouly, M.; Abida, F.; Hatim, Z. Silicon substituted hydroxyapatite: Preparation with solid-state reaction, characterization and dissolution properties. J. Mater. Environ. Sci. 2018, 9, 2322–2327. [Google Scholar]
- Harden, F.J.; Gibson, I.R.; Skakle, J.M.S. Simplification of the synthesis method for silicon-substituted hydroxyapatite: A Raman spectroscopy study. Key Eng. Mater. 2012, 529, 94–99. [Google Scholar] [CrossRef]
- Marchat, D.; Zymelka, M.; Coelho, C.; Gremillard, L.; Jolypottuz, L.; Babonneau, F.; Esnouf, C.; Chevalier, J.; Nernacheasollant, D. Accurate characterization of pure silicon-substituted hydroxyapatite powders synthesized by a new precipitation route. Acta Biomater. 2013, 9, 6992–7004. [Google Scholar] [CrossRef] [Green Version]
- Zou, S.; Huang, J.; Best, S.; Bonfield, W. Rietveld studies on silicon substituted hydroxyapatite. Key Eng. Mater. 2006, 309, 113–116. [Google Scholar] [CrossRef]
- Nakahira, A.; Nakata, K.; Numako, C.; Murata, H.; Matsunaga, K. Synthesis and evaluation of calcium-deficient hydroxyapatite with SiO2. Mater. Sci. Appl. 2011, 2, 1194–1198. [Google Scholar] [CrossRef] [Green Version]
- Outali, B.E.; Jamil, M.; Elouahli, A.; Ezzahmouly, M.; Abida, F.; Ilou, M.; Hatim, Z. Silicon substitution in biphasic calcium phosphate bioceramics: Crystal structure study. Int. J. Sci. Eng. Res. 2016, 7, 829–833. [Google Scholar]
- Yacoubi, A.E.; Massit, A.; Fathi, M.; Chafik, B.; Idrissi, E.; Yammi, K. Characterization of silicon-substituted hydroxyapatite powders synthesized by a wet precipitation method. IOSR J. Appl. Chem. 2014, 7, 24–29. [Google Scholar] [CrossRef]
- Aminian, A.; Solati, M.; Bakhshi, F.; Fazardi, A. Silicon substitution hydroxyapatite by hydrothermal method. Adv. Bioceram. Biotechnol. 2010, 11, 59–65. [Google Scholar]
- Gibson, I.R.; Best, S.M.; Bonfield, W. Chemical characterization of silicon-substituted hydroxyapatite. J. Biomed. Mater. Res. 1999, 44, 422–428. [Google Scholar] [CrossRef]
- Aminian, A.; Salti-Hashjin, M.; Samadikuchaksaraei, A.; Bakhshi, F.; Gorjipour, F.; Farzadi, A.; Motztarzadath, F.; Schmucker, M. Synthesis of silicon-substitute hydroxyapatite by a hydrothermal method with two different phosphorous sources. Ceram. Int. 2011, 37, 1219–1229. [Google Scholar] [CrossRef]
- Bulina, N.; Chaikina, M.V.; Ishchenko, A.V.; Prosanov, I.Y. Mechanochemical synthesis of SiO44−-substituted hydroxyapatite, part II—Reaction mechanism, structure, and substitution limit. Eur. J. Inorg. Chem. 2014, 28, 4803–4809. [Google Scholar] [CrossRef]
- Neira, I.S.; Kolen’ko, V.Y.; Lebedev, O.I.; Tendeloo, G.V.; Gupta, H.S.; Guitian, F.; Yoshimura, M. An effective morphology control of hydroxyapatite crystals via hydrothermal synthesis. Cryst. Growth Des. 2009, 9, 466–467. [Google Scholar] [CrossRef]
- Byrappa, K.; Yoshimura, M. Handbook of Hydrothermal Technology a Technology for Crystal Growth and Materials Processing; William Andrew Publishing, LLC: Norwich, NY, USA, 2010; pp. 755–780. ISBN 9781437778366. [Google Scholar]
