
Iron-Doped Bimodal Mesoporous Silica Nanomaterials as Sorbents for Solid-Phase Extraction of Perfluoroalkyl Substances in Environmental Water Samples

 ,
,  , ,
, ,  ,
,  and
and
Abstract
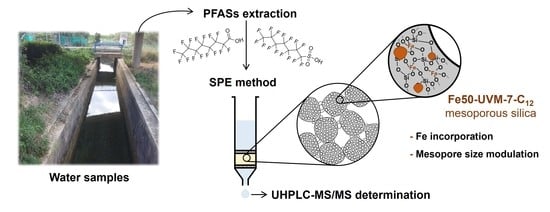
:
1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Instrumentation
2.3. Synthesis of Silica Nanomaterials Doped with Fe
2.4. SPE Optimization, Recommended Procedure, and Sample Analysis
2.5. Analytical Figures of Merit
3. Results and Discussion
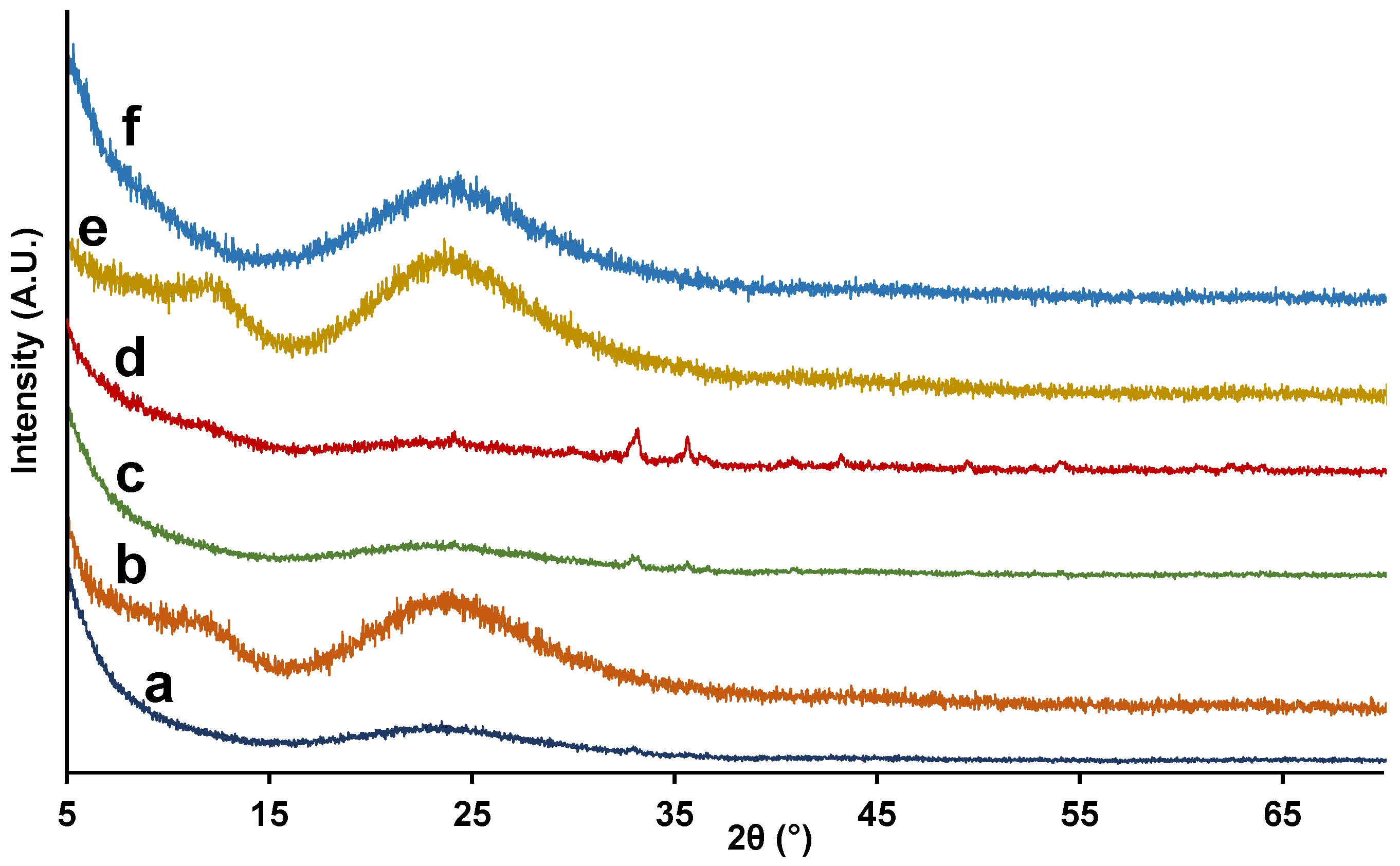
3.1. Synthesis and Characterization of Fe-Containing Silica Nanomaterials
3.2. Solid-Phase Evaluation and Optimization of SPE Parameters
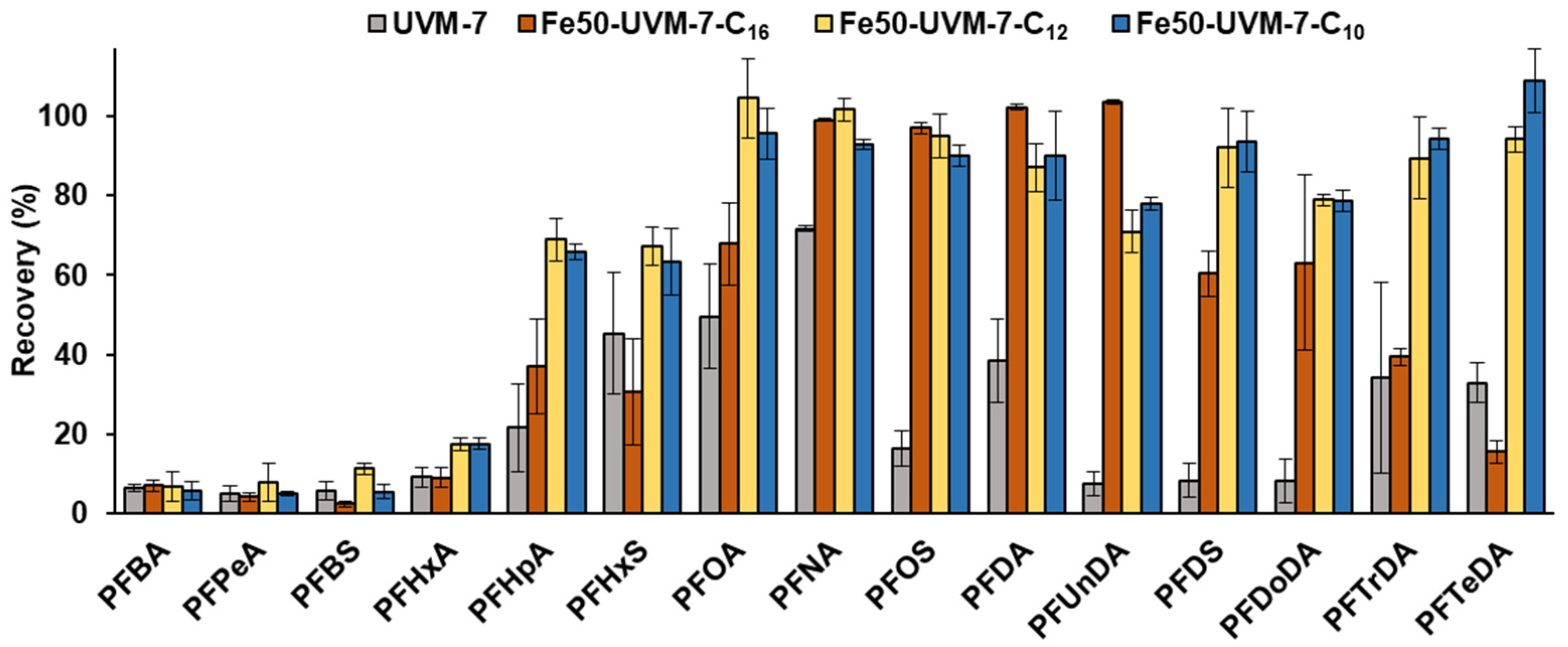
3.3. Method Performance
3.4. Water Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Buck, R.C.; Franklin, J.; Berger, U.; Conder, J.M.; Cousins, I.T.; de Voogt, P.; Jensen, A.A.; Kannan, K.; Mabury, S.A.; van Leeuwen, S.P.J. Perfluoroalkyl and polyfluoroalkyl substances in the environment: Terminology, classification, and origins. Integr. Environ. Assess. Manag. 2011, 7, 513–541. [Google Scholar] [CrossRef]
- Bach, C.; Boiteux, V.; Hemard, J.; Colin, A.; Rosin, C.; Munoz, J.F.; Dauchy, X. Simultaneous determination of perfluoroalkyl iodides, perfluoroalkane sulfonamides, fluorotelomer alcohols, fluorotelomer iodides and fluorotelomer acrylates and methacrylates in water and sediments using solid-phase microextraction-gas chromatography/mass spectrometry. J. Chromatogr. A 2016, 1448, 98–106. [Google Scholar] [CrossRef]
