
The Enhanced Hydrogen Storage Capacity of Carbon Fibers: The Effect of Hollow Porous Structure and Surface Modification

 , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
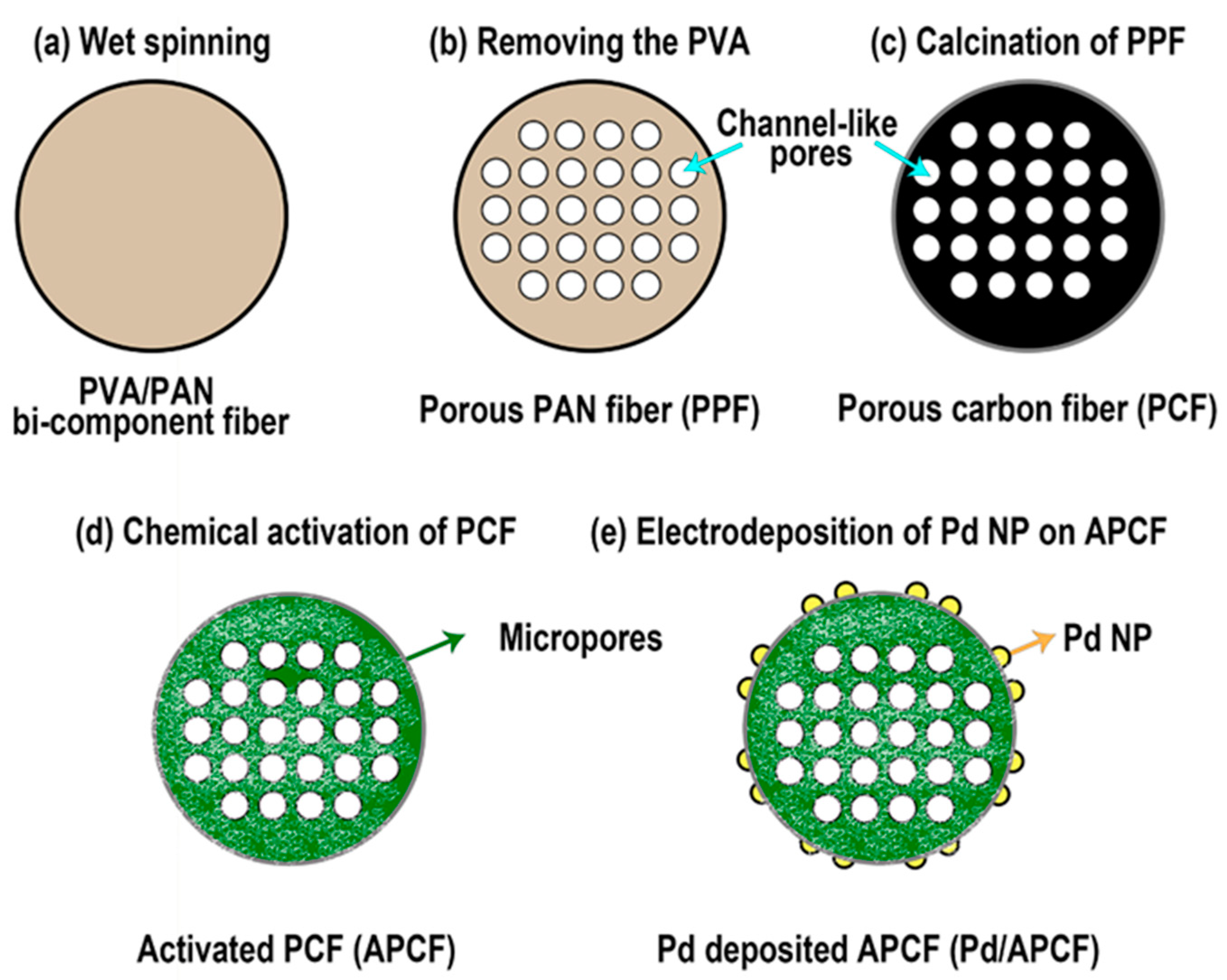
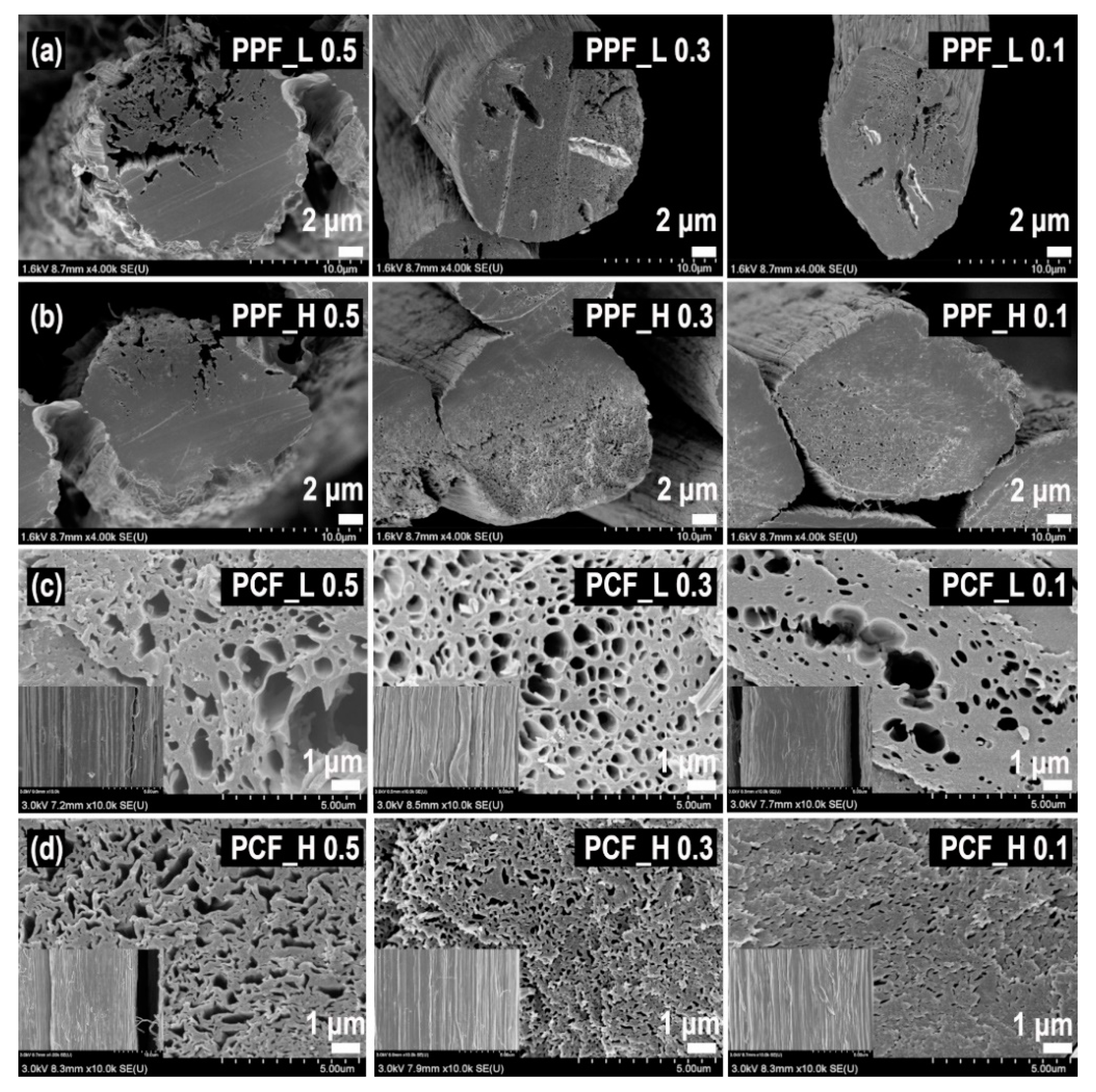
2.2. Preparation of PVA/PAN Bi-Component Fiber and Porous PAN Fiber (PPF)
2.3. Preparation of Porous Carbon Fiber (PCF) and Activated Porous Carbon Fiber (APCF)
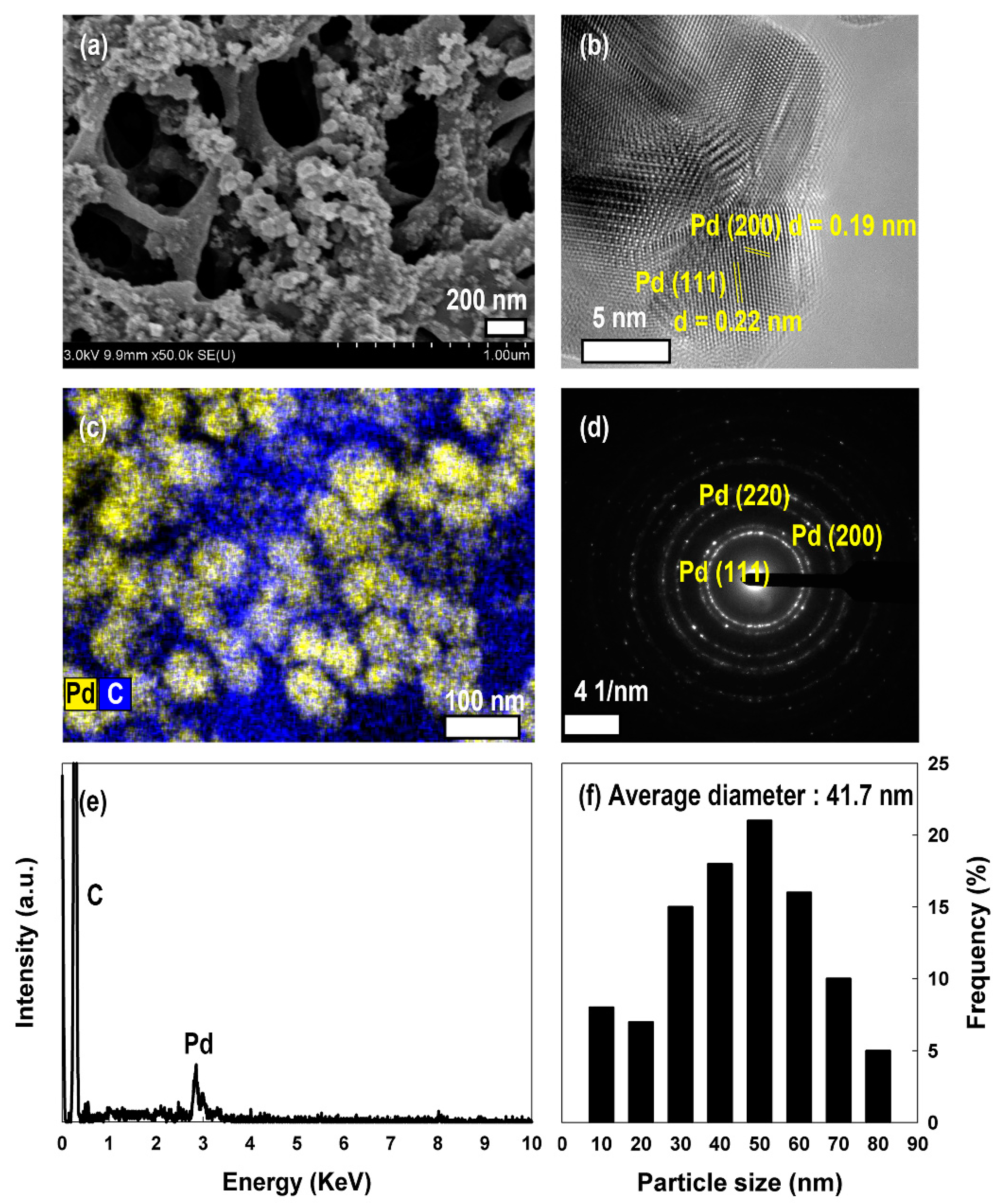
2.4. Electrodeposition of Pd Nanoparticles on APCF
2.5. Characterizations
3. Results and Discussion
3.1. Surface Characterization
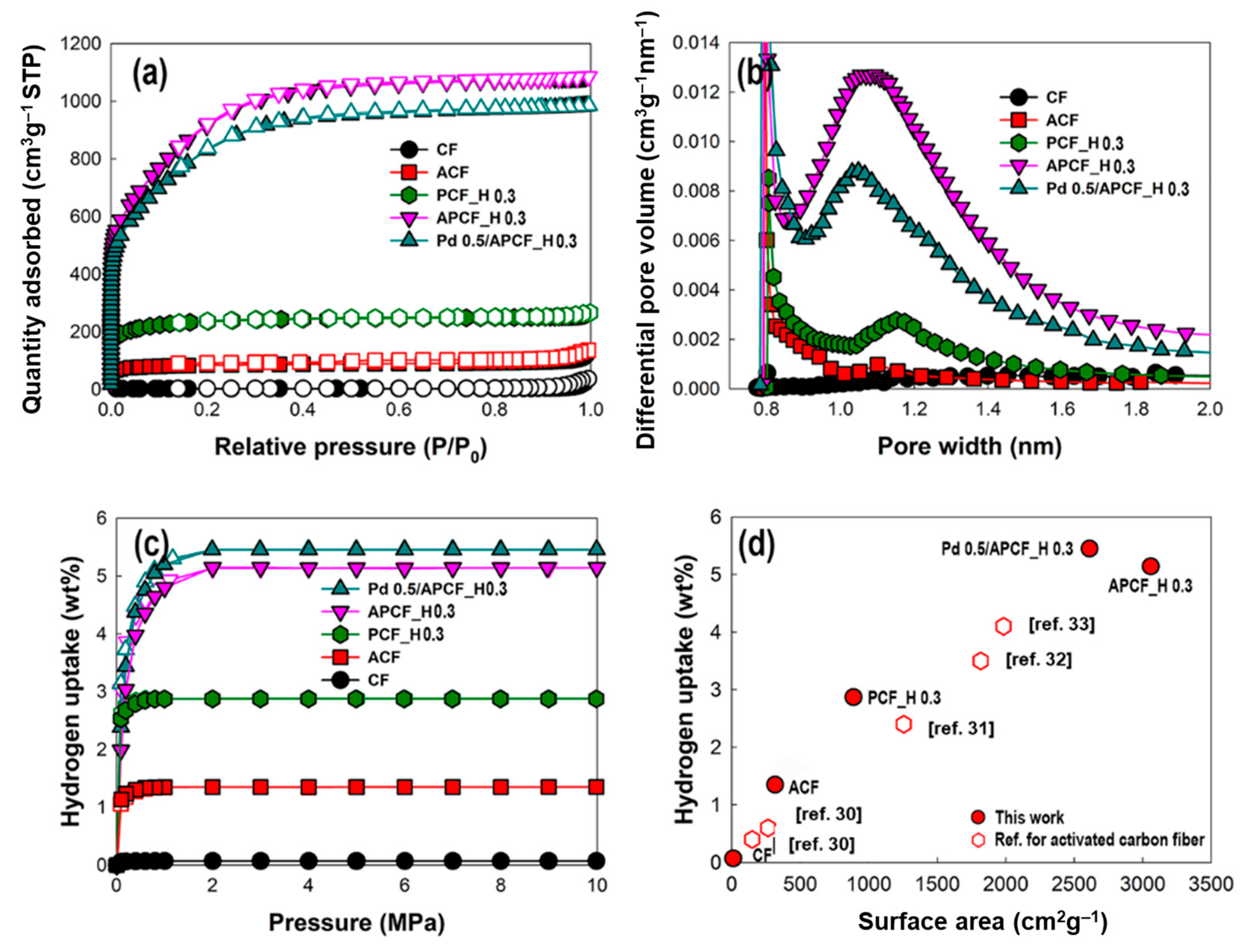
3.2. The Hydrogen Storage Capacity of PCF, APCF, and Pd Deposited APCF
3.3. APCF with an Excellent Hydrogen Storage Capacity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Park, J.H.; Ramasamy, P.; Kim, S.; Kim, Y.K.; Ahilan, V.; Shanmugam, S.; Lee, J.S. Hybrid metal—Cu2S nanostructures as efficient co-catalysts for photocatalytic hydrogen generation. Chem. Commun. 2017, 53, 3277–3280. [Google Scholar] [CrossRef] [PubMed]
- Moliner, R.; Lázaro, M.J.; Suelves, I. Analysis of the strategies for bridging the gap towards the hydrogen economy. Int. J. Hydrogen Energy 2016, 41, 19500–19508. [Google Scholar] [CrossRef]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid organic hydrogen carriers (LOHCs): Toward a hydrogen-free hydrogen economy. Acc. Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.K.; Piao, G.; Choi, W.; Park, H. Highly efficient hydrogen production using p-Si wire arrays and NiMoZn heterojunction photocathodes. Appl. Catal. B Environ. 2017, 217, 615–621. [Google Scholar] [CrossRef]
- Kim, Y.K.; Park, H. How and to what extent do carbon materials catalyze solar hydrogen production from water? Appl. Catal. B Environ. 2012, 125, 530–537. [Google Scholar] [CrossRef]
- Kim, Y.K.; Park, H. Light-harvesting multi-walled carbon nanotubes and CdS hybrids: Application to photocatalytic hydrogen production from water. Energy Environ. Sci. 2011, 4, 685–694. [Google Scholar] [CrossRef]
