

Machine Learning Electron Density Prediction Using Weighted Smooth Overlap of Atomic Positions


Abstract
:1. Introduction
2. Methods
2.1. Fingerprinting
2.2. NN Training
2.3. Data Generation with DFT
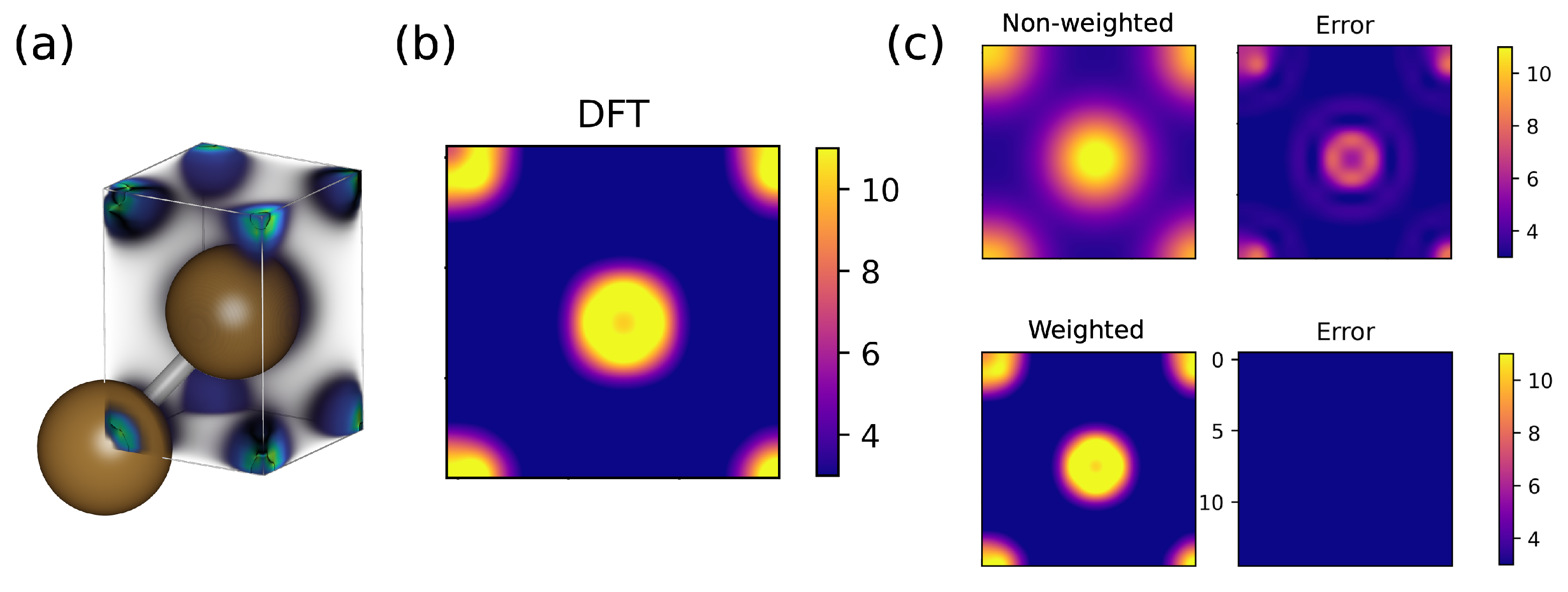
3. Results and Discussion
3.1. Bulk Cu
3.2. Bulk Si and LiF
3.3. Scaling with System Size
3.4. Water
3.5. Graphanol
3.5.1. Charge Tracking
3.5.2. Model Transferability
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| DeepCDP | Deep Learning for Charge Density Prediction |
| CDP | Charge Density Predictor |
| DFT | Density Functional Theory |
| MD | Molecular Dynamics |
| NN | Neural Network |
| SOAP | Smooth Overlap of Atomic Orbitals |
| MAE | Mean Absolute Error |
| MSE | Mean Squared Error |
| u24C | Uncharged Graphanol with 24 Carbon Atoms |
| c24C | Charged Graphanol with 24 Carbon Atoms |
References
- Langmann, J.; Kepenci, H.; Eickerling, G.; Batke, K.; Jesche, A.; Xu, M.; Canfield, P.; Scherer, W. Experimental X-Ray Charge-Density Studies-a Suitable Probe for Superconductivity? A Case Study on MgB2. J. Phys. Chem. A 2022, 126, 8494–8507. [Google Scholar] [CrossRef]
- Bartók, A.P.; Payne, M.C.; Kondor, R.; Csányi, G. Gaussian Approximation Potentials: The Accuracy of Quantum Mechanics, Without the Electrons. Phys. Rev. Lett. 2010, 104, 136403. [Google Scholar] [CrossRef] [Green Version]
- Wood, M.A.; Thompson, A.P. Extending the Accuracy of the SNAP Interatomic Potential Form. J. Chem. Phys. 2018, 148, 241721. [Google Scholar] [CrossRef] [Green Version]
- Botu, V.; Batra, R.; Chapman, J.; Ramprasad, R. Machine Learning Force Fields: Construction, Validation, and Outlook. J. Phys. Chem. C 2017, 121, 511–522. [Google Scholar] [CrossRef]
- Chen, M.S.; Morawietz, T.; Mori, H.; Markland, T.E.; Artrith, N. AENET–LAMMPS and AENET–TINKER: Interfaces for Accurate and Efficient Molecular Dynamics Simulations With Machine Learning Potentials. J. Chem. Phys. 2021, 155, 074801. [Google Scholar] [CrossRef]
- Khorshidi, A.; Peterson, A.A. Amp: A Modular Approach to Machine Learning in Atomistic Simulations. Comput. Phys. Commun. 2016, 207, 310–324. [Google Scholar] [CrossRef]
- Shapeev, A.V. Moment Tensor Potentials: A Class of Systematically Improvable Interatomic Potentials. Multiscale Model. Simul. 2016, 14, 1153–1173. [Google Scholar] [CrossRef]
- Wang, H.; Zhang, L.; Han, J.; E, W. DeePMD-Kit: A Deep Learning Package for Many-Body Potential Energy Representation and Molecular Dynamics. Comput. Phys. Commun. 2018, 228, 178–184. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Han, J.; Wang, H.; Car, R.; Weinan, E.J.P.R.L. Deep Potential Molecular Dynamics: A Scalable Model with the Accuracy of Quantum Mechanics. Phys. Rev. Lett. 2018, 120, 143001. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Han, J.; Wang, H.; Saidi, W.; Car, R.; Weinan, E. End-to-End Symmetry Preserving Inter-Atomic Potential Energy Model for Finite and Extended Systems. In Proceedings of the Advances in Neural Information Processing Systems, Montreal, QC, Canada, 2–8 December 2018; Bengio, S., Wallach, H., Larochelle, H., Grauman, K., Cesa-Bianchi, N., Garnett, R., Eds.; Curran Associates, Inc.: New York, NY, USA, 2018; Volume 31, pp. 4436–4446. [Google Scholar]