- Wang, Z.; Cousins, I.T.; Scheringer, M.; Buck, R.C.; Hungerbühler, K. Global emission inventories for C4-C14 perfluoroalkyl carboxylic acid (PFCA) homologues from 1951 to 2030, part I: Production and emissions from quantifiable sources. Environ. Int. 2014, 70, 62–75. [Google Scholar] [CrossRef]
- Gremmel, C.; Frömel, T.; Knepper, T.P. HPLC–MS/MS methods for the determination of 52 perfluoroalkyl and polyfluoroalkyl substances in aqueous samples. Anal. Bioanal. Chem. 2017, 409, 1643–1655. [Google Scholar] [CrossRef]
- Bull, S.; Burnett, K.; Vassaux, K.; Ashdown, L.; Brown, T.; Rushton, L. Extensive literature search and provision of summaries of studies related to the oral toxicity of perfluoroalkylated substances (PFASs), their precursors and potential replacements in experimental animals and humans. EFSA Support. Publ. 2014, 11, EN-572. [Google Scholar] [CrossRef]
- Lorenzo, M.; Campo, J.; Morales Suárez-Varela, M.; Picó, Y. Occurrence, distribution and behavior of emerging persistent organic pollutants (POPs) in a Mediterranean wetland protected area. Sci. Total Environ. 2019, 646, 1009–1020. [Google Scholar] [CrossRef]
- Secretariat of the Stockholm Convention—United Nations Environment Programme (UNEP) Stockholm Convention on Persistent Organic Pollutants (POPs). Revised in 2019. Available online: http://www.pops.int/TheConvention/Overview/TextoftheConvention/tabid/2232/Default.aspx (accessed on 14 January 2022).