- Yanagisawa, K.; Toya, H.; Feng, Q.; Yamasaki, N. In situ formation of hydroxyapatite crystals under hydrothermal conditions. Phosphorus Res. Bull. 1995, 5, 43. [Google Scholar] [CrossRef] [Green Version]
- Padmanabhan, S.K.; Haq, U.E.; Licciulli, A. Rapid synthesis and characterization of silicon substituted nano hydroxyapatite using microwave irradiation. Curr. Appl. Phys. 2014, 14, 87–92. [Google Scholar] [CrossRef]
- Montoya, K.L.; Rendón-Ángeles, J.C.; Matamoros-Veloza, Z.; Yanagisawa, K. Rapid synthesis and characterization of Zn substituted hydroxyapatite nanoparticles via a microwave-assisted hydrothermal method. Mater. Lett. 2017, 195, 5–9. [Google Scholar] [CrossRef]
- Lamkhao, S.; Phaya, M.; Jansakun, C.; Chandet, N.; Thongkorn, K.; Rujijanagul, G.; Bangrak, P.; Randorn, C. Synthesis of hydroxyapatite with antibacterial properties using a microwave-assisted combustion method. Sci. Rep. Springer Nat. 2019, 9, 4015. [Google Scholar] [CrossRef] [Green Version]
- Moreno Perez, B.; Matamoros Veloza, Z.; Rendon-Angeles, J.C.; Yanagisawa, K.; Onda, A.; Perez Terrazas, J.E.; Mejia Martinez, E.E.; Burciaga Diaz, O.; Rodriguez Reyes, M. Synthesis of silicon-substituted hydroxyapatite using hydrothermal process. Bol. De La Soc. Española De Cerámica Y Vidr. 2020, 59, 50–64. [Google Scholar] [CrossRef]
- Hasegawa, I.; Sakka, S.; Sugahara, Y.; Kuroda, K.; Kato, C. Silicate anions formed in tetramethylammonium silicate methanolic solution as studied by 29 Si nuclear magnetic resonance. J. Chem. Soc. Chem. Commun. 1989, 4, 208–210. [Google Scholar] [CrossRef]
- Yoichiro, M.; Masateru, H.; Masahiko, O.; Toshihiro, K.; Masayuki, N. Large-sized hydroxyapatite whiskers derived from calcium tripolyphosphate gel. J. Euro. Ceram. Soc. 2005, 25, 3181–3185. [Google Scholar] [CrossRef]
- Shing, H.Y.; Jung, J.Y.; Kim, S.W.; Lee, W.K. XPS analysis on chemical properties of calcium phosphate thin films and osteoblastic HOS cell responses. J. Ind. Eng. Chem. 2006, 12, 476–483. [Google Scholar]
- Bianco, A.; Cacciotti, I.; Lombardi, M.; Montanaro, L. Si-substituted hydroxyapatite nanopowders: Synthesis, thermal stability and sinterability. Mater. Res. Bull. 2009, 44, 345–354. [Google Scholar] [CrossRef]
- Younes, B.; Meriame, B.; Ferreira, J.; El Mabrouk, K. Hydrothermal synthesis of Si-doped hydroxyapatite nanopowders: Mechanical and bioactivity evaluation. Int. J. Appl. Ceram. Technol. 2015, 12, 329–340. [Google Scholar] [CrossRef]
- Botelho, C.M.; Lopes, M.A.; Gibson, I.R.; Best, S.M.; Santos, J.D. Structural analysis of Si-substituted hydroxyapatite: Zeta potential and X-ray photoelectron spectroscopy. J. Mater. Sci. Mater. Med. 2002, 13, 1123–1127. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample * Id Reference | Molar Concentration (±0.001) of Si4+ | Nominal Mol % | Chemical Compositon a | Molar Ratio Ca/P | GOF χ2 b | Lattice Parameter b | Strain b | Rwp | RBragg | Crystallite Size (nm) | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PO4 | SiO4 | a0 (Å) | c0 (Å) | Cell Volume (Å3) | |||||||||