- Kim, Y.K.; Lim, S.K.; Park, H.; Hoffmann, M.R.; Kim, S. Trilayer CdS/carbon nanofiber (CNF) mat/Pt-TiO2 composite structures for solar hydrogen production: Effects of CNF mat thickness. Appl. Catal. B Environ. 2016, 196, 216–222. [Google Scholar] [CrossRef]
- Hwang, S.-H.; Choi, W.M.; Lim, S.K. Hydrogen storage characteristics of carbon fibers derived from rice straw and paper mulberry. Mater. Lett. 2016, 167, 18–21. [Google Scholar] [CrossRef]
- Liu, C.; Chen, Y.; Wu, C.-Z.; Xu, S.-T.; Cheng, H.-M. Hydrogen storage in carbon nanotubes revisited. Carbon 2010, 48, 452–455. [Google Scholar] [CrossRef]
- Isidro-Ortega, F.J.; Pacheco-Sánchez, J.H.; Desales-Guzmán, L.A. Hydrogen storage on lithium decorated zeolite templated carbon, DFT study. Int. J. Hydrog. Energy 2017, 42, 30704–30717. [Google Scholar] [CrossRef]
- Wang, Q. Transportation of hydrogen molecules using carbon nanotubes in torsion. Carbon 2009, 47, 1870–1873. [Google Scholar] [CrossRef]
- Zhang, F.; Zhao, P.; Niu, M.; Maddy, J. The survey of key technologies in hydrogen energy storage. Int. J. Hydrog. Energy 2016, 41, 14535–14552. [Google Scholar] [CrossRef]
- Xia, Y.; Yang, Z.; Zhu, Y. Porous carbon-based materials for hydrogen storage: Advancement and challenges. J. Mater. Chem. A 2013, 1, 9365–9381. [Google Scholar] [CrossRef]
- Rochat, S.; Polak-Krasna, K.; Tian, M.; Holyfield, L.T.; Mays, T.J.; Bowen, C.R.; Burrows, A.D. Hydrogen storage in polymer-based processable microporous composites. J. Mater. Chem. A 2017, 5, 18752–18761. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Wang, X.; Xu, H.; Zhao, Q.; Li, J. Hydrogen storage in several microporous zeolites. Int. J. Hydrog. Energy 2007, 32, 4998–5004. [Google Scholar] [CrossRef]
