

Self-Healing Iron Oxide Polyelectrolyte Nanocomposites: Influence of Particle Agglomeration and Water on Mechanical Properties

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Characterization
2.3. Syntheses
- Optimization of polymerization conditions:
3. Results and Discussion
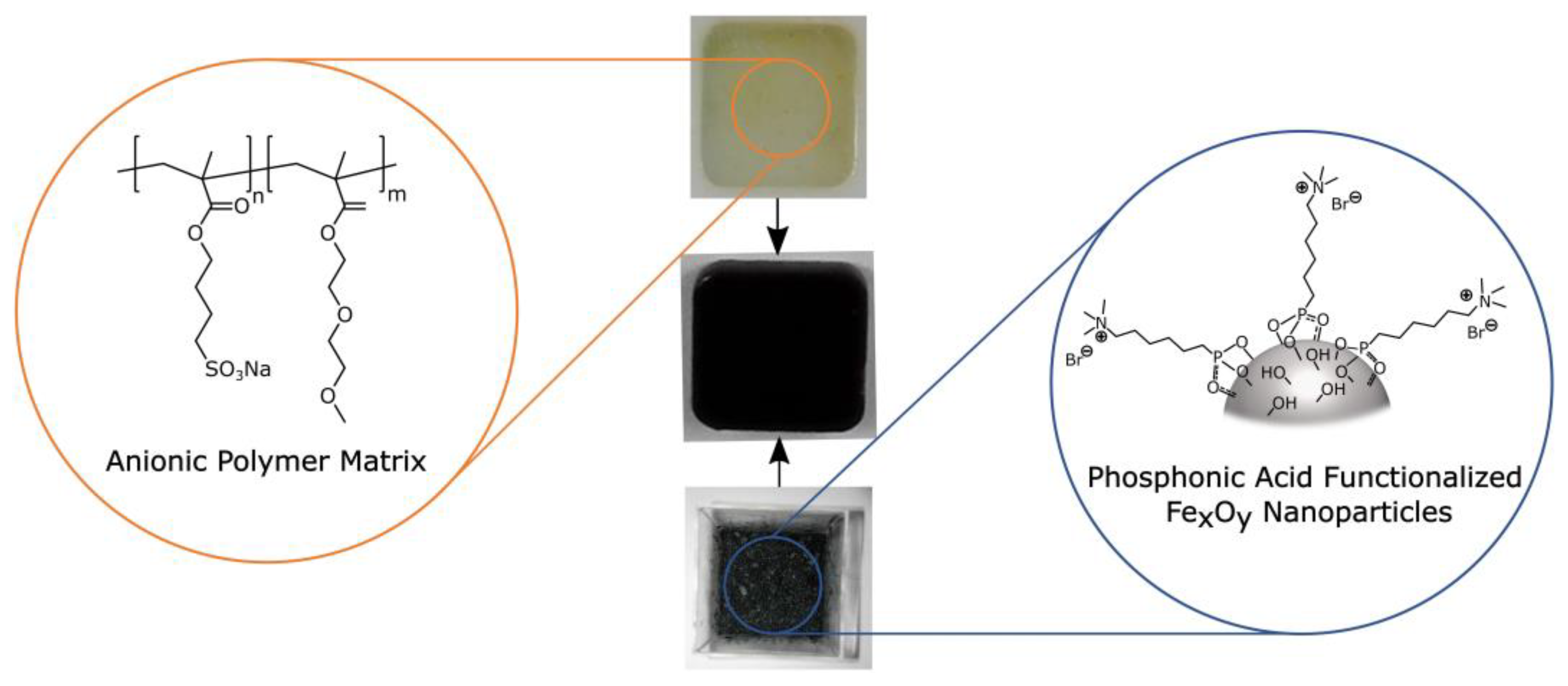
3.1. General Synthesis of the Self-Healing Nanocomposites
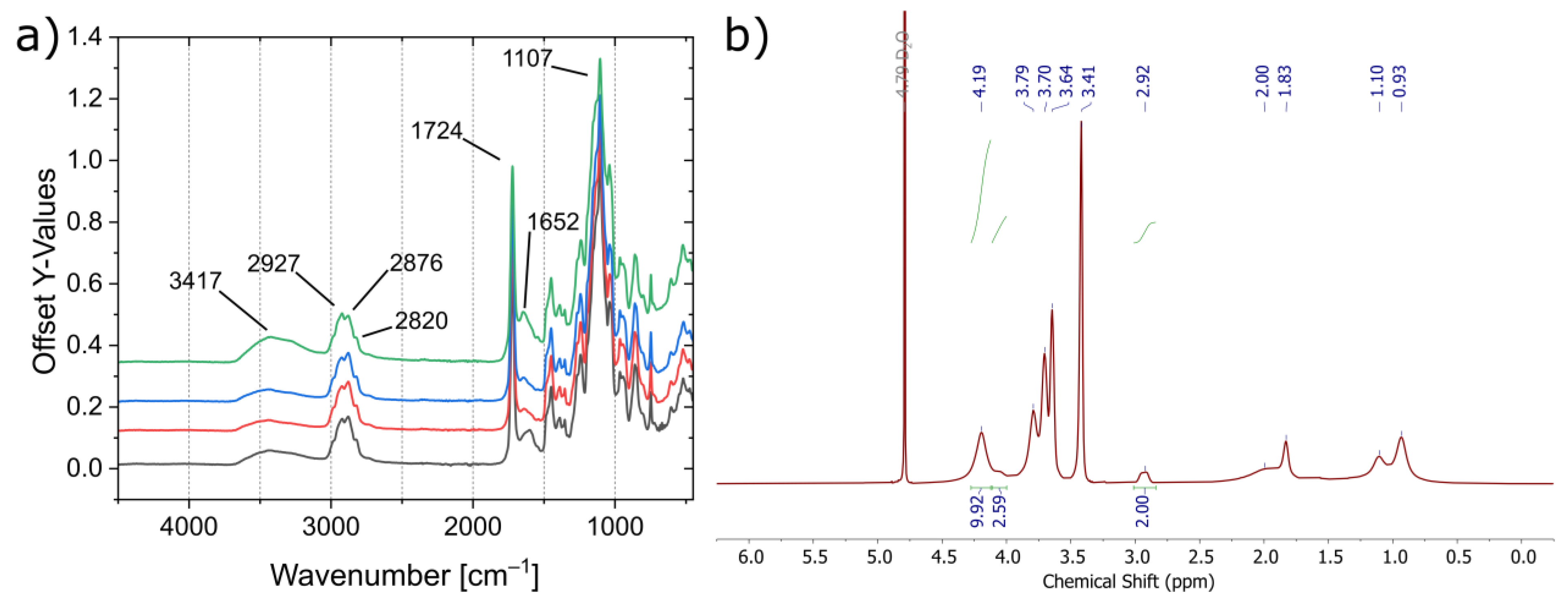
3.2. Polymer Synthesis
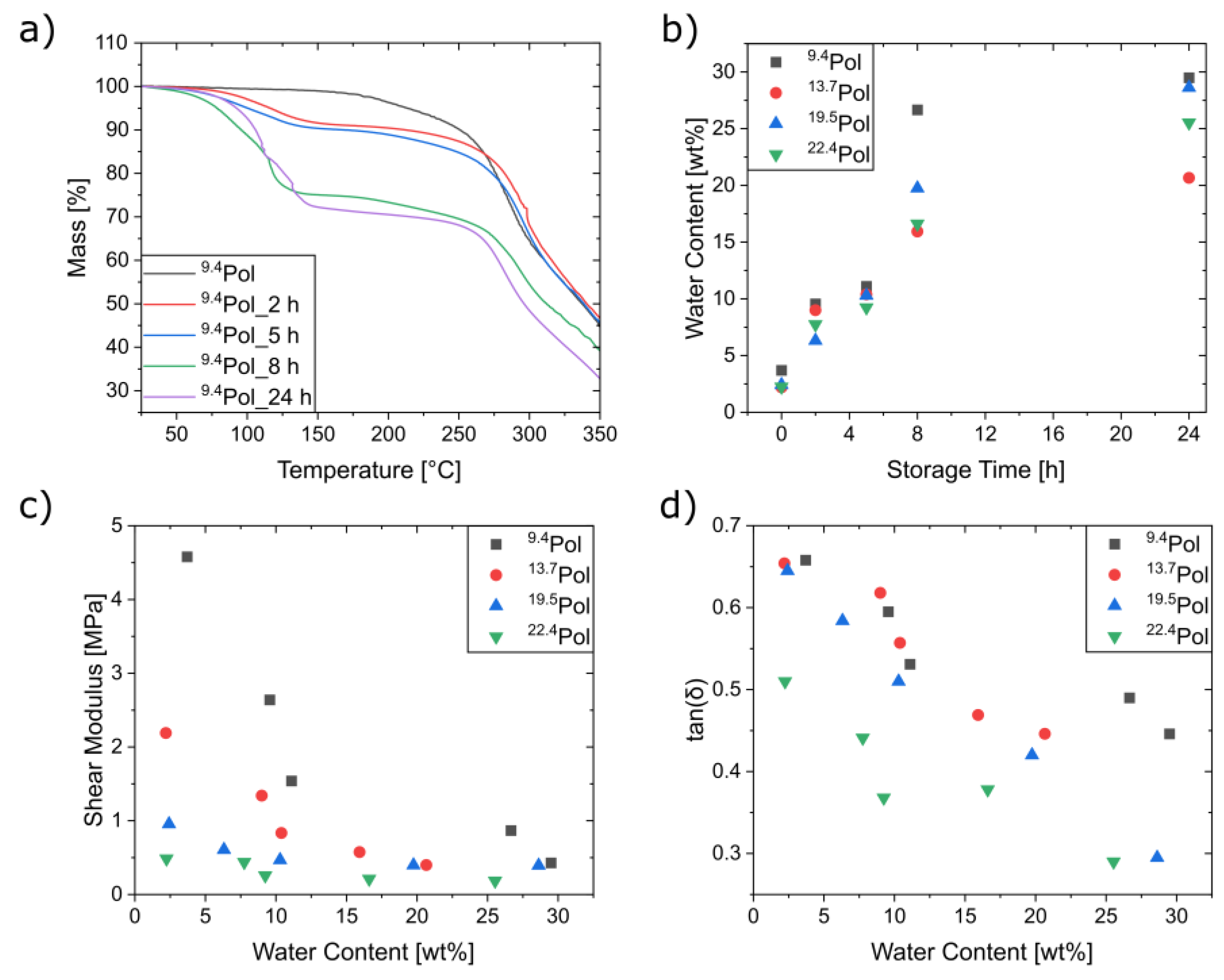
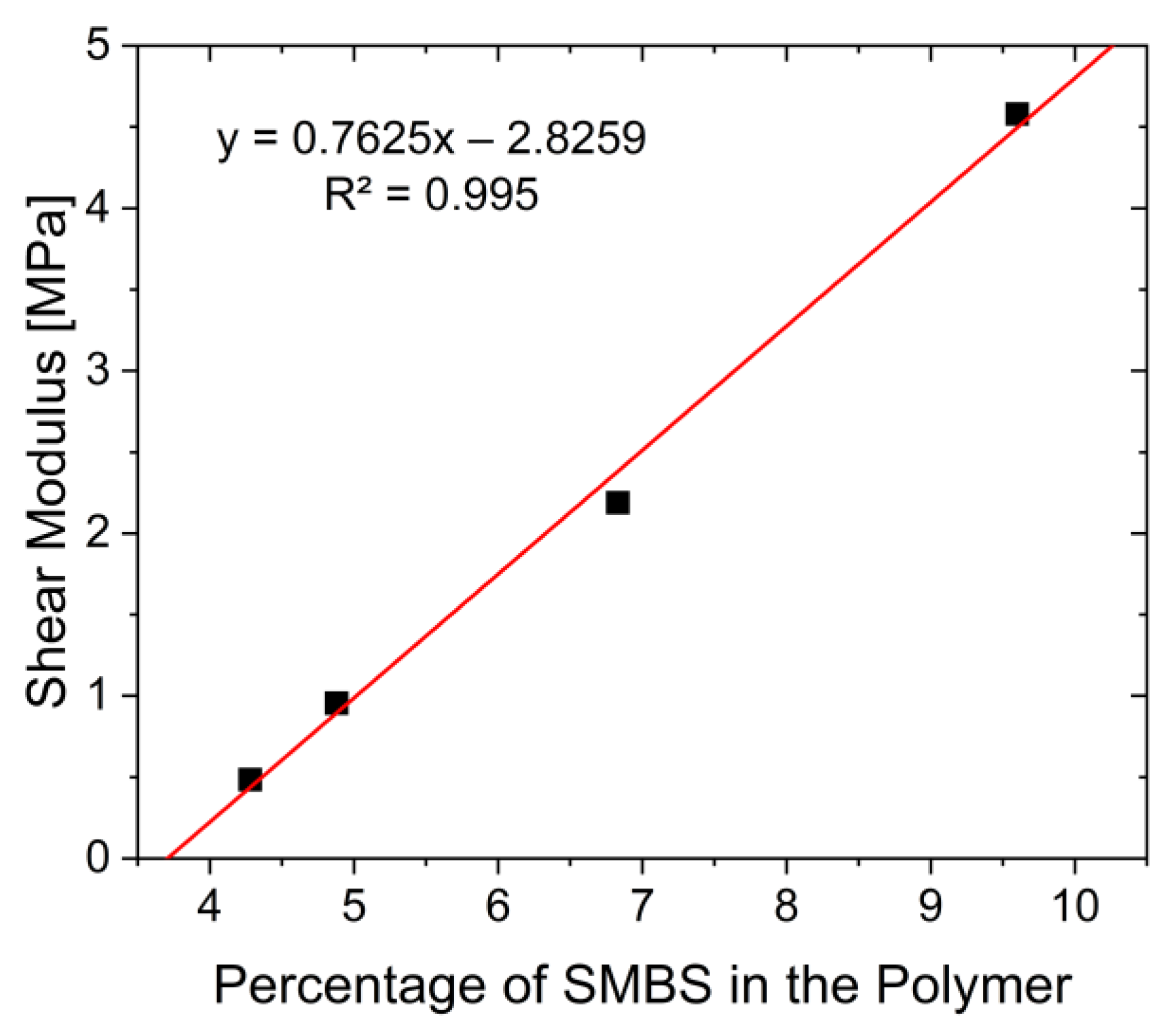
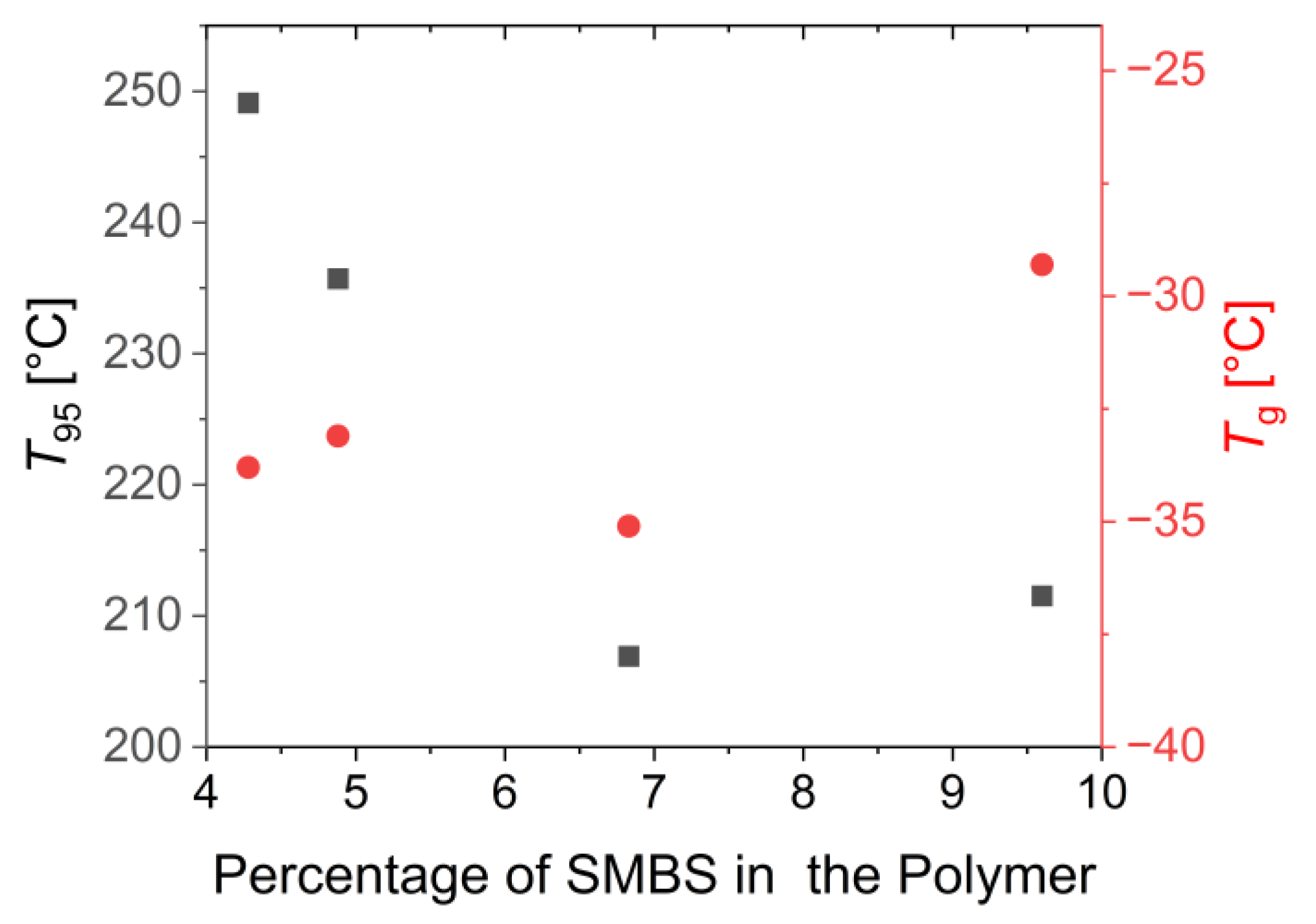
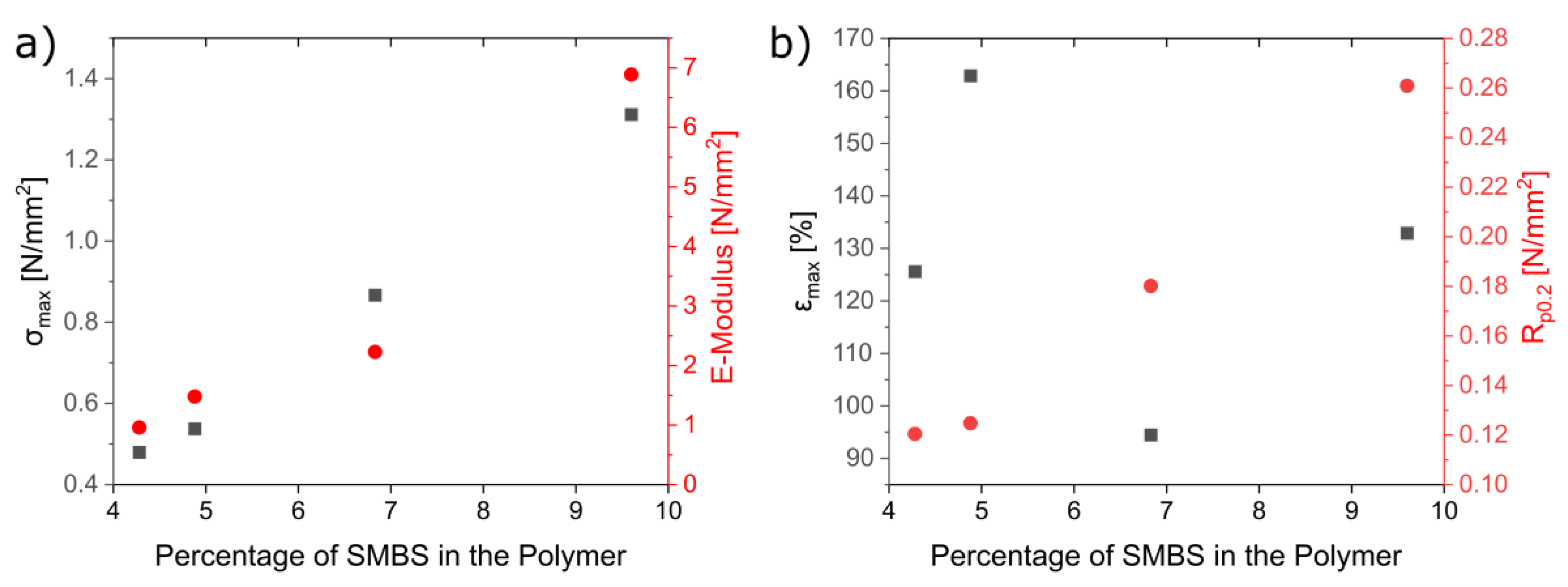
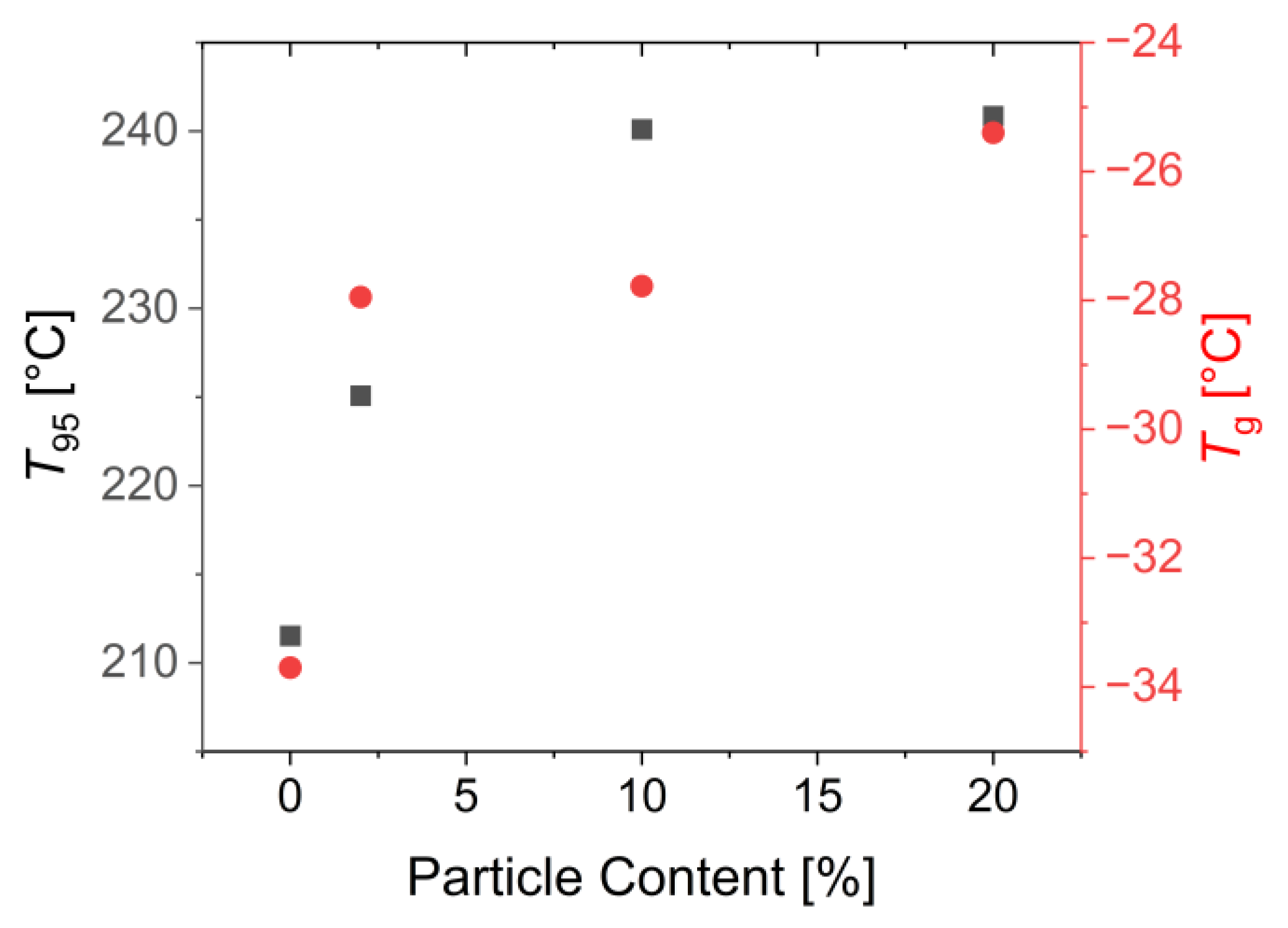
3.3. Characterization of the Mechanical and Thermal Properties of the Polymers
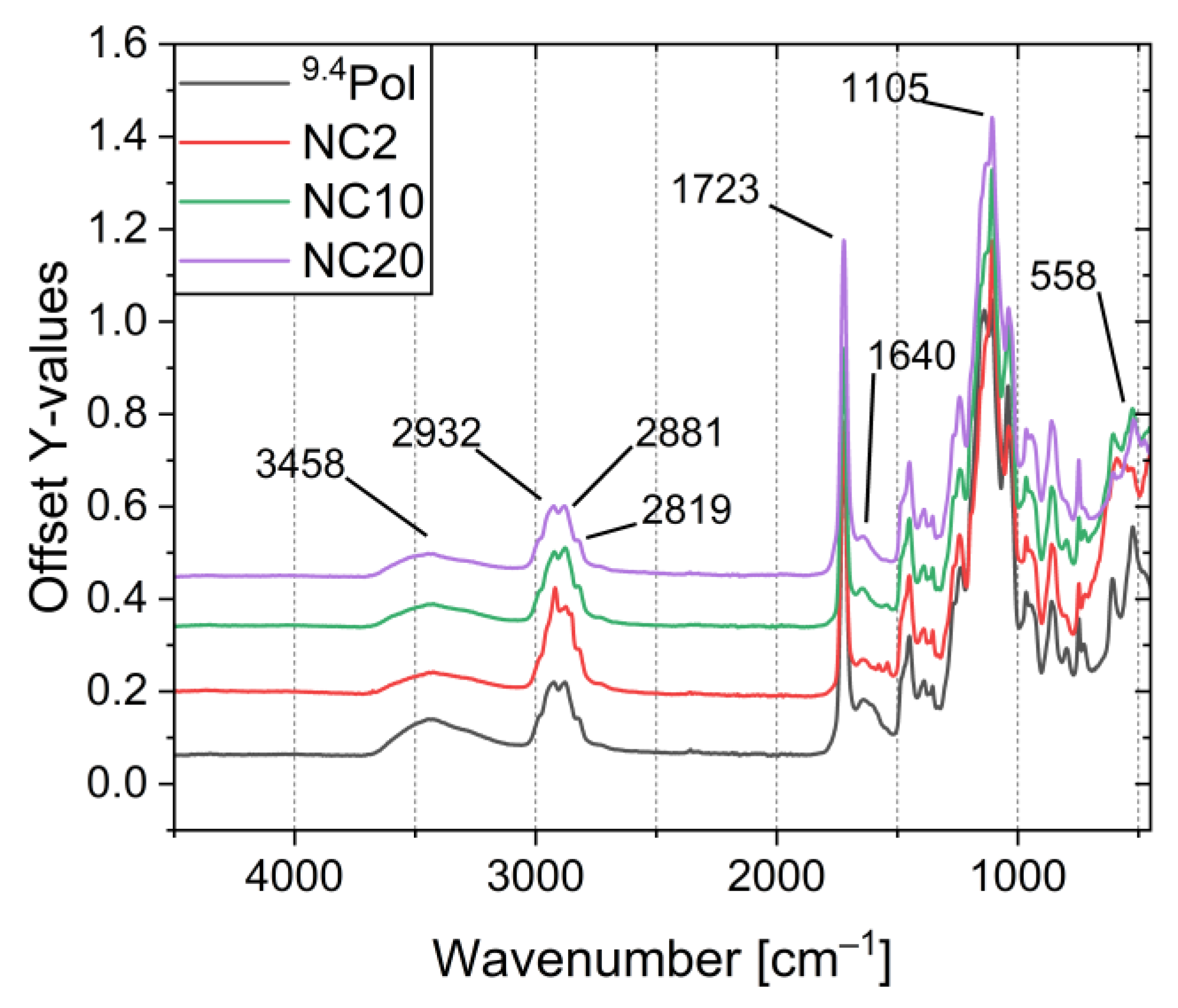
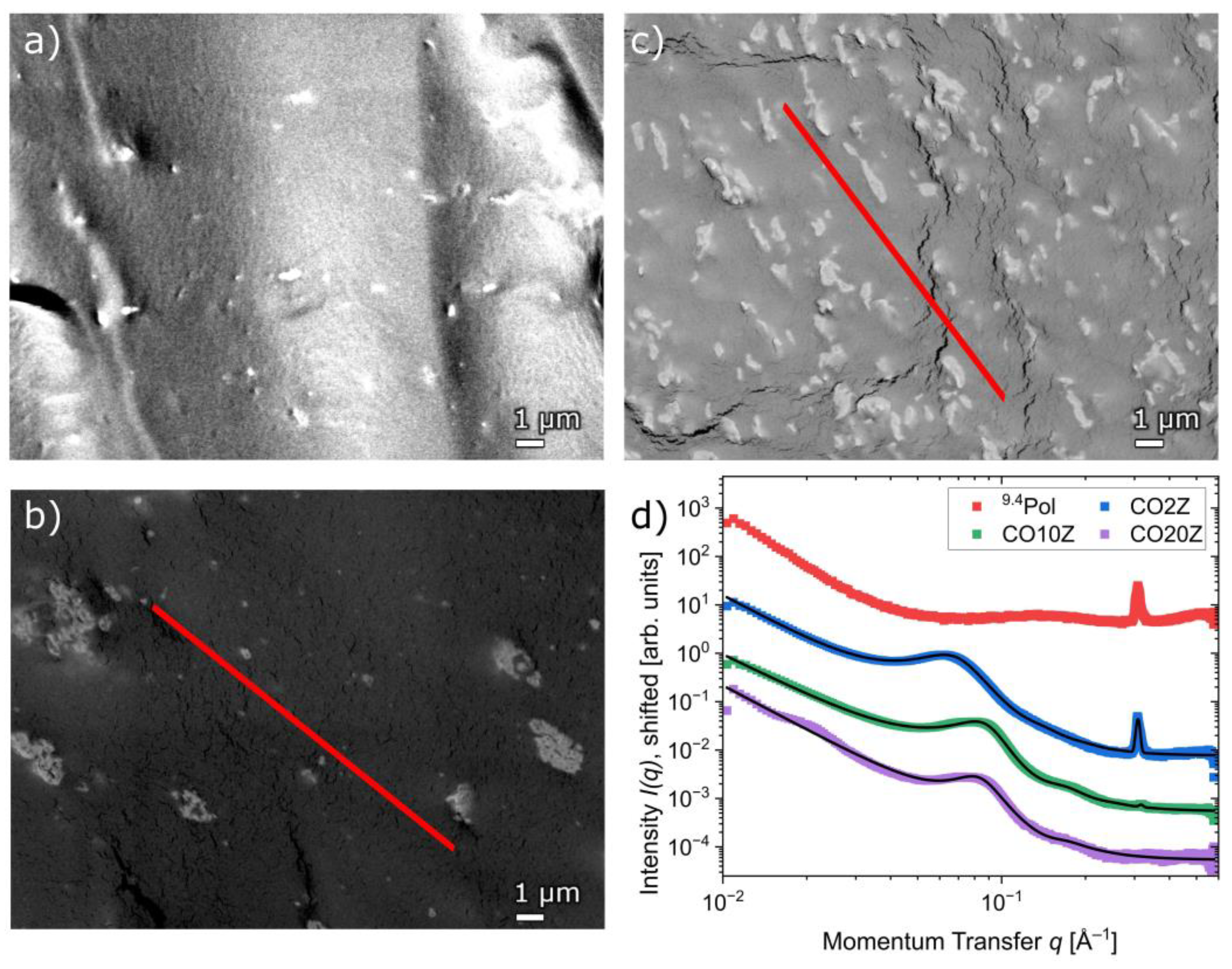
3.4. Composite Synthesis and Characterization
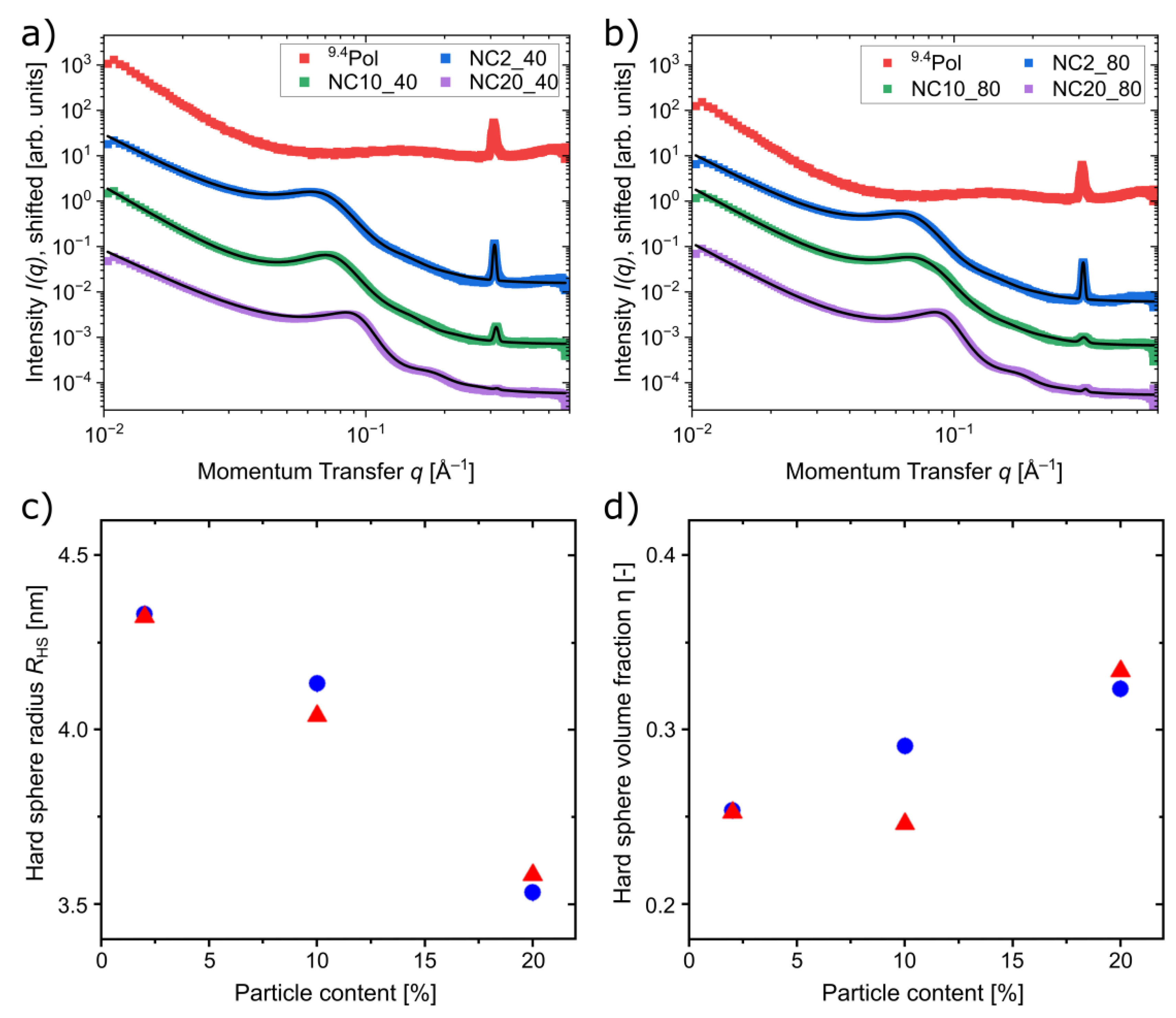
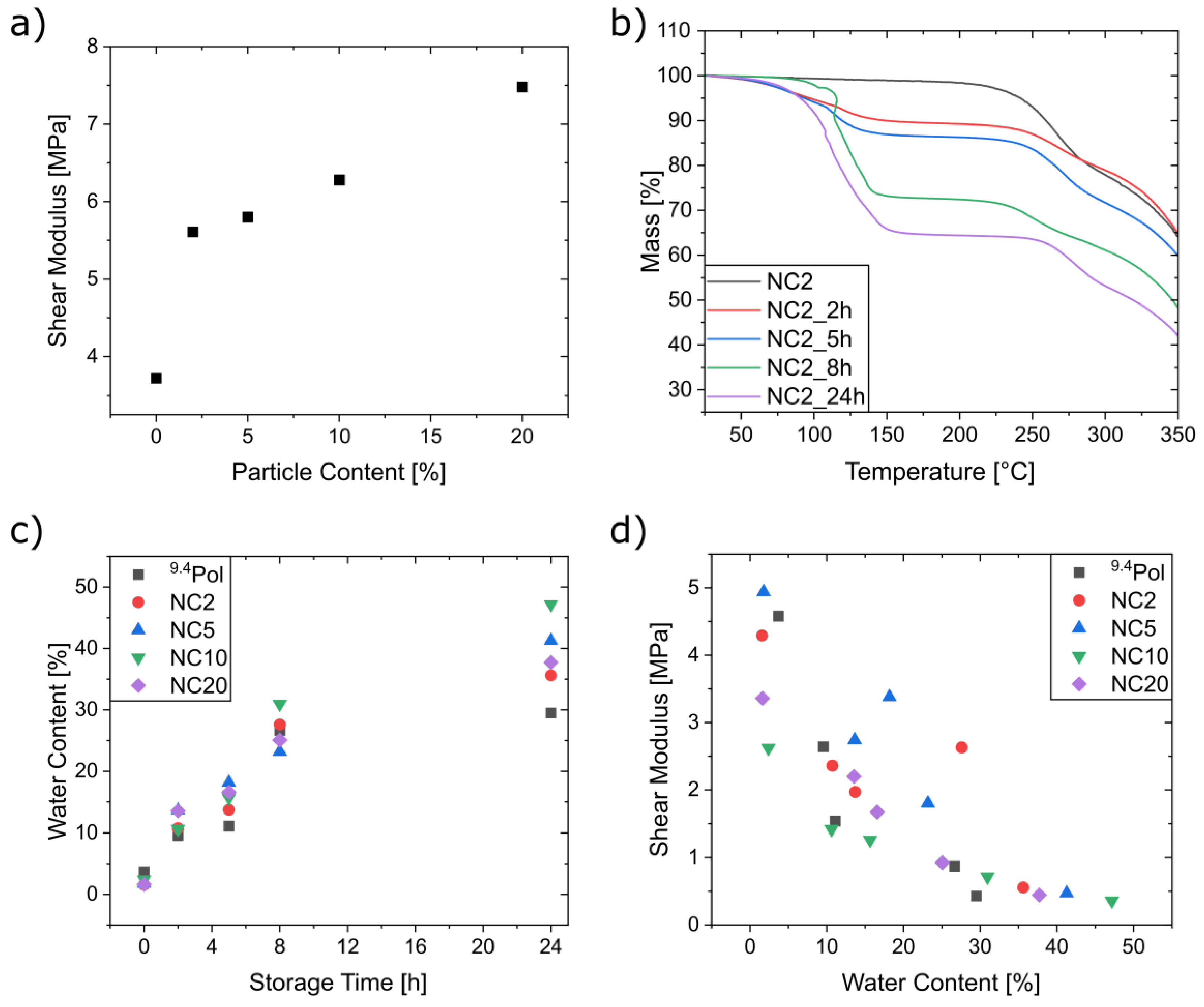
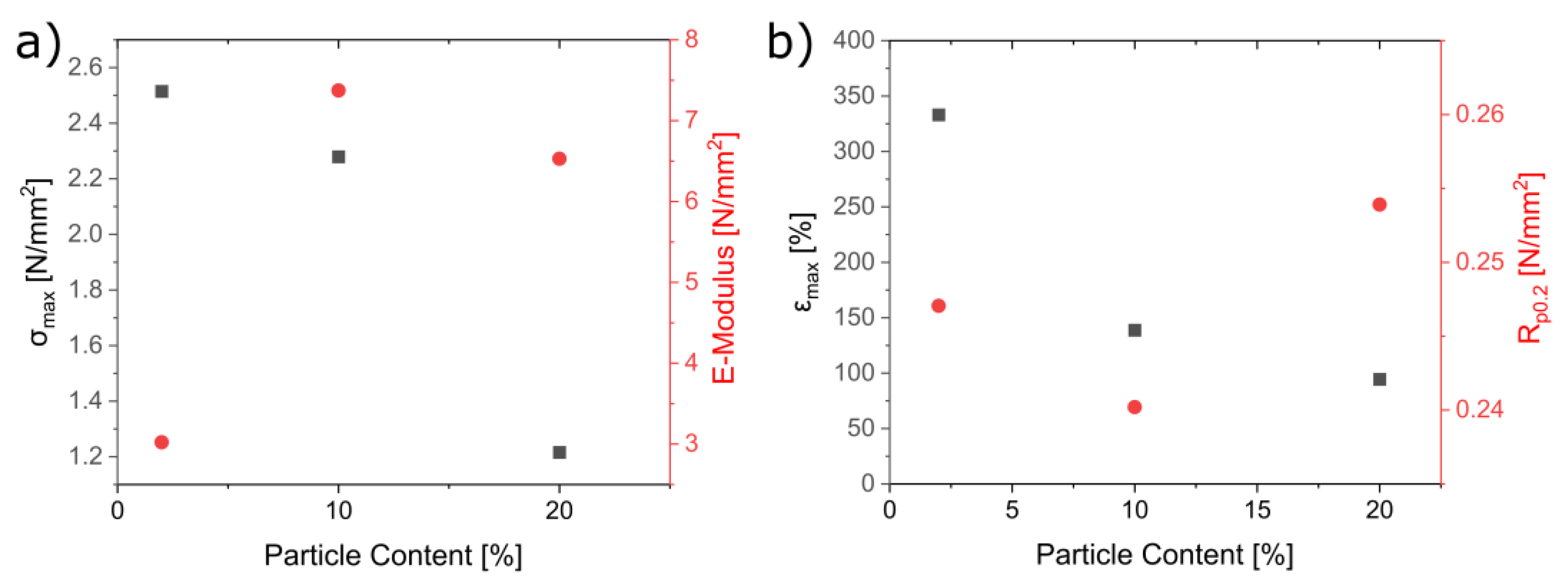
3.5. Mechanical Characterization of the Composites
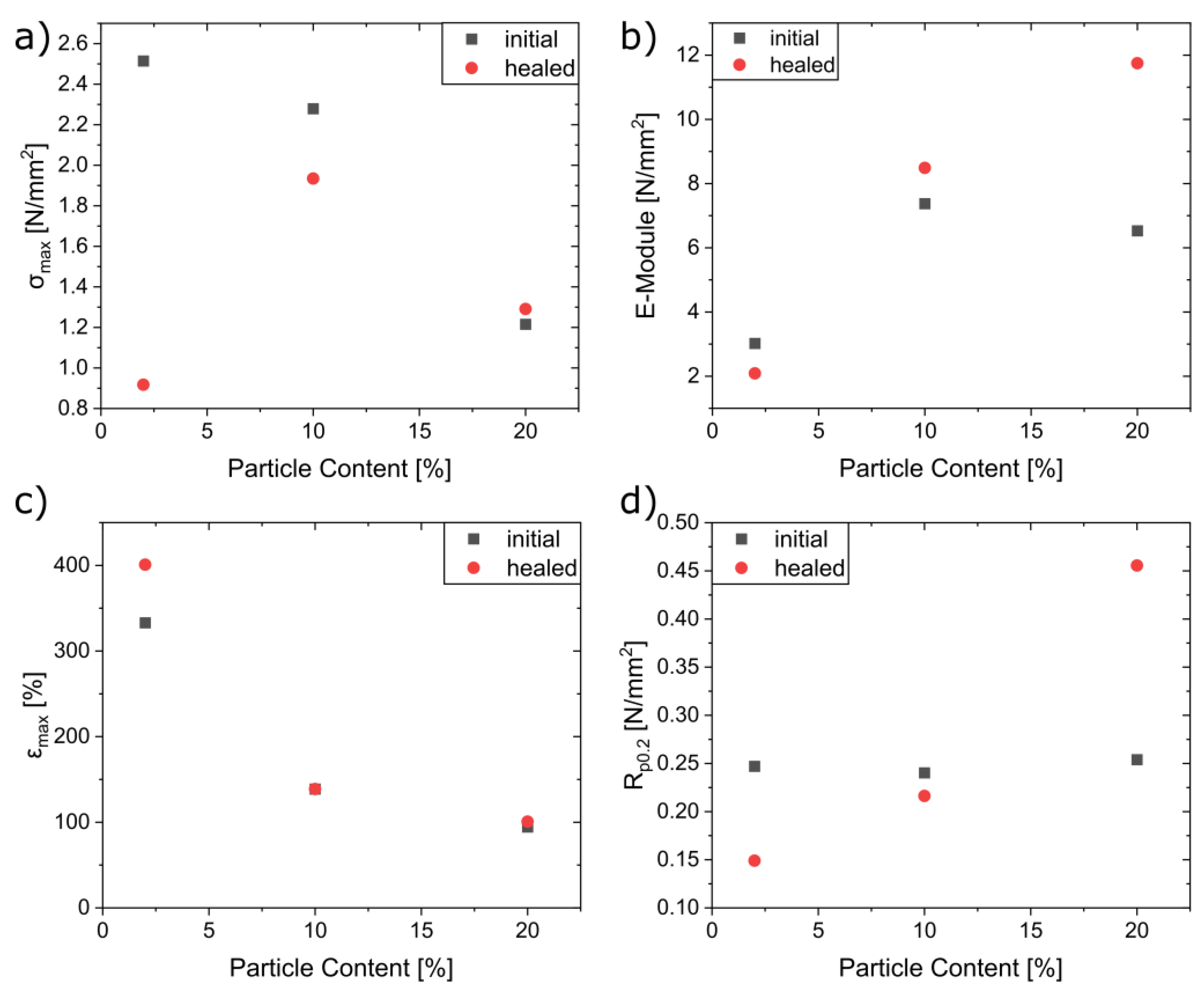
3.6. Self-Healing Experiments
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Thostenson, E.T.; Li, C.; Chou, T.W. Nanocomposites in Context. Compos. Sci. Technol. 2005, 65, 491–516. [Google Scholar] [CrossRef]
- Schaefer, D.W.; Justice, R.S. How Nano Are Nanocomposites? Macromolecules 2007, 40, 8501–8517. [Google Scholar] [CrossRef]
- Harito, C.; Bavykin, D.V.; Yuliarto, B.; Dipojono, H.K.; Walsh, F.C. Polymer Nanocomposites Having a High Filler Content: Synthesis, Structures, Properties, and Applications. Nanoscale 2019, 11, 4653–4682. [Google Scholar] [CrossRef] [PubMed]
- Ates, B.; Koytepe, S.; Ulu, A.; Gurses, C.; Thakur, V.K. Chemistry, Structures, and Advanced Applications of Nanocomposites from Biorenewable Resources. Chem. Rev. 2020, 120, 9304–9362. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.T.; Liu, H.Y.; Saintier, N.; Mai, Y.W. Cyclic Fatigue of Polymer Nanocomposites. Eng. Fail. Anal. 2009, 16, 2635–2645. [Google Scholar] [CrossRef]
- Paladugu, S.R.M.; Sreekanth, P.S.R.; Sahu, S.K.; Naresh, K.; Karthick, S.A.; Venkateshwaran, N.; Ramoni, M.; Mensah, R.A.; Das, O.; Shanmugam, R. A Comprehensive Review of Self-Healing Polymer, Metal, and Ceramic Matrix Composites and Their Modeling Aspects for Aerospace Applications. Materials 2022, 15, 8521. [Google Scholar] [CrossRef]
- Akhan, S.; Oktay, B.; Özdemir, O.K.; Madakbaş, S.; Kayaman Apohan, N. Polyurethane Graphene Nanocomposites with Self-Healing Properties by Azide-Alkyne Click Reaction. Mater. Chem. Phys. 2020, 254, 123315. [Google Scholar] [CrossRef]
- Tao, H.Q.; Yue, D.W.; Li, C.H. A Fast Self-Healing Magnetic Nanocomposite for Magnetic Actuators. Macromol. Mater. Eng. 2022, 307, 2100649. [Google Scholar] [CrossRef]