- Deringer, V.L.; Caro, M.A.; Csányi, G. Machine Learning Interatomic Potentials as Emerging Tools for Materials Science. Adv. Mater. 2019, 31, 1902765. [Google Scholar] [CrossRef] [PubMed]
- Bogojeski, M.; Vogt-Maranto, L.; Tuckerman, M.E.; Müller, K.R.; Burke, K. Quantum chemical accuracy from density functional approximations via machine learning. Nat. Commun. 2020, 11, 5223. [Google Scholar] [CrossRef] [PubMed]
- Fabrizio, A.; Grisafi, A.; Meyer, B.; Ceriotti, M.; Corminboeuf, C. Electron Density Learning of Non-Covalent Systems. Chem. Sci. 2019, 10, 9424–9432. [Google Scholar] [CrossRef] [Green Version]
- Grisafi, A.; Fabrizio, A.; Meyer, B.; Wilkins, D.M.; Corminboeuf, C.; Ceriotti, M. Transferable Machine-Learning Model of the Electron Density. ACS Cent. Sci. 2018, 5, 57–64. [Google Scholar] [CrossRef]
- Chandrasekaran, A.; Kamal, D.; Batra, R.; Kim, C.; Chen, L.; Ramprasad, R. Solving the Electronic Structure Problem With Machine Learning. npj Comput. Mater. 2019, 5, 22. [Google Scholar] [CrossRef] [Green Version]
- Rackers, J.; Tecot, L.; Geiger, M.; Smidt, T. A Recipe for Cracking the Quantum Scaling Limit with Machine Learned Electron Densities. Mach. Learn. Sci. Technol. 2023, 4, 015027. [Google Scholar] [CrossRef]
- Cuevas-Zuviría, B.; Pacios, L.F. Machine Learning of Analytical Electron Density in Large Molecules Through Message-Passing. J. Chem. Inf. Model. 2021, 61, 2658–2666. [Google Scholar] [CrossRef]
- Gong, S.; Xie, T.; Zhu, T.; Wang, S.; Fadel, E.R.; Li, Y.; Grossman, J.C. Predicting Charge Density Distribution of Materials Using a Local-Environment-Based Graph Convolutional Network. Phys. Rev. B 2019, 100, 184103. [Google Scholar] [CrossRef] [Green Version]
- del Rio, B.G.; Kuenneth, C.; Tran, H.D.; Ramprasad, R. An Efficient Deep Learning Scheme to Predict the Electronic Structure of Materials and Molecules: The Example of Graphene-Derived Allotropes. J. Phys. Chem. A 2020, 124, 9496–9502. [Google Scholar] [CrossRef]
- Kamal, D.; Chandrasekaran, A.; Batra, R.; Ramprasad, R. A Charge Density Prediction Model for Hydrocarbons Using Deep Neural Networks. Mach. Learn. Sci. Technol. 2020, 1, 025003. [Google Scholar] [CrossRef] [Green Version]
- Achar, S.; Bernasconi, L.; DeMaio, R.; Howard, K.; Johnson, J.K. In Silico Demonstration of Fast Anhydrous Proton Conduction on Graphanol. ACS Appl. Mater. Interfaces 2023, 15, 25873–25883. [Google Scholar] [CrossRef]