- Gygi, D.; Bloch, E.D.; Mason, J.A.; Hudson, M.R.; Gonzalez, M.I.; Siegelman, R.L.; Darwish, T.A.; Queen, W.L.; Brown, C.M.; Long, J.R. Hydrogen storage in the expanded pore metal–organic frameworks M2(dobpdc) (M = Mg, Mn, Fe, Co, Ni, Zn). Chem. Mater. 2016, 28, 1128–1138. [Google Scholar] [CrossRef]
- Kabbour, H.; Baumann, T.F.; Satcher, J.H.; Saulnier, A.; Ahn, C.C. Toward new candidates for hydrogen storage: High-surface-area carbon aerogels. Chem. Mater. 2006, 18, 6085–6087. [Google Scholar] [CrossRef]
- Zhang, L.; Liu, X.; Dou, Y.; Zhang, B.; Yang, H.; Dou, S.; Liu, H.; Huang, Y.; Hu, X. Mass production and pore size control of holey carbon microcages. Angew. Chem. Int. Ed. 2017, 56, 13790–13794. [Google Scholar] [CrossRef]
- Fang, D.Z.; Striemer, C.C.; Gaborski, T.R.; McGrath, J.L.; Fauchet, P.M. Pore size control of ultrathin silicon membranes by rapid thermal carbonization. Nano Lett. 2010, 10, 3904–3908. [Google Scholar] [CrossRef] [Green Version]
- Konda, S.K.; Chen, A. Palladium based nanomaterials for enhanced hydrogen spillover and storage. Mater. Today 2016, 19, 100–108. [Google Scholar] [CrossRef]
- Boudart, M.; Vannice, M.A.; Benson, J.E. Adlineation, portholes and spillover. Z. Für Phys. Chem. 1969, 64, 171–177. [Google Scholar] [CrossRef]
- Mohamed, K. Dope solution. In Encyclopedia of Membranes; Drioli, E., Giorno, L., Eds.; Springer: Berlin/Heidelberg, Germany, 2015; pp. 1–3. [Google Scholar] [CrossRef]
- Horváth, G.; Kawazoe, K. Method for the calculation of effective pore size distribution in molecular sieve carbon. J. Chem. Eng. Jpn. 1983, 16, 470–475. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.K.; Seo, H.-J.; Kim, S.; Hwang, S.-H.; Park, H.; Lim, S.K. Effect of ZnO electrodeposited on carbon film and decorated with metal nanoparticles for solar hydrogen production. J. Mater. Sci. Technol. 2016, 32, 1059–1065. [Google Scholar] [CrossRef]
- Yue, Z.; Economy, J.; Mangun, C.L. Preparation of fibrous porous materials by chemical activation 2. H3PO4 activation of polymer coated fibers. Carbon 2003, 41, 1809–1817. [Google Scholar] [CrossRef]