- United States Environmental Protection Agency (US EPA) Fact Sheet: 2010/2015 PFOA Stewardship Program. Available online: https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/fact-sheet-20102015-pfoa-stewardship-program (accessed on 14 January 2022).
- Directive 2008/105/EC of the European Parliament and of the Council of 16 December 2008 on environmental quality standards in the field of water policy amending and subsequently repealing Council Directives 82/176/EEC, 83/513/EEC, 84/156/EEC, 84/491/EEC, 86/280/EEC and amending Directive 2000/60/EC of the European Parliament and of the Council. Off. J. Eur. Union 2008, L348, 84–97.
- Joerss, H.; Apel, C.; Ebinghaus, R. Emerging per-and polyfluoroalkyl substances (PFASs) in surface water and sediment of the North and Baltic Seas. Sci. Total Environ. 2019, 686, 360–369. [Google Scholar] [CrossRef]
- European Chemicals Agency (ECHA) Candidate List of Substances of Very High Concern for Authorization. Available online: https://echa.europa.eu/es/candidate-list-table (accessed on 14 January 2022).
- Secretariat of the Basel Rotterdam and Stockholm Conventions—United Nations Environment Programme (UNEP) Persistent Organic Pollutants Review Committee (POPRC) Recommendations for Listing Chemicals—Chemicals under Review. Available online: http://chm.pops.int/Convention/POPsReviewCommittee/Chemicals/tabid/243/Default.aspx (accessed on 14 January 2022).