- Osada, T.; Kamoda, K.; Mitome, M.; Hara, T.; Abe, T.; Tamagawa, Y.; Nakao, W.; Ohmura, T. A Novel Design Approach for Self-Crack-Healing Structural Ceramics with 3D Networks of Healing Activator. Sci. Rep. 2017, 7, 17853. [Google Scholar] [CrossRef]
- Grabowski, B.; Tasan, C.C. Self-Healing Metals. Adv. Polym. Sci. 2016, 273, 387–407. [Google Scholar]
- Thakur, V.K.; Kessler, M.R. Self-Healing Polymer Nanocomposite Materials: A Review. Polymer 2015, 69, 369–383. [Google Scholar] [CrossRef]
- Idumah, C.I. Recent Advancements in Self-Healing Polymers, Polymer Blends, and Nanocomposites. Polym. Polym. Compos. 2021, 29, 246–258. [Google Scholar] [CrossRef]
- Liu, Y.L.; Chuo, T.W. Self-Healing Polymers Based on Thermally Reversible Diels-Alder Chemistry. Polym. Chem. 2013, 4, 2194–2205. [Google Scholar] [CrossRef]
- Kuhl, N.; Bode, S.; Hager, M.D.; Schubert, U.S. Self-Healing Polymers Based on Reversible Covalent Bonds. Adv. Polym. Sci. 2015, 273, 1–58. [Google Scholar]
- Engel, T.; Kickelbick, G. Thermoreversible Reactions on Inorganic Nanoparticle Surfaces: Diels–Alder Reactions on Sterically Crowded Surfaces. Chem. Mater. 2013, 25, 149–157. [Google Scholar] [CrossRef]
- Engel, T.; Kickelbick, G. Self-Healing Nanocomposites from Silica-Polymer Core-Shell Nanoparticles. Polym. Int. 2014, 63, 915–923. [Google Scholar] [CrossRef]
- He, S.; Kickelbick, G. Reversible Diels–Alder Reactions with a Fluorescent Dye on the Surface of Magnetite Nanoparticles. Molecules 2021, 26, 877. [Google Scholar] [CrossRef] [PubMed]
- Pratama, P.A.; Sharifi, M.; Peterson, A.M.; Palmese, G.R. Room Temperature Self-Healing Thermoset Based on the Diels-Alder Reaction. ACS Appl. Mater. Interfaces 2013, 5, 12425–12431. [Google Scholar] [CrossRef]
- Turkenburg, D.H.; Fischer, H.R. Diels-Alder Based, Thermo-Reversible Cross-Linked Epoxies for Use in Self-Healing Composites. Polymer 2015, 79, 187–194. [Google Scholar] [CrossRef]
- Park, J.S.; Darlington, T.; Starr, A.F.; Takahashi, K.; Riendeau, J.; Thomas Hahn, H.T. Multiple Healing Effect of Thermally Activated Self-Healing Composites Based on Diels-Alder Reaction. Compos. Sci. Technol. 2010, 70, 2154–2159. [Google Scholar] [CrossRef]
- Lee, Y.H.; Zhuang, Y.N.; Wang, H.T.; Wei, M.F.; Ko, W.C.; Chang, W.J.; Way, T.F.; Rwei, S.P. Fabrication of Self-Healable Magnetic Nanocomposites via Diels-Alder Click Chemistry. Appl. Sci. 2019, 9, 506. [Google Scholar] [CrossRef]
- Behera, P.K.; Mondal, P.; Singha, N.K. Self-Healable and Ultrahydrophobic Polyurethane-POSS Hybrids by Diels-Alder “Click” Reaction: A New Class of Coating Material. Macromolecules 2018, 51, 4770–4781. [Google Scholar] [CrossRef]
- Wu, S.; Li, J.; Zhang, G.; Yao, Y.; Li, G.; Sun, R.; Wong, C. Ultrafast Self-Healing Nanocomposites via Infrared Laser and Their Application in Flexible Electronics. ACS Appl. Mater. Interfaces 2017, 9, 3040–3049. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, S.; Kickelbick, G. Self-Healing Polymer Nanocomposites Based on Diels-Alder-Reactions with Silica Nanoparticles: The Role of the Polymer Matrix. Polymer 2015, 69, 357–368. [Google Scholar] [CrossRef]