- Lee, J.S.; Choi, K.H.; Ghim, H.D.; Kim, S.S.; Chun, D.H.; Kim, H.Y.; Lyoo, W.S. Role of molecular weight of atactic poly(vinyl alcohol) (PVA) in the structure and properties of PVA nanofabric prepared by electrospinning. J. Appl. Polym. Sci. 2004, 93, 1638–1646. [Google Scholar] [CrossRef]
- Wang, J.; Kaskel, S. KOH activation of carbon-based materials for energy storage. J. Mater. Chem. 2012, 22, 23710–23725. [Google Scholar] [CrossRef]
- Diez, N.; Alvarez, P.; Granda, M.; Blanco, C.; Santamaria, R.; Menendez, R. A novel approach for the production of chemically activated carbon fibers. Chem. Eng. J. 2015, 260, 463–468. [Google Scholar] [CrossRef]
- Lillo-Ródenas, M.A.; Cazorla-Amorós, D.; Linares-Solano, A. Understanding chemical reactions between carbons and NaOH and KOH: An insight into the chemical activation mechanism. Carbon 2003, 41, 267–275. [Google Scholar] [CrossRef]
- Jordá-Beneyto, M.; Suárez-García, F.; Lozano-Castelló, D.; Cazorla-Amorós, D.; Linares-Solano, A. Hydrogen storage on chemically activated carbons and carbon nanomaterials at high pressures. Carbon 2007, 45, 293–303. [Google Scholar] [CrossRef]
- Xu, W.C.; Takahashi, K.; Matsuo, Y.; Hattori, Y.; Kumagai, M.; Ishiyama, S.; Kaneko, K.; Iijima, S. Investigation of hydrogen storage capacity of various carbon materials. Int. J. Hydrog. Energy 2007, 32, 2504–2512. [Google Scholar] [CrossRef]
- Purewal, J.J.; Kabbour, H.; Vajo, J.J.; Ahn, C.C.; Fultz, B. Pore size distribution and supercritical hydrogen adsorption in activated carbon fibers. Nanotechnology 2009, 20, 204012. [Google Scholar] [CrossRef] [PubMed]
- Gao, F.; Zhao, D.-L.; Li, Y.; Li, X.-G. Preparation and hydrogen storage of activated rayon-based carbon fibers with high specific surface area. J. Phys. Chem. Solids 2010, 71, 444–447. [Google Scholar] [CrossRef]