- Pico, Y.; Blasco, C.; Farré, M.; Barceló, D. Occurrence of perfluorinated compounds in water and sediment of L’Albufera Natural Park (València, Spain). Environ. Sci. Pollut. Res. 2012, 19, 946–957. [Google Scholar] [CrossRef]
- Pignotti, E.; Casas, G.; Llorca, M.; Tellbüscher, A.; Almeida, D.; Dinelli, E.; Farré, M.; Barceló, D. Seasonal variations in the occurrence of perfluoroalkyl substances in water, sediment and fish samples from Ebro Delta (Catalonia, Spain). Sci. Total Environ. 2017, 607–608, 933–943. [Google Scholar] [CrossRef]
- Lenka, S.P.; Kah, M.; Padhye, L.P. A review of the occurrence, transformation, and removal of poly-and perfluoroalkyl substances (PFAS) in wastewater treatment plants. Water Res. 2021, 199, 117187. [Google Scholar] [CrossRef]
- Xu, B.; Liu, S.; Zhou, J.L.; Zheng, C.; Weifeng, J.; Chen, B.; Zhang, T.; Qiu, W. PFAS and their substitutes in groundwater: Occurrence, transformation and remediation. J. Hazard. Mater. 2021, 412, 125159. [Google Scholar] [CrossRef]
- Baluyot, J.C.; Reyes, E.M.; Velarde, M.C. Per-and polyfluoroalkyl substances (PFAS) as contaminants of emerging concern in Asia’s freshwater resources. Environ. Res. 2021, 197, 111122. [Google Scholar] [CrossRef]
- Sharifan, H.; Bagheri, M.; Wang, D.; Burken, J.G.; Higgins, C.P.; Liang, Y.; Liu, J.; Schaefer, C.E.; Blotevogel, J. Fate and transport of per-and polyfluoroalkyl substances (PFASs) in the vadose zone. Sci. Total Environ. 2021, 771, 145427. [Google Scholar] [CrossRef]
- Joerss, H.; Xie, Z.; Wagner, C.C.; Von Appen, W.J.; Sunderland, E.M.; Ebinghaus, R. Transport of legacy perfluoroalkyl substances and the replacement compound HFPO-DA through the Atlantic gateway to the Arctic ocean—Is the Arctic a sink or a source? Environ. Sci. Technol. 2020, 54, 9958–9967. [Google Scholar] [CrossRef]
- Wang, Z.; Xie, Z.; Mi, W.; Möller, A.; Wolschke, H.; Ebinghaus, R. Neutral poly/per-fluoroalkyl substances in air from the Atlantic to the Southern ocean and in Antarctic snow. Environ. Sci. Technol. 2015, 49, 7770–7775. [Google Scholar] [CrossRef]
- Portolés, T.; Rosales, L.E.; Sancho, J.V.; Santos, F.J.; Moyano, E. Gas chromatography-tandem mass spectrometry with atmospheric pressure chemical ionization for fluorotelomer alcohols and perfluorinated sulfonamides determination. J. Chromatogr. A 2015, 1413, 107–116. [Google Scholar] [CrossRef] [Green Version]
- Monteleone, M.; Naccarato, A.; Sindona, G.; Tagarelli, A. A rapid and sensitive assay of perfluorocarboxylic acids in aqueous matrices by headspace solid phase microextraction-gas chromatography-triple quadrupole mass spectrometry. J. Chromatogr. A 2012, 1251, 160–168. [Google Scholar] [CrossRef]
- Lorenzo, M.; Campo, J.; Picó, Y. Analytical challenges to determine emerging persistent organic pollutants in aquatic ecosystems. TrAC-Trends Anal. Chem. 2018, 103, 137–155. [Google Scholar] [CrossRef]
- Berger, U.; Kaiser, M.A.; Kärrman, A.; Barber, J.L.; van Leeuwen, S.P.J. Recent developments in trace analysis of poly-and perfluoroalkyl substances. Anal. Bioanal. Chem. 2011, 400, 1625–1635. [Google Scholar] [CrossRef]
- González-Barreiro, C.; Martínez-Carballo, E.; Sitka, A.; Scharf, S.; Gans, O. Method optimization for determination of selected perfluorinated alkylated substances in water samples. Anal. Bioanal. Chem. 2006, 386, 2123–2132. [Google Scholar] [CrossRef]
