Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts

Abstract
:
1. Introduction
2. Experiment
2.1. Preparation of the Au/TiO2 and AuM/TiO2 Photocatalysts
2.2. Characterization of the as-Prepared Aux/TiO2 and Au3M3/TiO2 Photocatalysts
2.3. Photocatalytic Activity Test
3. Results and Discussion
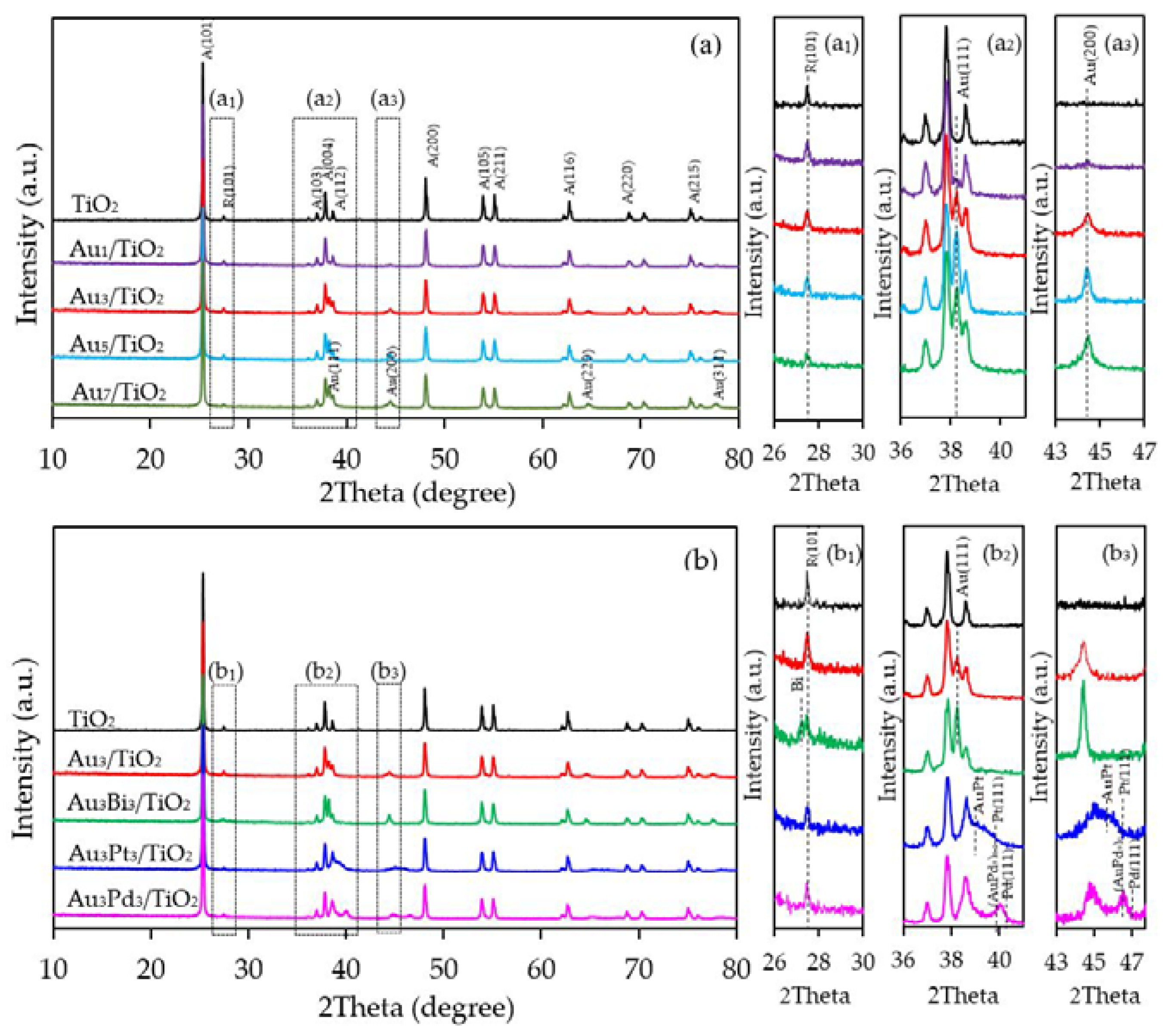
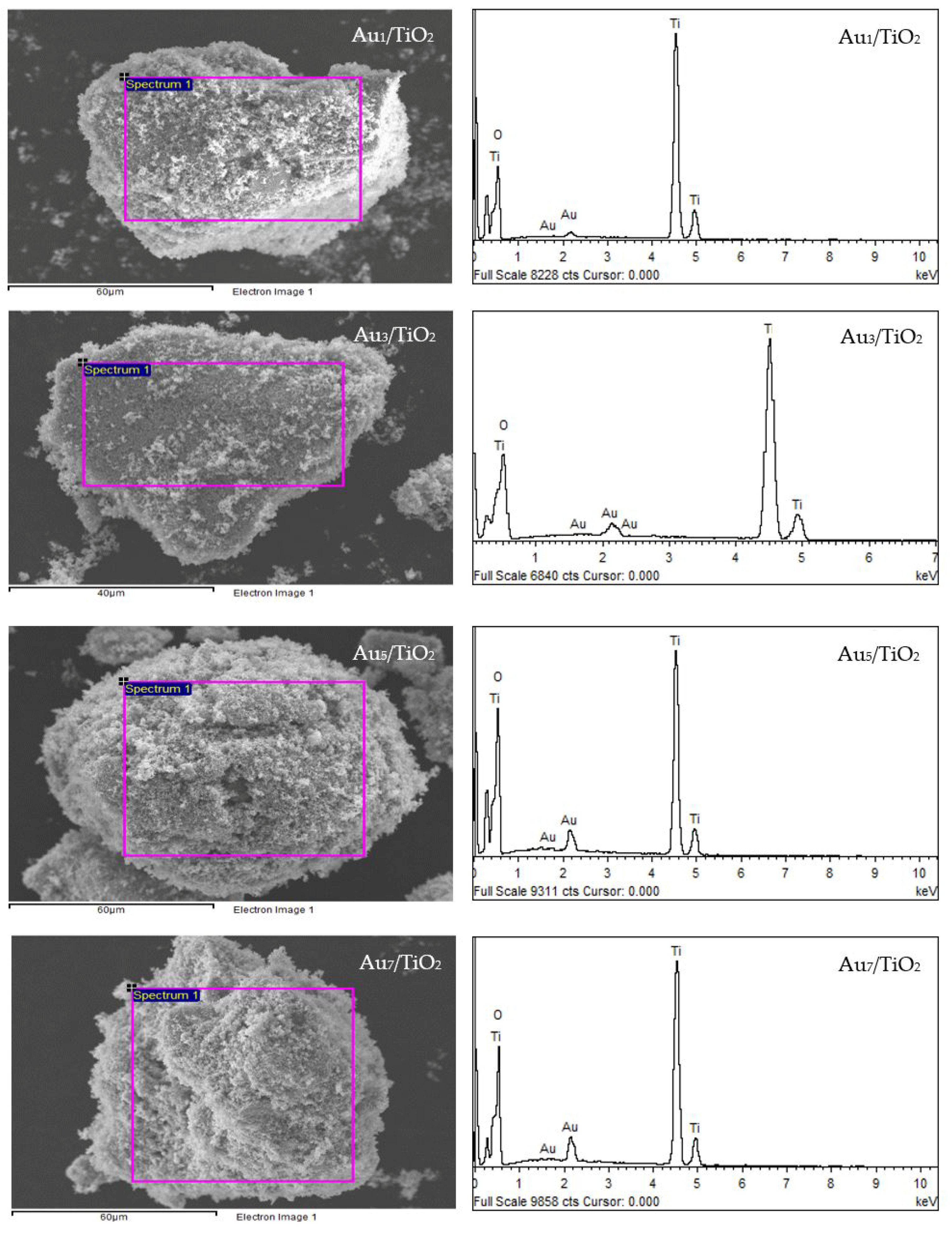