- Sevilla, M.; Sangchoom, W.; Balahmar, N.; Fuertes, A.B.; Mokaya, R. Highly porous renewable carbons for enhanced storage of energy-related gases (H2 and CO2) at high pressures. ACS Sustain. Chem. Eng. 2016, 4, 4710–4716. [Google Scholar] [CrossRef] [Green Version]
- Goldsmith, J.; Wong-Foy, A.G.; Cafarella, M.J.; Siegel, D.J. Theoretical limits of hydrogen storage in metal–organic frameworks: Opportunities and trade-offs. Chem. Mater. 2013, 25, 3373–3382. [Google Scholar] [CrossRef]
- Züttel, A.; Sudan, P.; Mauron, P.; Wenger, P. Model for the hydrogen adsorption on carbon nanostructures. Appl. Phys. A 2004, 78, 941–946. [Google Scholar] [CrossRef]
- Wang, Q.; Johnson, J.K. Molecular simulation of hydrogen adsorption in single-walled carbon nanotubes and idealized carbon slit pores. J. Chem. Phys. 1999, 110, 577–586. [Google Scholar] [CrossRef]
- Kadono, K.; Kajiura, H.; Shiraishi, M. Dense hydrogen adsorption on carbon subnanopores at 77 K. Appl. Phys. Lett. 2003, 83, 3392–3394. [Google Scholar] [CrossRef]
- Felderhoff, M.; Weidenthaler, C.; von Helmolt, R.; Eberle, U. Hydrogen storage: The remaining scientific and technological challenges. Phys. Chem. Chem. Phsy. 2007, 9, 2643–2653. [Google Scholar] [CrossRef]
- Czakkel, O.; Nagy, B.; Dobos, G.; Fouquet, P.; Bahn, E.; László, K. Static and dynamic studies of hydrogen adsorption on nanoporous carbon gels. Int. J. Hydrog. Energy 2019, 44, 18169–18178. [Google Scholar] [CrossRef]
- Im, J.S.; Park, S.-J.; Kim, T.J.; Kim, Y.H.; Lee, Y.-S. The study of controlling pore size on electrospun carbon nanofibers for hydrogen adsorption. J. Colloid Interface Sci. 2008, 318, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Patchkovskii, S.; Tse, J.S.; Yurchenko, S.N.; Zhechkov, L.; Heine, T.; Seifert, G. Graphene nanostructures as tunable storage media for molecular hydrogen. Proc. Natl. Acad. Sci. USA 2005, 102, 10439–10444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogotsi, Y.; Portet, C.; Osswald, S.; Simmons, J.M.; Yildirim, T.; Laudisio, G.; Fischer, J.E. Importance of pore size in high-pressure hydrogen storage by porous carbons. Int. J. Hydrog. Energy 2009, 34, 6314–6319. [Google Scholar] [CrossRef]