- Schäfer, S.; Kickelbick, G. Diels–Alder Reactions on Surface-Modified Magnetite/Maghemite Nanoparticles: Application in Self-Healing Nanocomposites. ACS Appl. Nano Mater. 2018, 1, 2640–2652. [Google Scholar] [CrossRef]
- Shao, C.; Chang, H.; Wang, M.; Xu, F.; Yang, J. High-Strength, Tough, and Self-Healing Nanocomposite Physical Hydrogels Based on the Synergistic Effects of Dynamic Hydrogen Bond and Dual Coordination Bonds. ACS Appl. Mater. Interfaces 2017, 9, 28305–28318. [Google Scholar] [CrossRef]
- Lu, C.H.; Yeh, Y.C. Fabrication of Multiresponsive Magnetic Nanocomposite Double-Network Hydrogels for Controlled Release Applications. Small 2021, 17, 2105997. [Google Scholar] [CrossRef]
- Heidarian, P.; Kouzani, A.Z. A Self-Healing Nanocomposite Double Network Bacterial Nanocellulose/Gelatin Hydrogel for Three Dimensional Printing. Carbohydr. Polym. 2023, 313, 120879. [Google Scholar] [CrossRef]
- Tsai, T.-Y.; Shen, K.-H.; Chang, C.-W.; Jovanska, L.; Wang, R.; Yeh, Y.-C. In Situ Formation of Nanocomposite Double-Network Hydrogels with Shear-Thinning and Self-Healing Properties. Biomater. Sci. 2021, 9, 985–999. [Google Scholar] [CrossRef]
- Schäfer, S.; Kickelbick, G. Double Reversible Networks: Improvement of Self-Healing in Hybrid Materials via Combination of Diels–Alder Cross-Linking and Hydrogen Bonds. Macromolecules 2018, 51, 6099–6110. [Google Scholar] [CrossRef]
- Kalista, S.J.; Ward, T.C.; Oyetunji, Z. Self-Healing of Poly(Ethylene-Co-Methacrylic Acid) Copolymers Following Projectile Puncture. Mech. Adv. Mater. Struct. 2007, 14, 391–397. [Google Scholar] [CrossRef]
- Varley, R.J.; van der Zwaag, S. Towards an Understanding of Thermally Activated Self-Healing of an Ionomer System During Ballistic Penetration. Acta Mater. 2008, 56, 5737–5750. [Google Scholar] [CrossRef]
- Wang, X.; Wang, Y.; Bi, S.; Wang, Y.; Chen, X.; Qiu, L.; Sun, J. Optically Transparent Antibacterial Films Capable of Healing Multiple Scratches. Adv. Funct. Mater. 2014, 24, 403–411. [Google Scholar] [CrossRef]
- Reisch, A.; Roger, E.; Phoeung, T.; Antheaume, C.; Orthlieb, C.; Boulmedais, F.; Lavalle, P.; Schlenoff, J.B.; Frisch, B.; Schaaf, P. On the Benefits of Rubbing Salt in the Cut: Self-Healing of Saloplastic PAA/PAH Compact Polyelectrolyte Complexes. Adv. Mater. 2014, 26, 2547–2551. [Google Scholar] [CrossRef]
- Cuthbert, T.J.; Jadischke, J.J.; de Bruyn, J.R.; Ragogna, P.J.; Gillies, E.R. Self-Healing Polyphosphonium Ionic Networks. Macromolecules 2017, 50, 5253–5260. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, C.; Zhu, G.; Zacharia, N.S. Self-Healing of Bulk Polyelectrolyte Complex Material as a Function of PH and Salt. ACS Appl. Mater. Interfaces 2016, 8, 26258–26265. [Google Scholar] [CrossRef]