- van Leeuwen, S.P.J.; de Boer, J. Extraction and clean-up strategies for the analysis of poly-and perfluoroalkyl substances in environmental and human matrices. J. Chromatogr. A 2007, 1153, 172–185. [Google Scholar] [CrossRef]
- Majors, R.E. Solid-phase extraction. In Handbook of Sample Preparation; Pawliszyn, J., Lord, H.L., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010; pp. 53–79. [Google Scholar]
- Mahmoodi, N.M. Synthesis of magnetic carbon nanotube and photocatalytic dye degradation ability. Environ. Monit. Assess. 2014, 186, 5595–5604. [Google Scholar] [CrossRef]
- Mahmoodi, N.M.; Taghizadeh, A.; Taghizadeh, M.; Shahali, M.A. Surface modified montmorillonite with cationic surfactants: Preparation, characterization, and dye adsorption from aqueous solution. J. Environ. Chem. Eng. 2019, 7, 103243. [Google Scholar] [CrossRef]
- Taniyasu, S.; Kannan, K.; Yeung, L.W.Y.; Kwok, K.Y.; Lam, P.K.S.; Yamashita, N. Analysis of trifluoroacetic acid and other short-chain perfluorinated acids (C2-C4) in precipitation by liquid chromatography-tandem mass spectrometry: Comparison to patterns of long-chain perfluorinated acids (C5-C18). Anal. Chim. Acta 2008, 619, 221–230. [Google Scholar] [CrossRef]
- Janda, J.; Nödler, K.; Brauch, H.J.; Zwiener, C.; Lange, F.T. Robust trace analysis of polar (C2-C8) perfluorinated carboxylic acids by liquid chromatography-tandem mass spectrometry: Method development and application to surface water, groundwater and drinking water. Environ. Sci. Pollut. Res. 2019, 26, 7326–7336. [Google Scholar] [CrossRef]
- Munoz, G.; Vo Duy, S.; Budzinski, H.; Labadie, P.; Liu, J.; Sauvé, S. Quantitative analysis of poly-and perfluoroalkyl compounds in water matrices using high resolution mass spectrometry: Optimization for a laser diode thermal desorption method. Anal. Chim. Acta 2015, 881, 98–106. [Google Scholar] [CrossRef]
- Boiteux, V.; Bach, C.; Sagres, V.; Hemard, J.; Colin, A.; Rosin, C.; Munoz, J.F.; Dauchy, X. Analysis of 29 per-and polyfluorinated compounds in water, sediment, soil and sludge by liquid chromatography–tandem mass spectrometry. Int. J. Environ. Anal. Chem. 2016, 96, 705–728. [Google Scholar] [CrossRef]
- Zacs, D.; Bartkevics, V. Trace determination of perfluorooctane sulfonate and perfluorooctanoic acid in environmental samples (surface water, wastewater, biota, sediments, and sewage sludge) using liquid chromatography—Orbitrap mass spectrometry. J. Chromatogr. A 2016, 1473, 109–121. [Google Scholar] [CrossRef]
- Campo, J.; Pérez, F.; Masiá, A.; Picó, Y.; la Farré, M.; Barceló, D. Perfluoroalkyl substance contamination of the Llobregat River ecosystem (Mediterranean area, NE Spain). Sci. Total Environ. 2015, 503–504, 48–57. [Google Scholar] [CrossRef]
- Liu, Y.; Bao, J.; Hu, X.M.; Lu, G.L.; Yu, W.J.; Meng, Z.H. Optimization of extraction methods for the analysis of PFOA and PFOS in the salty matrices during the wastewater treatment. Microchem. J. 2020, 155, 104673. [Google Scholar] [CrossRef]
- Lashgari, M.; Basheer, C.; Kee Lee, H. Application of surfactant-templated ordered mesoporous material as sorbent in micro-solid phase extraction followed by liquid chromatography-triple quadrupole mass spectrometry for determination of perfluorinated carboxylic acids in aqueous media. Talanta 2015, 141, 200–206. [Google Scholar] [CrossRef]