- Achar, S.; Bernasconi, L.; Alvarez, J.; Johnson, J.K. Deep-Learning Potentials for Proton Transport in Double-Sided Graphanol. ChemRxiv 2023. [Google Scholar] [CrossRef]
- De, S.; Bartók, A.P.; Csányi, G.; Ceriotti, M. Comparing Molecules and Solids Across Structural and Alchemical Space. Phys. Chem. Chem. Phys. 2016, 18, 13754–13769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caro, M.A. Optimizing Many-Body Atomic Descriptors for Enhanced Computational Performance of Machine Learning Based Interatomic Potentials. Phys. Rev. B 2019, 100, 024112. [Google Scholar] [CrossRef] [Green Version]
- Willatt, M.J.; Musil, F.; Ceriotti, M. Feature Optimization for Atomistic Machine Learning Yields a Data-Driven Construction of the Periodic Table of the Elements. Phys. Chem. Chem. Phys. 2018, 20, 29661–29668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Himanen, L.; Jäger, M.O.J.; Morooka, E.V.; Federici Canova, F.; Ranawat, Y.S.; Gao, D.Z.; Rinke, P.; Foster, A.S. DScribe: Library of Descriptors for Machine Learning in Materials Science. Comput. Phys. Commun. 2020, 247, 106949. [Google Scholar] [CrossRef]
- Paszke, A.; Gross, S.; Massa, F.; Lerer, A.; Bradbury, J.; Chanan, G.; Killeen, T.; Lin, Z.; Gimelshein, N.; Antiga, L.; et al. PyTorch: An Imperative Style, High-Performance Deep Learning Library. In Advances in Neural Information Processing Systems 32; Curran Associates, Inc.: New York, NY, USA, 2019; pp. 8024–8035. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-Learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar] [CrossRef] [Green Version]
- Agarap, A.F. Deep Learning Using Rectified Linear Units (ReLu). arXiv 2018, arXiv:1803.08375. [Google Scholar]
- Ioffe, S.; Szegedy, C. Batch Normalization: Accelerating Deep Network Training by Reducing Internal Covariate Shift. In Proceedings of the International Conference on Machine Learning, PMLR, Lille, France, 6–11 July 2015; pp. 448–456. [Google Scholar] [CrossRef] [Green Version]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Bagusetty, A.; Choudhury, P.; Saidi, W.A.; Derksen, B.; Gatto, E.; Johnson, J.K. Facile Anhydrous Proton Transport on Hydroxyl Functionalized Graphane. Phys. Rev. Lett. 2017, 118, 186101. [Google Scholar] [CrossRef] [Green Version]
- Bagusetty, A.; Johnson, J.K. Unraveling Anhydrous Proton Conduction in Hydroxygraphane. J. Phys. Chem. Lett. 2019, 10, 518–523. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef] [Green Version]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. CP2K: Atomistic Simulations of Condensed Matter Systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula Into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed With a Long-Range Dispersion Correction. J. Comput. Chem 2006, 27, 1787–1799. [Google Scholar] [CrossRef]