| HAp | 100 | 0 | Ca10(PO4)6(OH)2 | 1.667 | 1.36 | 9.4248(7) | 6.8764(5) | 528.97(0.09) | 0.5 (0.02) | 9.43 | 1.44 | 51.69 (2.3) | |
| MM62 | 0.3 | 94 | 6 | Ca10(PO4)6(SiO4)0.0(OH)2 | 1.667 | 1.28 | 9.4433(10) | 6.8886 (7) | 532.00(0.12) | 0.68 (0.03) | 8.58 | 1.63 | 46.14 (3.4) |
| MM66 | 0.3 | 90 | 10 | Ca10(PO4)5.99(SiO4)0.01(OH)1.99 | 1.667 | 1.66 | 9.4447(18) | 6.8869 (8) | 532.10(0.22) | 0.53 (0.08) | 10.89 | 3.08 | 46.64 (4.9) |
| MM63 | 0.3 | 80 | 20 | Ca10(PO4)5.98(SiO4)0.02(OH)1.99 | 1.667 | 1.74 | 9.4439(16) | 6.8872(7) | 531.89(0.12) | 0.38(0.04) | 11.15 | 3.93 | 46.18(2.9) |
| MM50 | 0.9 | 94 | 6 | Ca10(PO4)5.975(SiO4)0.025(OH)1.975 | 1.674 | 1.64 | 9.4443(13) | 6.8866(11) | 531.97(0.19) | 0.56(0.07) | 10.86 | 3.18 | 45.03(3.7) |
| MM51 | 0.9 | 90 | 10 | Ca10(PO4)5.946(SiO4)0.054(OH)1.946 | 1.682 | 1.77 | 9.444413) | 6.8866(10) | 532.00(0.17) | 031 (0.05) | 11.54 | 4.11 | 36.96(2.1) |
| MM53 | 0.9 | 80 | 20 | Ca10(PO4)5.699(SiO4)0.301(OH)1.699 | 1.755 | 1.27 | 9.4424(9) | 6.8863(6) | 531.73(0.11) | 0.30(0.03) | 8.24 | 1.71 | 35.94(1.2) |
| MM64 | 1.8 | 94 | 6 | Ca10(PO4)5.908(SiO4)0.097(OH)1.908 | 1.692 | 1.64 | 9.4475(16) | 6.8884(11) | 532.46(0.20) | 0.61(0.06) | 10.56 | 2.95 | 48.82(5.1) |
| MM68 | 1.8 | 90 | 10 | Ca10(PO4)5.633(SiO4)0.367(OH)1.633 | 1.775 | 1.30 | 9.4447(11) | 6.8872(8) | 532.09(0.14) | 0.68(0.04) | 8.44 | 1.81 | 48.64(3.4) |
| MM65 | 1.8 | 80 | 20 | Ca10(PO4)5.271(SiO4)0.729(OH)1.271 | 1.897 | 1.11 | 9.4365(9) | 6.8824(7) | 530.75(0.11) | 1.68(0.09) | 2.63 | 1.15 | 39.87(3.6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matamoros-Veloza, Z.; Rendon-Angeles, J.C.; Yanagisawa, K.; Ueda, T.; Zhu, K.; Moreno-Perez, B. Preparation of Silicon Hydroxyapatite Nanopowders under Microwave-Assisted Hydrothermal Method. Nanomaterials 2021, 11, 1548. https://doi.org/10.3390/nano11061548
Matamoros-Veloza Z, Rendon-Angeles JC, Yanagisawa K, Ueda T, Zhu K, Moreno-Perez B. Preparation of Silicon Hydroxyapatite Nanopowders under Microwave-Assisted Hydrothermal Method. Nanomaterials. 2021; 11(6):1548. https://doi.org/10.3390/nano11061548
Chicago/Turabian StyleMatamoros-Veloza, Zully, Juan Carlos Rendon-Angeles, Kazumichi Yanagisawa, Tadaharu Ueda, Kongjun Zhu, and Benjamin Moreno-Perez. 2021. "Preparation of Silicon Hydroxyapatite Nanopowders under Microwave-Assisted Hydrothermal Method" Nanomaterials 11, no. 6: 1548. https://doi.org/10.3390/nano11061548
APA StyleMatamoros-Veloza, Z., Rendon-Angeles, J. C., Yanagisawa, K., Ueda, T., Zhu, K., & Moreno-Perez, B. (2021). Preparation of Silicon Hydroxyapatite Nanopowders under Microwave-Assisted Hydrothermal Method. Nanomaterials, 11(6), 1548. https://doi.org/10.3390/nano11061548