- Kharbouche, L.; Gil García, M.D.; Lozano, A.; Hamaizi, H.; Galera, M.M. Solid phase extraction of pesticides from environmental waters using an MSU-1 mesoporous material and determination by UPLC-MS/MS. Talanta 2019, 199, 612–619. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C.; Beck, J.S. Ordered mesoporous molecular sieves synthesized by a liquid-crystal template mechanism. Nature 1992, 359, 710. [Google Scholar] [CrossRef]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C. Synthetic Mesoporous Crystalline Material. U.S. Patent 5098684, 24 March 1992. [Google Scholar]
- Kresge, C.T.; Leonowicz, M.E.; Roth, W.J.; Vartuli, J.C. Composition of Synthetic Porous Crystalline Material, Its Synthesis. U.S. Patent 5102643, 7 April 1992. [Google Scholar]
- El Haskouri, J.; Morales, J.M.; Ortiz de Zárate, D.; Ferna, L.; Latorre, J.; Guillem, C.; Beltrán, A.; Beltrán, D.; Amorós, P. Nanoparticulated silicas with bimodal porosity: Chemical control of the pore sizes. Inorg. Chem. 2008, 47, 8267–8277. [Google Scholar] [CrossRef]
- Pérez-Cabero, M.; Esteve-Turrillas, F.A.; Beltrán, D.; Amorós, P. Hierarchical porous carbon with designed pore architecture and study of its adsorptive properties. Solid State Sci. 2010, 12, 15–25. [Google Scholar] [CrossRef]
- Cabrera, S.; El Haskouri, J.; Guillem, C.; Latorre, J.; Beltrán-Porter, A.; Beltrán-Porter, D.; Marcos, M.D.; Amorós, P. Generalised syntheses of ordered mesoporous oxides: The atrane route. Solid State Sci. 2000, 2, 405–420. [Google Scholar] [CrossRef]
- Pellicer-Castell, E.; Belenguer-Sapiña, C.; Amorós, P.; El Haskouri, J.; Herrero-Martínez, J.M.; Mauri-Aucejo, A. Study of silica-structured materials as sorbents for organophosphorus pesticides determination in environmental water samples. Talanta 2018, 189, 560–567. [Google Scholar] [CrossRef]
- Shirkhanloo, H.; Falahnejad, M.; Zavvar Mousavi, H. Mesoporous silica nanoparticles as an adsorbent for preconcentration and determination of trace amount of nickel in environmental samples by atom trap flame atomic absorption spectrometry. J. Appl. Spectrosc. 2016, 82, 1072–1077. [Google Scholar] [CrossRef]
- Pellicer-Castell, E.; Belenguer-Sapiña, C.; Amorós, P.; El Haskouri, J.; Herrero-Martínez, J.M.; Mauri-Aucejo, A.R. Mesoporous silica sorbent with gold nanoparticles for solid-phase extraction of organochlorine pesticides in water samples. J. Chromatogr. A 2021, 1662, 462729. [Google Scholar] [CrossRef]
- Rayne, S.; Forest, K. Perfluoroalkyl sulfonic and carboxylic acids: A critical review of physicochemical properties, levels and patterns in waters and wastewaters, and treatment methods. J. Environ. Sci. Health Part A 2009, 44, 1145–1199. [Google Scholar] [CrossRef] [PubMed]
- Johnson, R.L.; Anschutz, A.J.; Smolen, J.M.; Simcik, M.F.; Penn, R.L. The adsorption of perfluorooctane sulfonate onto sand, clay, and iron oxide surfaces. J. Chem. Eng. Data 2007, 52, 1165–1170. [Google Scholar] [CrossRef]
- Carmona, E.; Andreu, V.; Picó, Y. Occurrence of acidic pharmaceuticals and personal care products in Turia River Basin: From waste to drinking water. Sci. Total Environ. 2014, 484, 53–63. [Google Scholar] [CrossRef]
- Olivieri, A.C.; Faber, N.M.; Ferré, J.; Boqué, R.; Kalivas, J.H.; Mark, H. Uncertainty estimation and figures of merit for multivariate calibration (IUPAC Technical Report). Pure Appl. Chem. 2006, 78, 633–661. [Google Scholar] [CrossRef] [Green Version]