- Rzepka, M.; Lamp, P.; de la Casa-Lillo, M.A. Physisorption of hydrogen on microporous carbon and carbon nanotubes. J. Phys. Chem. B 1998, 102, 10894–10898. [Google Scholar] [CrossRef] [Green Version]
- Georgiev, P.A.; Ross, D.K.; Albers, P.; Ramirez-Cuesta, A.J. The rotational and translational dynamics of molecular hydrogen physisorbed in activated carbon: A direct probe of microporosity and hydrogen storage performance. Carbon 2006, 44, 2724–2738. [Google Scholar] [CrossRef]
- Cabria, I.; López, M.J.; Alonso, J.A. Simulation of the hydrogen storage in nanoporous carbons with different pore shapes. Int. J. Hydrog. Energy 2011, 36, 10748–10759. [Google Scholar] [CrossRef]
- Sevilla, M.; Alam, N.; Mokaya, R. Enhancement of hydrogen storage capacity of zeolite-templated carbons by chemical activation. J. Phys. Chem. C 2010, 114, 11314–11319. [Google Scholar] [CrossRef]
- de la Casa-Lillo, M.A.; Lamari-Darkrim, F.; Cazorla-Amorós, D.; Linares-Solano, A. Hydrogen storage in activated carbons and activated carbon fibers. J. Phys. Chem. B 2002, 106, 10930–10934. [Google Scholar] [CrossRef]
- Wenelska, K.; Michalkiewicz, B.; Chen, X.; Mijowska, E. Pd nanoparticles with tunable diameter deposited on carbon nanotubes with enhanced hydrogen storage capacity. Energy 2014, 75, 549–554. [Google Scholar] [CrossRef]
- Zhang, T.; Nakagawa, Y.; Wakasugi, T.; Isobe, S.; Wang, Y.; Hashimoto, N.; Ohnuki, S. Hydrogen absorption of palladium thin films observed by in situ transmission electron microscopy with an environmental cell. ACS Appl. Mater. Interfaces 2016, 8, 14548–14551. [Google Scholar] [CrossRef]
- Lachawiec, A.J.; Qi, G.; Yang, R.T. Hydrogen storage in nanostructured carbons by spillover: Bridge-building enhancement. Langmuir 2005, 21, 11418–11424. [Google Scholar] [CrossRef]