- Lin, I.C.; Seitsonen, A.P.; Tavernelli, I.; Rothlisberger, U. Structure and Dynamics of Liquid Water From Ab Initio Molecular Dynamics—Comparison of BLYP, PBE, and revPBE Density Functionals with and without Van Der Waals Corrections. J. Chem. Theory Comput. 2012, 8, 3902–3910. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lippert, G.; Hutter, J.; Parrinello, M. The Gaussian and Augmented-Plane-Wave Density Functional Method for Ab-Initio Molecular Dynamics Simulations. Theor. Chem. Acc. 1999, 103, 124–140. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian Basis Sets for Accurate Calculations on Molecular Systems in Gas and Condensed Phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef] [Green Version]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [Green Version]
- Ceriotti, M.; Bussi, G.; Parrinello, M. Langevin Equation with Colored Noise for Constant-Temperature Molecular Dynamics Simulations. Phys. Rev. Lett. 2009, 102, 020601. [Google Scholar] [CrossRef] [Green Version]
- Rudberg, E.; Rubensson, E.H.; Sałek, P.; Kruchinina, A. Ergo: An Open-Source Program for Linear-Scaling Electronic Structure Calculations. SoftwareX 2018, 7, 107–111. [Google Scholar] [CrossRef]
- Manz, T.A.; Limas, N.G. Introducing DDEC6 Atomic Population Analysis: Part 1. Charge Partitioning Theory and Methodology. RSC Adv. 2016, 6, 47771–47801. [Google Scholar] [CrossRef] [Green Version]
- Limas, N.G.; Manz, T.A. Introducing DDEC6 Atomic Population Analysis: Part 2. Computed Results for a Wide Range of Periodic and Nonperiodic Materials. RSC Adv. 2016, 6, 45727–45747. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | MSE ( Å) | |
|---|---|---|
| Non-weighted SOAP | 0.619 | |
| Weighted SOAP | 0.991 |
| Model | (MSE) on u24C Data | (MSE) on c24C Data | Total DFT Valence Electrons (DeepCDP Valence Electrons) in u24C | Total DFT Valence Electrons (DeepCDP Valence Electrons) in c24C |
|---|---|---|---|---|
| u24C | 0.993 () | 0.989 () | 192.0 (192.1) | 192.0 (193.2) |
| c24C | 0.992 () | 0.994 () | 192.0 (191.8) | 192.0 (192.2) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Achar, S.K.; Bernasconi, L.; Johnson, J.K. Machine Learning Electron Density Prediction Using Weighted Smooth Overlap of Atomic Positions. Nanomaterials 2023, 13, 1853. https://doi.org/10.3390/nano13121853
Achar SK, Bernasconi L, Johnson JK. Machine Learning Electron Density Prediction Using Weighted Smooth Overlap of Atomic Positions. Nanomaterials. 2023; 13(12):1853. https://doi.org/10.3390/nano13121853
Chicago/Turabian StyleAchar, Siddarth K., Leonardo Bernasconi, and J. Karl Johnson. 2023. "Machine Learning Electron Density Prediction Using Weighted Smooth Overlap of Atomic Positions" Nanomaterials 13, no. 12: 1853. https://doi.org/10.3390/nano13121853
APA StyleAchar, S. K., Bernasconi, L., & Johnson, J. K. (2023). Machine Learning Electron Density Prediction Using Weighted Smooth Overlap of Atomic Positions. Nanomaterials, 13(12), 1853. https://doi.org/10.3390/nano13121853