- Laughlin, R.G. The Aqueous Phase Behavior of Surfactants; Academic Press: San Diego, CA, USA, 1994. [Google Scholar]
- Schwertmann, U.; Cornell, R.M. Iron Oxides in the Laboratory, Preparation and Characterization; Wiley-VCH: Weinheim, Germany, 2000; ISBN 3-527-29669-7. [Google Scholar]
- Baes, C.F.; Mesmer, R.S. The Hydrolysis of Cations; John Wiley & Sons, Inc.: New York, NY, USA, 1976. [Google Scholar]
- Iler, R.K. The Chemistry of Silica; John Wiley & Sons, Inc.: New York, NY, USA, 1979; ISBN 978-0-471-02404-0. [Google Scholar]
- Dutta, P.; Pal, S.; Seehra, M.S.; Shah, N.; Huffman, G.P. Size dependence of magnetic parameters and surface disorder in magnetite nanoparticles. J. Appl. Phys. 2009, 105, 10–13. [Google Scholar] [CrossRef]
- Sun, S.; Zeng, H. Size-controlled synthesis of magnetite nanoparticles. J. Am. Chem. Soc. 2002, 124, 8204–8205. [Google Scholar] [CrossRef]
- Park, M.; Wu, S.; Lopez, I.J.; Chang, J.Y.; Karanfil, T.; Snyder, S.A. Adsorption of perfluoroalkyl substances (PFAS) in groundwater by granular activated carbons: Roles of hydrophobicity of PFAS and carbon characteristics. Water Res. 2020, 170, 115364. [Google Scholar] [CrossRef]
- Wang, L.; Gong, X.; Wang, R.; Gan, Z.; Lu, Y.; Sun, H. Application of an immobilized ionic liquid for the passive sampling of perfluorinated substances in water. J. Chromatogr. A 2017, 1515, 45–53. [Google Scholar] [CrossRef]
- Igarashi, Y.; Takahashi, M.; Tsutsumi, T.; Inoue, K.; Akiyama, H. Monitoring analysis of perfluoroalkyl substances and F-53B in bottled water, tea and juice samples by LC-MS/MS. Chem. Pharm. Bull. 2021, 69, 286–290. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Material | Si/Fe (Real Molar Ratio) | Surface Area (m2 g−1) | d100 (nm) | Mesopore Size (nm) | Mesopore Volume (cm3 g−1) | Large Pore Size (nm) | Large Pore Volume (cm3 g−1) | |
|---|---|---|---|---|---|---|---|---|
| Fe100-UVM-7-C16 | 63 | 1109 | 4.1 | 2.81 | 0.82 a | 0.77 b | 27.4 | 1.20 |
| Fe50-UVM-7-C16 | 29 | 1067 | 4.2 | 2.83 | 0.77 a | 0.70 b | 44.4 | 1.75 |
| Fe25-UVM-7-C16 | 14 | 907 | 4.2 | 2.58 | 0.45 a | 0.39 b | 36.4 | 1.05 |
| Fe10-UVM-7-C16 | 6 | 931 | 4.2 | 2.55 | 0.57 a | 0.56 b | 34.9 | 0.55 |
| Fe50-UVM-7-C12 | 24 | 1171 | 3.0 | 2.26 | 0.48 a | 0.44 b | 43.3 | 1.47 |
| Fe50-UVM-7-C10 | 18 | 1046 | 2.7 | 2.01 | 0.28 a | 0.27 b | 34.7 | 1.28 |
| Compound | Linearity a (μg L−1) | Sensitivity (ng L−1) | Extraction Efficiency (%) | EF | Repeatability RSD (%) | ||
|---|---|---|---|---|---|---|---|
| LOD | LOQ | Intra-Day | Inter-Day | ||||
| PFOA | 2–50 | 4 | 12 | 66 ± 20 | 132 | 22 | 38 |
| PFNA | 4–50 | 7 | 22 | 110 ± 30 | 212 | 16 | 28 |
| PFOS | 2–50 | 3 | 9 | 110 ± 30 | 212 | 19 | 29 |
| PFDA | 5–50 | 8 | 24 | 77 ± 16 | 154 | 15 | 21 |
| PFUnDA | 4–50 | 7 | 21 | 73 ± 15 | 145 | 17 | 21 |
| PFDS | 2–50 | 3 | 11 | 67 ± 14 | 134 | 21 | 21 |
| PFDoDA | 4–50 | 6 | 18 | 75 ± 13 | 151 | 13 | 17 |
| PFTrDA | 5–50 | 8 | 25 | 68 ± 17 | 136 | 14 | 25 |
| PFTeDA | 2–50 | 3 | 11 | 61 ± 14 | 121 | 18 | 23 |
| Sample | Sorbent | LOD (ng L−1) | LOQ (ng L−1) | Recovery (%) | RSD (%) | EF (EFmax) | Solvents | Instrumental Determination | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Well water (100 mL) | Fe-UVM-7-C12 (300 mg) | 3–8 | 9–25 | 61–110 | 14–29 (38) a | 121–212 (200) | 11 mL MeOH | UPLC-MS/MS | This work |