- Chen, Z.; Li, P.; Anderson, R.; Wang, X.; Zhang, X.; Robison, L.; Redfern, L.R.; Moribe, S.; Islamoglu, T.; Gómez-Gualdrón, D.A.; et al. Balancing volumetric and gravimetric uptake in highly porous materials for clean energy. Science 2020, 368, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Blankenship, L.S.; Mokaya, R. Cigarette butt-derived carbons have ultra-high surface area and unprecedented hydrogen storage capacity. Energy Environ. Sci. 2017, 10, 2552–2562. [Google Scholar] [CrossRef]
- Gadipelli, S.; Guo, Z.X. Graphene-based materials: Synthesis and gas sorption, storage and separation. Prog. Mater. Sci. 2015, 69, 1–60. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Zhou, Y.; Luo, L.; Wu, X.; Li, Z.; Fan, M.; Zhao, W. Preparation and characterization of heteroatom self-doped activated biocarbons as hydrogen storage and supercapacitor electrode materials. Electrochim. Acta 2019, 325, 134941. [Google Scholar] [CrossRef]
- Gadipelli, S.; Howard, C.A.; Guo, J.; Skipper, N.T.; Zhang, H.; Shearing, P.R.; Brett, D.J.L. Superior Multifunctional Activity of Nanoporous Carbons with Widely Tunable Porosity: Enhanced Storage Capacities for Carbon-Dioxide, Hydrogen, Water, and Electric Charge. Adv. Energy Mater. 2020, 10, 1903649. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Dope Solution | Weight Ratio | SBET 1 (m2 g−1) | Vp 2 (cm3 g−1) | Vmicro 3 (cm3 g−1) |
|---|---|---|---|---|---|
| PCF_L 0.5 | PAN/PVA_L | 5:5 | 201 | 0.25 | 0.12 (48.0) |
| PCF_L 0.3 | PAN/PVA_L | 7:3 | 424 | 0.27 | 0.16 (59.2) |
| PCF_L 0.1 | PAN/PVA_L | 9:1 | 110 | 0.06 | 0.02 (33.3) |
| PCF_H 0.5 | PAN/PVA_H | 5:5 | 377 | 0.27 | 0.14 (51.9) |
| PCF_H 0.3 | PAN/PVA_H | 7:3 | 889 | 0.47 | 0.34 (72.3) |
| PCF_H 0.1 | PAN/PVA_H | 9:1 | 798 | 0.46 | 0.25 (54.3) |
| Samples | SBET 1 (m2 g−1) | Vp 2 (cm3 g−1) | Vmicro 3 (cm3 g−1) | Waverage 4 (nm) | H2 Uptake (wt%) | Reference |
|---|---|---|---|---|---|---|
| CF | 11 | 0.06 | 0.01 (16.7) | 1.81 | 0.07 | This work |