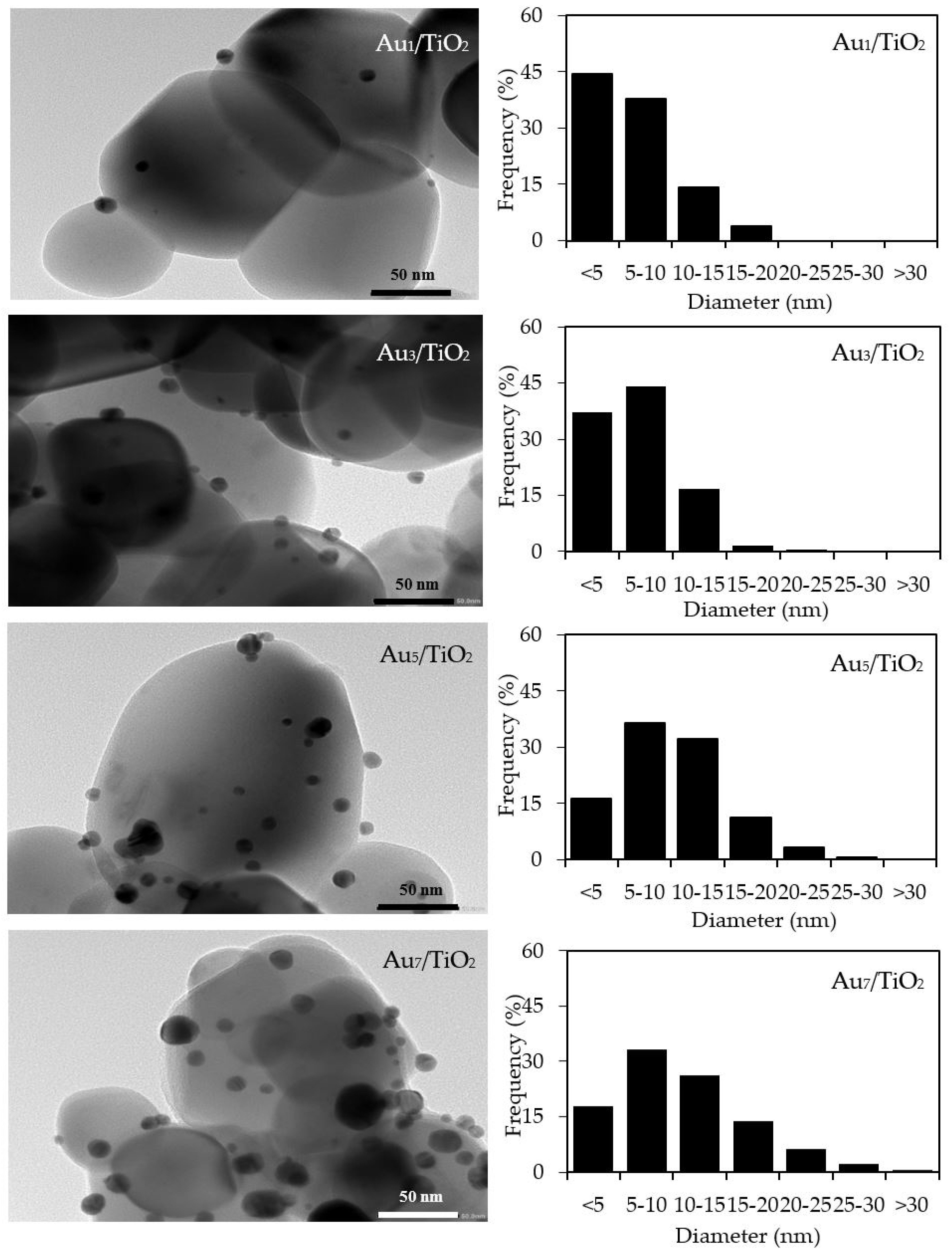
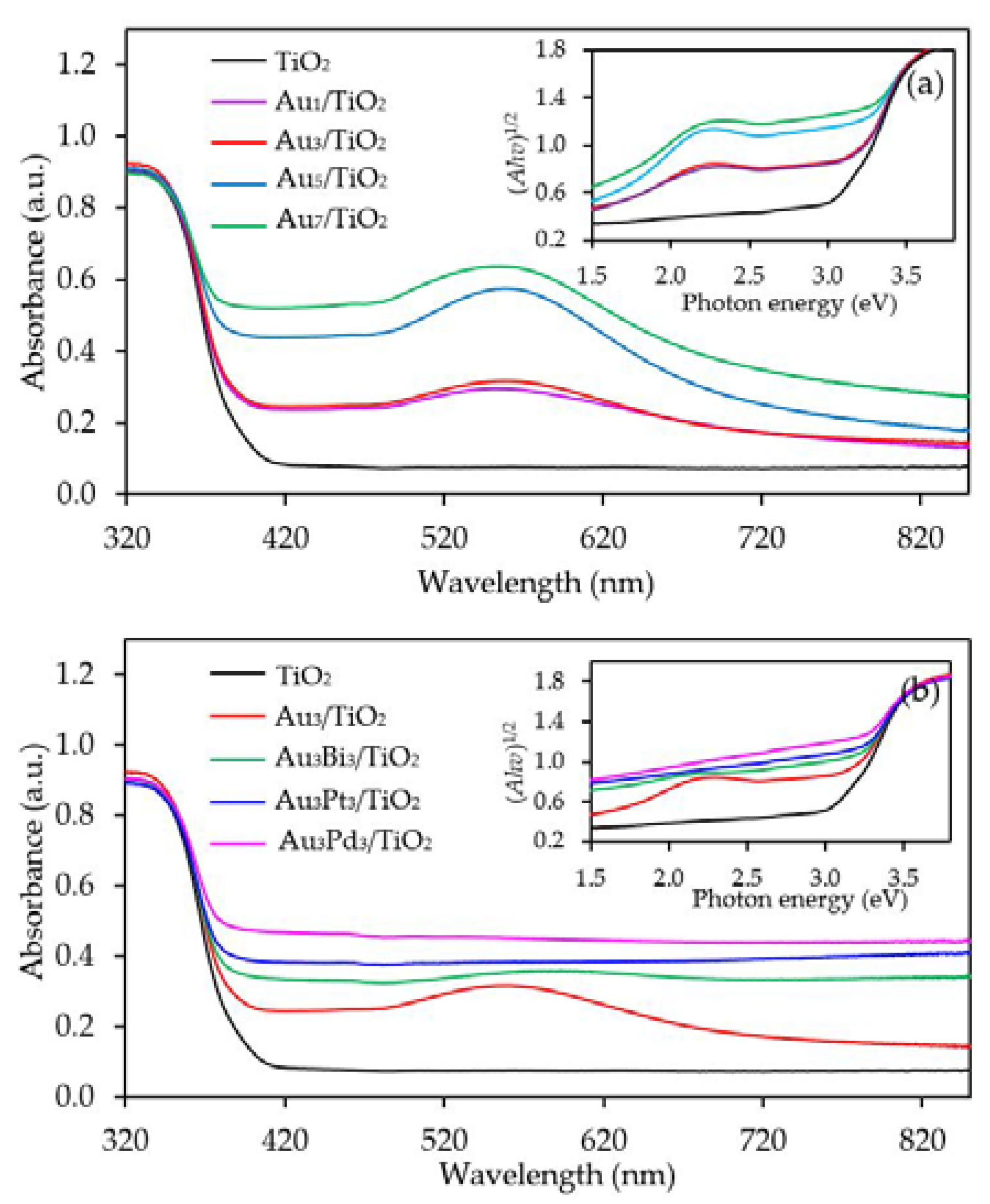