| Wastewater (200 mL) | Oasis WAX (60 mg) | 0.2–5.8 | 1.7–11 | 19–99 | 2–22 | 76–396 b (400) | 8 mL MeOH | HPLC-MS/MS | [4] |
| Rainwater (200 mL) | Oasis WAX (150 mg) | - c | 0.02–0.1 | 97–132 | 6–15 | 24–66 b (25–50) | 12 mL MeOH | LC-MS/MS | [30] |
| Surface and tap water (50 mL) | Oasis WAX (150 mg) | 0.3–0.5 | 1.5–2.5 | 83–102 d | 0.7–6.2 | 104–127 b (125) | 15 mL MeOH | LC-MS/MS | [31] |
| Surface, tap, and wastewater (250 mL−1 L) | Strata-X AW (200 mg) (+carbon clean-up) | 0.3–3 | - c | 38–104 | 4–33 | 380–1040 b (1000) | 26 mL MeOH | LDTD-HRMS | [32] |
| River, ground, and drinking water (250 mL) | Strata-X AW (200 mg) | - c | 4–10 | (15) a 49–103 d | 7–35 (180) a | (375) a 1225–2575 b (2500) | 15 mL MeOH 1.4 mL DCM 0.6 mL IPA | UHPLC-MS/MS | [33] |
| Surface and wastewater e (200 mL) | Strata-X AW (200 mg) (+carbon clean-up) | - c | - c | 98–113 | 1.8–7.3 | 1960–2260 b (2000) | 15 mL MTBE 3 mL MeOH | HPLC-MS/MS | [34] |
| River water (250 mL) | Strata (200 mg) | - c | 0.01–2 | 60–92 d | 10–18 | 600–920 b (1000) | 12 mL MeOH | LC-MS/MS | [35] |
| Surface water e (10 mL) | Oasis HLB (225 mg) | 0.1 | 0.5 | 61–83 | 3–10 | 6.1–8.3 b (10) | 7 mL MeOH | LC-MS/MS | [36] |
| Wastewater (1 L) | Imidazolium-based IL (passive) (30 mg) | 0.2–0.3 | 0.7–1 | 53.7–110 | <13 | - c | 6 mL MeOH | HPLC-MS/MS | [59] |
| Drinking water (35 mL) | Presep PFC-11 (60 mg) | - c | 5–25 | 83.2–112.4 | 0.1–0.4 | 58–79 b (70) | 11.5 mL ACN | LC-MS/MS | [60] |
| Compound | M1S | M2S | |
|---|---|---|---|
| Fe50-UVM-7-C12 | Oasis WAX | Fe50-UVM-7-C12 | |
| PFOA | 165 ± 15 | 150 ± 9 | 152 ± 5 |
| PFNA | 152 ± 15 | 149 ± 15 | 150 ± 2 |
| PFOS | 161 ± 6 | 155 ± 15 | 147 ± 12 |
| PFDA | 152 ± 12 | 151 ± 15 | 147 ± 3 |
| PFUnDA | 147 ± 8 | 157 ± 10 | 148 ± 7 |
| PFDS | 160 ± 13 | 141 ± 5 | 140 ± 14 |
| PFDoDA | 138 ± 18 | 156 ± 8 | 150 ± 18 |
| PFTrDA | 130 ± 20 | 139 ± 6 | 138 ± 12 |
| PFTeDA | 153 ± 18 | 164 ± 3 | 152 ± 8 |
| PFOA | 165 ± 15 | 150 ± 9 | 152 ± 5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pellicer-Castell, E.; Belenguer-Sapiña, C.; El Haskouri, J.; Amorós, P.; Herrero-Martínez, J.M.; Mauri-Aucejo, A.R. Iron-Doped Bimodal Mesoporous Silica Nanomaterials as Sorbents for Solid-Phase Extraction of Perfluoroalkyl Substances in Environmental Water Samples. Nanomaterials 2022, 12, 1441. https://doi.org/10.3390/nano12091441
Pellicer-Castell E, Belenguer-Sapiña C, El Haskouri J, Amorós P, Herrero-Martínez JM, Mauri-Aucejo AR. Iron-Doped Bimodal Mesoporous Silica Nanomaterials as Sorbents for Solid-Phase Extraction of Perfluoroalkyl Substances in Environmental Water Samples. Nanomaterials. 2022; 12(9):1441. https://doi.org/10.3390/nano12091441
Chicago/Turabian StylePellicer-Castell, Enric, Carolina Belenguer-Sapiña, Jamal El Haskouri, Pedro Amorós, José Manuel Herrero-Martínez, and Adela R. Mauri-Aucejo. 2022. "Iron-Doped Bimodal Mesoporous Silica Nanomaterials as Sorbents for Solid-Phase Extraction of Perfluoroalkyl Substances in Environmental Water Samples" Nanomaterials 12, no. 9: 1441. https://doi.org/10.3390/nano12091441
APA StylePellicer-Castell, E., Belenguer-Sapiña, C., El Haskouri, J., Amorós, P., Herrero-Martínez, J. M., & Mauri-Aucejo, A. R. (2022). Iron-Doped Bimodal Mesoporous Silica Nanomaterials as Sorbents for Solid-Phase Extraction of Perfluoroalkyl Substances in Environmental Water Samples. Nanomaterials, 12(9), 1441. https://doi.org/10.3390/nano12091441