| ACF | 316 | 0.21 | 0.12 (57.1) | 1.13 | 1.35 | This work |
| PCF_H 0.3 | 889 | 0.47 | 0.34 (72.3) | 1.12 | 2.87 | This work |
| APCF_H 0.3 | 3058 | 1.55 | 1.18 (76.1) | 1.05 | 5.14 | This work |
| Pd 0.5/ APCF_H 0.3 | 2611 | 1.43 | 1.03 (72.0) | 1.03 | 5.45 | This work |
| NF | 150 | - | - | - | 0.4 | [30] |
| ANF | 265 | - | - | - | 0.6 | [30] |
| ARCF1 | 1256 | - | - | - | 2.4 | [31] |
| ACF A20 | 1817 | - | - | - | 3.5 | [32] |
| ACF A20 | 1984 | - | - | - | 4.1 | [33] |
| Sample | [PdCl2] (mM) | SBET 1 (m2 g−1) | Vp 2 (cm3 g−1) | Vmicro 3 (cm3 g−1) | H2 Storage (wt%) |
|---|---|---|---|---|---|
| APCF_H 0.3 | 0 | 3058 | 1.55 | 1.18 (76.1) | 5.14 |
| Pd 0.1/APCF_H 0.3 | 0.1 | 2760 | 1.44 | 1.05 (72.9) | 5.30 |
| Pd 0.5/APCF_H 0.3 | 0.5 | 2611 | 1.43 | 1.03 (72.0) | 5.45 |
| Pd 1/APCF_H 0.3 | 1 | 2503 | 1.40 | 1.00 (71.4) | 5.17 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.-H.; Kim, Y.K.; Seo, H.-J.; Jeong, S.M.; Kim, J.; Lim, S.K. The Enhanced Hydrogen Storage Capacity of Carbon Fibers: The Effect of Hollow Porous Structure and Surface Modification. Nanomaterials 2021, 11, 1830. https://doi.org/10.3390/nano11071830
Hwang S-H, Kim YK, Seo H-J, Jeong SM, Kim J, Lim SK. The Enhanced Hydrogen Storage Capacity of Carbon Fibers: The Effect of Hollow Porous Structure and Surface Modification. Nanomaterials. 2021; 11(7):1830. https://doi.org/10.3390/nano11071830
Chicago/Turabian StyleHwang, Sung-Ho, Young Kwang Kim, Hye-Jin Seo, Soon Moon Jeong, Jongwon Kim, and Sang Kyoo Lim. 2021. "The Enhanced Hydrogen Storage Capacity of Carbon Fibers: The Effect of Hollow Porous Structure and Surface Modification" Nanomaterials 11, no. 7: 1830. https://doi.org/10.3390/nano11071830
APA StyleHwang, S. -H., Kim, Y. K., Seo, H. -J., Jeong, S. M., Kim, J., & Lim, S. K. (2021). The Enhanced Hydrogen Storage Capacity of Carbon Fibers: The Effect of Hollow Porous Structure and Surface Modification. Nanomaterials, 11(7), 1830. https://doi.org/10.3390/nano11071830