- Zhang, W.; Wu, B.; Sun, S.; Wu, P. Skin-like Mechanoresponsive Self-Healing Ionic Elastomer from Supramolecular Zwitterionic Network. Nat. Commun. 2021, 12, 4082. [Google Scholar] [CrossRef]
- Ihsan, A.B.; Sun, T.L.; Kurokawa, T.; Karobi, S.N.; Nakajima, T.; Nonoyama, T.; Roy, C.K.; Luo, F.; Gong, J.P. Self-Healing Behaviors of Tough Polyampholyte Hydrogels. Macromolecules 2016, 49, 4245–4252. [Google Scholar] [CrossRef]
- Zheng, J.; Xiao, P.; Liu, W.; Zhang, J.; Huang, Y.; Chen, T. Mechanical Robust and Self-Healable Supramolecular Hydrogel. Macromol. Rapid Commun. 2016, 37, 265–270. [Google Scholar] [CrossRef]
- Odent, J.; Raquez, J.-M.; Dubois, P.; Giannelis, E.P. Ultra-Stretchable Ionic Nanocomposites: From Dynamic Bonding to Multi-Responsive Behavior. J. Mater. Chem. A 2017, 5, 13357–13363. [Google Scholar] [CrossRef]
- Mugemana, C.; Moghimikheirabadi, A.; Arl, D.; Addiego, F.; Schmidt, D.F.; Kröger, M.; Karatrantos, A.V. Ionic Poly(Dimethylsiloxane)–Silica Nanocomposites: Dispersion and Self-Healing. MRS Bull. 2022, 47, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Mohapatra, S.; Pramanik, P. Synthesis and Stability of Functionalized Iron Oxide Nanoparticles Using Organophosphorus Coupling Agents. Colloids Surfaces A Physicochem. Eng. Asp. 2009, 339, 35–42. [Google Scholar] [CrossRef]
- Boyer, C.; Whittaker, M.R.; Bulmus, V.; Liu, J.; Davis, T.P. The Design and Utility of Polymer-Stabilized Iron-Oxide Nanoparticles for Nanomedicine Applications. NPG Asia Mater. 2010, 2, 23–30. [Google Scholar] [CrossRef]
- Queffélec, C.; Petit, M.; Janvier, P.; Knight, D.A.; Bujoli, B. Surface Modification Using Phosphonic Acids and Esters. Chem. Rev. 2012, 112, 3777–3807. [Google Scholar] [CrossRef]
- Raza, M.; Bachinger, A.; Zahn, N.; Kickelbick, G. Interaction and UV-Stability of Various Organic Capping Agents on the Surface of Anatase Nanoparticles. Materials 2014, 7, 2890–2912. [Google Scholar] [CrossRef]
- Brodard-Severac, F.; Guerrero, G.; Maquet, J.; Florian, P.; Gervais, C.; Mutin, P.H. High-Field 17O MAS NMR Investigation of Phosphonic Acid Monolayers on Titania. Chem. Mater. 2008, 20, 5191–5196. [Google Scholar] [CrossRef]
- Dydio, P.; Detz, R.J.; Reek, J.N.H. Precise Supramolecular Control of Selectivity in the Rh-Catalyzed Hydroformylation of Terminal and Internal Alkenes. J. Am. Chem. Soc. 2013, 135, 10817–10828. [Google Scholar] [CrossRef]
- Mancini, G.; Bouda, M.; Gamrat, J.M.; Tomsho, J.W. Synthesis and Antimicrobial Evaluation of Î-Borono Phosphonate Compounds in Escherichia Coli and Mycobacterium Smegmatis. ACS Omega 2019, 4, 14551–14559. [Google Scholar] [CrossRef]
- Wang, Y.M.; Pan, M.; Liang, X.Y.; Li, B.J.; Zhang, S. Electromagnetic Wave Absorption Coating Material with Self-Healing Properties. Macromol. Rapid Commun. 2017, 38, 1700447. [Google Scholar] [CrossRef]
- Soroka, M.; Goldeman, W. Process for Preparation of Ureidoalkylphosphonic. Polish Patent PL201311B1, 31 March 2009. [Google Scholar]
- Heinrich, C.; Niedner, L.; Oberhausen, B.; Kickelbick, G. Surface-Charged Zirconia Nanoparticles Prepared by Organophosphorus Surface Functionalization with Ammonium or Sulfonate Groups. Langmuir 2019, 35, 11369–11379. [Google Scholar] [CrossRef]
- Guo, Z.; Lei, K.; Li, Y.; Ng, H.W.; Prikhodko, S.; Hahn, H.T. Fabrication and Characterization of Iron Oxide Nanoparticles Reinforced Vinyl-Ester Resin Nanocomposites. Compos. Sci. Technol. 2008, 68, 1513–1520. [Google Scholar] [CrossRef]
- Ali, S.; Khan, S.A.; Yamani, Z.H.; Qamar, M.T.; Morsy, M.A.; Sarfraz, S. Shape- and Size-Controlled Superparamagnetic Iron Oxide Nanoparticles Using Various Reducing Agents and Their Relaxometric Properties by Xigo Acorn Area. Appl. Nanosci. 2019, 9, 479–489. [Google Scholar] [CrossRef]
- Torres-Lugo, M.; Rinaldi, C. Thermal Potentiation of Chemotherapy by Magnetic Nanoparticles. Nanomedicine 2013, 8, 1689–1707. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, K.; Ono, K.; Suzuki, H.; Sawada, M.; Moriya, M.; Sakamoto, W.; Yogo, T. High-Frequency, Magnetic-Field-Responsive Drug Release from Magnetic Nanoparticle/Organic Hybrid Based on Hyperthermic Effect. ACS Appl. Mater. Interfaces 2010, 2, 1903–1911. [Google Scholar] [CrossRef] [PubMed]
- Lucia, O.; Maussion, P.; Dede, E.J.; Burdio, J.M. Induction Heating Technology and Its Applications: Past Developments, Current Technology, and Future Challenges. IEEE Trans. Ind. Electron. 2014, 61, 2509–2520. [Google Scholar] [CrossRef]
- Xiang, Z.; Ducharne, B.; Della Schiava, N.; Capsal, J.F.; Cottinet, P.J.; Coativy, G.; Lermusiaux, P.; Le, M.Q. Induction Heating-Based Low-Frequency Alternating Magnetic Field: High Potential of Ferromagnetic Composites for Medical Applications. Mater. Des. 2019, 174, 107804. [Google Scholar] [CrossRef]
- Xiang, Z.; Jakkpat, K.I.; Ducharne, B.; Capsal, J.F.; Mogniotte, J.F.; Lermusiaux, P.; Cottinet, P.J.; Schiava, N.D.; Le, M.Q. Enhancing the Low-Frequency Induction Heating Effect of Magnetic Composites for Medical Applications. Polymers 2020, 12, 386. [Google Scholar] [CrossRef]
- Corten, C.C.; Urban, M.W. Repairing Polymers Using Oscillating Magnetic Field. Adv. Mater. 2009, 21, 5011–5015. [Google Scholar] [CrossRef]
- Hohlbein, N.; Shaaban, A.; Schmidt, A.M. Remote-Controlled Activation of Self-Healing Behavior in Magneto-Responsive Ionomeric Composites. Polymer 2015, 69, 301–309. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Wen, Y.; Zhong, Q.; Zhao, Y. Self-Healable Magnetic Structural Color Hydrogels. ACS Appl. Mater. Interfaces 2020, 12, 7486–7493. [Google Scholar] [CrossRef]
- Oberhausen, B.; Kickelbick, G. Induction Heating Induced Self-Healing of Nanocomposites Based on Surface-Functionalized Cationic Iron Oxide Particles and Polyelectrolytes. Nanoscale Adv. 2021, 3, 5589–5604. [Google Scholar] [CrossRef] [PubMed]
- Neuhaus, S.; Seibert, H.; Diebels, S. Investigation of the Damage Behavior of Polyurethane in Stress Relaxation Experiments and Estimation of the Stress-at-Break σb with a Failure Envelope. In Engineering Design Applications II Structures, Materials and Processes; Öchsner, A., Altenbach, H., Eds.; Springer Nature Switzerland: Basel, Switzerland, 2020; pp. 1–15. [Google Scholar]
- Karlsson, J.O.; Gatenholm, P. Solid-Supported Wettable Hydrogels Prepared by Ozone Induced Grafting. Polymer 1996, 37, 4251–4256. [Google Scholar] [CrossRef]
- Kittle, J.; Levin, J.; Levin, N. Water Content of Polyelectrolyte Multilayer Films Measured by Quartz Crystal Microbalance and Deuterium Oxide Exchange. Sensors 2021, 21, 771. [Google Scholar] [CrossRef] [PubMed]
- An, H.; Habib, T.; Shah, S.; Gao, H.; Patel, A.; Echols, I.; Zhao, X.; Radovic, M.; Green, M.J.; Lutkenhaus, J.L. Water Sorption in MXene/Polyelectrolyte Multilayers for Ultrafast Humidity Sensing. ACS Appl. Nano Mater. 2019, 2, 948–955. [Google Scholar] [CrossRef]
- Ostendorf, A.; Schönhoff, M.; Cramer, C. Ionic Conductivity of Solid Polyelectrolyte Complexes with Varying Water Content: Application of the Dynamic Structure Model. Phys. Chem. Chem. Phys. 2019, 21, 7321–7329. [Google Scholar] [CrossRef]
- Liu, T.; An, Q.; Zhao, Q.; Wu, J.; Song, Y.; Zhu, B.; Gao, C. Synergistic Strenghening of Polyelectrolyte Complex Membranes by Functionalized Carbon Nanotubes and Metal Ions. Sci. Rep. 2015, 5, 7782. [Google Scholar] [CrossRef]
- Hariri, H.H.; Lehaf, A.M.; Schlenoff, J.B. Mechanical Properties of Osmotically Stressed Polyelectrolyte Complexes and Multilayers: Water as a Plasticizer. Macromolecules 2012, 45, 9364–9372. [Google Scholar] [CrossRef]
- Ollivier-Lamarque, L.; Livi, S.; Uchimoto, T.; Mary, N. Water Uptake in Epoxy Ionic Liquid Free Film Polymer by Gravimetric Analysis and Comparison with Nondestructive Dielectric Analysis. Nanomaterials 2022, 12, 651. [Google Scholar] [CrossRef]
- Rakowska, J.; Węgrzyn, M.; Rudnik, E. Impact of Ionic Liquids on Absorption Behaviour of Natural Fibers/Biopolyethylene Biocomposites. Sci. Rep. 2021, 11, 20483. [Google Scholar] [CrossRef]
- Wu, G.; Lin, S.J.; Hsu, I.C.; Su, J.Y.; Chen, D.W. Study of High Performance Sulfonated Polyether Ether Ketone Composite Electrolyte Membranes. Polymers 2019, 11, 1177. [Google Scholar] [CrossRef]
- Osman, M.A.; Atallah, A. Effect of the Particle Size on the Viscoelastic Properties of Filled Polyethylene. Polymer 2006, 47, 2357–2368. [Google Scholar] [CrossRef]
- Dabbaghi, M.; Namjoshi, S.; Panchal, B.; Grice, J.E.; Prakash, S.; Roberts, M.S.; Mohammed, Y. Viscoelastic and Deformation Characteristics of Structurally Different Commercial Topical Systems. Pharmaceutics 2021, 13, 1351. [Google Scholar] [CrossRef] [PubMed]
- Capponi, S.; Alvarez, F.; Račko, D. Free Volume in a PVME Polymer-Water Solution. Macromolecules 2020, 53, 4770–4782. [Google Scholar] [CrossRef]
- Johnson, J.A.; Jones, D.W. The Mechanical Properties of PMMA and Its Copolymers with Ethyl Methacrylate and Butyl Methacrylate. J. Mater. Sci. 1994, 29, 870–876. [Google Scholar] [CrossRef]
- Liu, G.; Zhang, X.; Li, X.; Chen, H.; Walton, K.; Wang, D. Correlation of Miscibility and Mechanical Properties of Polypropylene/Olefin Block Copolymers: Effect of Chain Composition. J. Appl. Polym. Sci. 2012, 125, 666–675. [Google Scholar] [CrossRef]
- Chen, R.S.; Mohd Amran, N.A.; Ahmad, S. Reinforcement Effect of Nanocomposites with Single/Hybrid Graphene Nanoplatelets and Magnesium Hydroxide: Thermal Stability, Flame Retardancy and Mechanical Performance. J. Therm. Anal. Calorim. 2019, 137, 79–92. [Google Scholar] [CrossRef]
- Sattar, A.M. Interface Structure and Dynamics in Polymer-Nanoparticle Hybrids: A Review on Molecular Mechanisms Underlying the Improved Interfaces. Chem. Sel. 2021, 6, 5068–5096. [Google Scholar] [CrossRef]
- Huang, J.; Zhou, J.; Liu, M. Interphase in Polymer Nanocomposites. J. Am. Chem. Soc. Au 2022, 2, 280–291. [Google Scholar] [CrossRef]
- Pandey, Y.N.; Papakonstantopoulos, G.J.; Doxastakis, M. Polymer/Nanoparticle Interactions: Bridging the Gap. Macromolecules 2013, 46, 5097–5106. [Google Scholar] [CrossRef]
- Fekete, E.; Molnár, S.; Kim, G.-M.; Michler, G.H.; Pukánszky, B. Aggregation, Fracture Initiation, and Strength of PP/CaCO3 Composites. J. Macromol. Sci. Part B 1999, 38, 885–899. [Google Scholar] [CrossRef]
- Zare, Y.; Rhee, K.Y.; Hui, D. Influences of Nanoparticles Aggregation/Agglomeration on the Interfacial/Interphase and Tensile Properties of Nanocomposites. Compos. Part B Eng. 2017, 122, 41–46. [Google Scholar] [CrossRef]
- Shin, H.; Yang, S.; Choi, J.; Chang, S.; Cho, M. Effect of Interphase Percolation on Mechanical Behavior of Nanoparticle-Reinforced Polymer Nanocomposite with Filler Agglomeration: A Multiscale Approach. Chem. Phys. Lett. 2015, 635, 80–85. [Google Scholar] [CrossRef]
- Hamley, I.W.; Castelletto, V. Small-Angle Scattering of Block Copolymers. Prog. Polym. Sci. 2004, 29, 909–948. [Google Scholar] [CrossRef]
- Eisenberg, A.; Hird, B.; Moore, R.B. A New Multiplet-Cluster Model for the Morphology of Random Ionomers. Macromolecules 1990, 23, 4098–4107. [Google Scholar] [CrossRef]
- Lindner, P.; Zemb, T. Neutrons, X-rays and Light: Scattering Methods Applied to Soft Condensed Matter; Elsevier: Amsterdam, The Netherlands, 2002. [Google Scholar]
- Percus, J.K.; Yevick, G.J. Analysis of Classical Statistical Mechanics by Means of Collective Coordinates. Phys. Rev. 1958, 110, 1–13. [Google Scholar] [CrossRef]
- Jordan, J.; Jacob, K.I.; Tannenbaum, R.; Sharaf, M.A.; Jasiuk, I. Experimental Trends in Polymer Nanocomposites—A Review. Mater. Sci. Eng. A 2005, 393, 1–11. [Google Scholar] [CrossRef]
- Richard, S.; Rajadurai, J.S.; Manikandan, V. Influence of Particle Size and Particle Loading on Mechanical and Dielectric Properties of Biochar Particulate-Reinforced Polymer Nanocomposites. Int. J. Polym. Anal. Charact. 2016, 21, 462–477. [Google Scholar] [CrossRef]
- Chan, C.-M.; Wu, J.; Li, J.-X.; Cheung, Y.-K. Polypropylene/Calcium Carbonate Nanocomposites. Polymer 2002, 43, 2981–2992. [Google Scholar] [CrossRef]
- Sun, S.; Zeng, H.; Robinson, D.B.; Raoux, S.; Rice, P.M.; Wang, S.X.; Li, G. Monodisperse MFe2O4 (M = Fe, Co, Mn) Nanoparticles. J. Am. Chem. Soc. 2004, 126, 273–279. [Google Scholar] [CrossRef]
- Niederhauser, W.D.; Broderick, E.; Owings, F.F. Method for Preparing Salts of Sulfoalkyl Methacrylates. U.S. Patent US2964557, 13 December 1960. [Google Scholar]
- Minoura, Y.; Ueda, M.; Mizunuma, S.; Oba, M. The Reaction of Polypropylene with Maleic Anhydride. J. Appl. Polym. Sci. 1969, 13, 1625–1640. [Google Scholar] [CrossRef]
- Stickler, M. Free-Radical Polymerization Kinetics of Methyl Methacrylate at Very High Conversions. Die Makromol. Chemie 1983, 184, 2563–2579. [Google Scholar] [CrossRef]
- Saadat, Y.; Kim, K.; Foudazi, R. Initiator-Dependent Kinetics of Lyotropic Liquid Crystal-Templated Thermal Polymerization. Polym. Chem. 2021, 12, 2236–2252. [Google Scholar] [CrossRef]
- Eisenberg, A. Clustering of Ions in Organic Polymers. A Theoretical Approach. Macromolecules 1970, 3, 147–154. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | DEGMA/SMBStheo | DEGMA/SMBSNMR | DEGMA/SMBSCHN |
|---|---|---|---|
| 22.4Pol | 10 | 9.5 | 22.4 |
| 19.5Pol | 8 | 5.4 | 19.5 |
| 13.7Pol | 5 | 7.3 | 13.7 |
| 9.4Pol | 3 | 6.6 | 9.4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Oberhausen, B.; Plohl, A.; Niebuur, B.-J.; Diebels, S.; Jung, A.; Kraus, T.; Kickelbick, G. Self-Healing Iron Oxide Polyelectrolyte Nanocomposites: Influence of Particle Agglomeration and Water on Mechanical Properties. Nanomaterials 2023, 13, 2983. https://doi.org/10.3390/nano13232983
Oberhausen B, Plohl A, Niebuur B-J, Diebels S, Jung A, Kraus T, Kickelbick G. Self-Healing Iron Oxide Polyelectrolyte Nanocomposites: Influence of Particle Agglomeration and Water on Mechanical Properties. Nanomaterials. 2023; 13(23):2983. https://doi.org/10.3390/nano13232983
Chicago/Turabian StyleOberhausen, Bastian, Ajda Plohl, Bart-Jan Niebuur, Stefan Diebels, Anne Jung, Tobias Kraus, and Guido Kickelbick. 2023. "Self-Healing Iron Oxide Polyelectrolyte Nanocomposites: Influence of Particle Agglomeration and Water on Mechanical Properties" Nanomaterials 13, no. 23: 2983. https://doi.org/10.3390/nano13232983
APA StyleOberhausen, B., Plohl, A., Niebuur, B. -J., Diebels, S., Jung, A., Kraus, T., & Kickelbick, G. (2023). Self-Healing Iron Oxide Polyelectrolyte Nanocomposites: Influence of Particle Agglomeration and Water on Mechanical Properties. Nanomaterials, 13(23), 2983. https://doi.org/10.3390/nano13232983