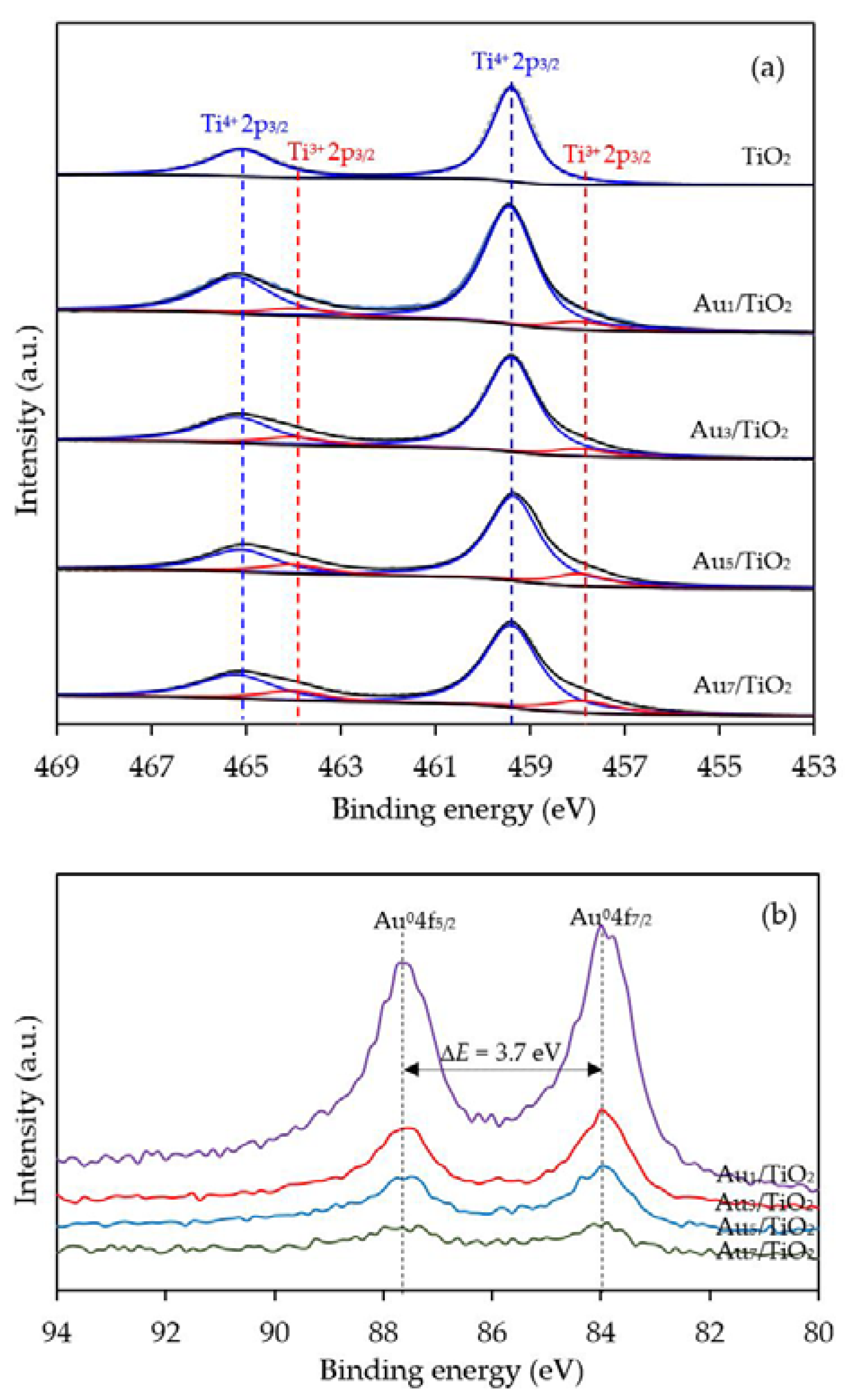
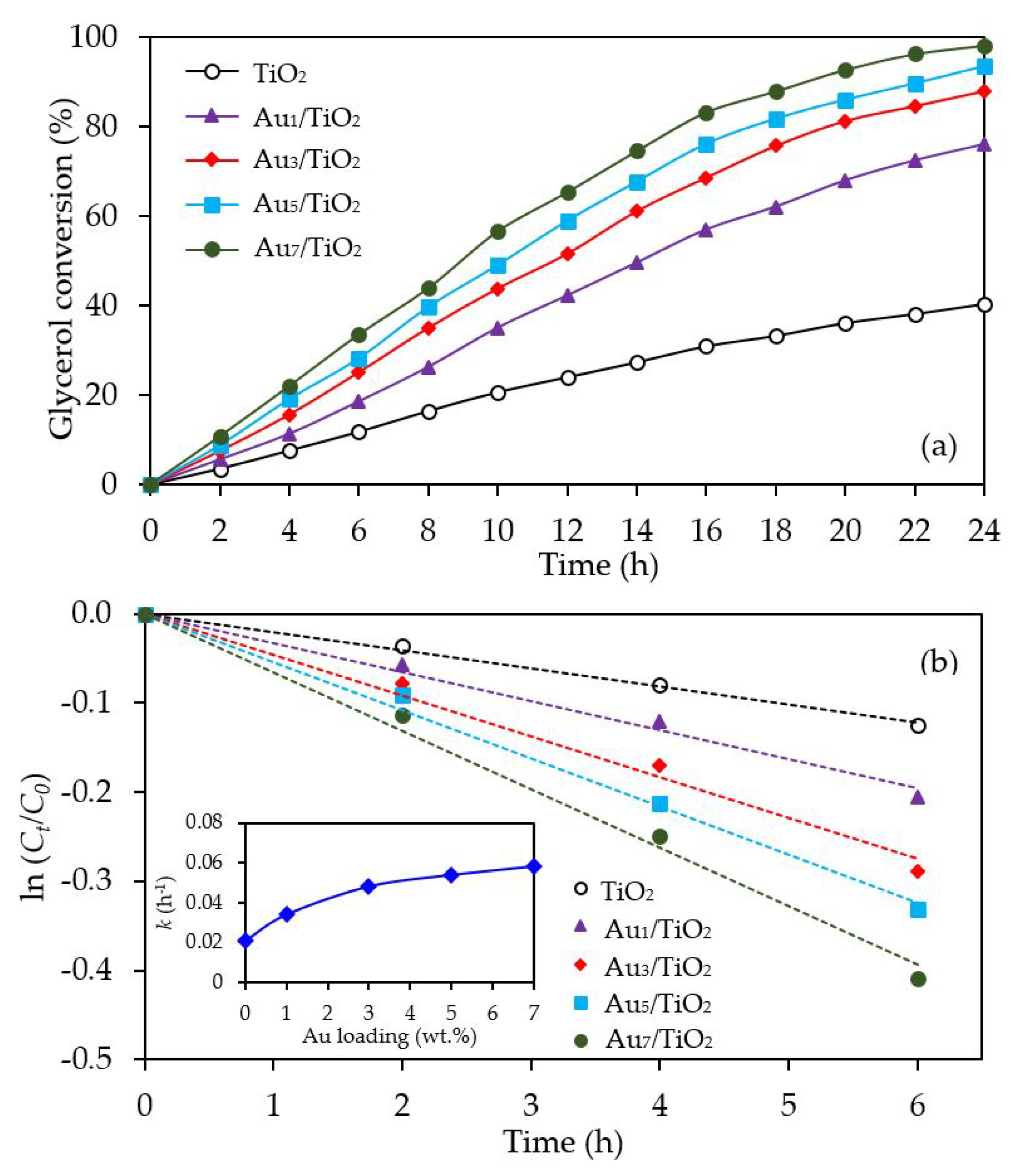
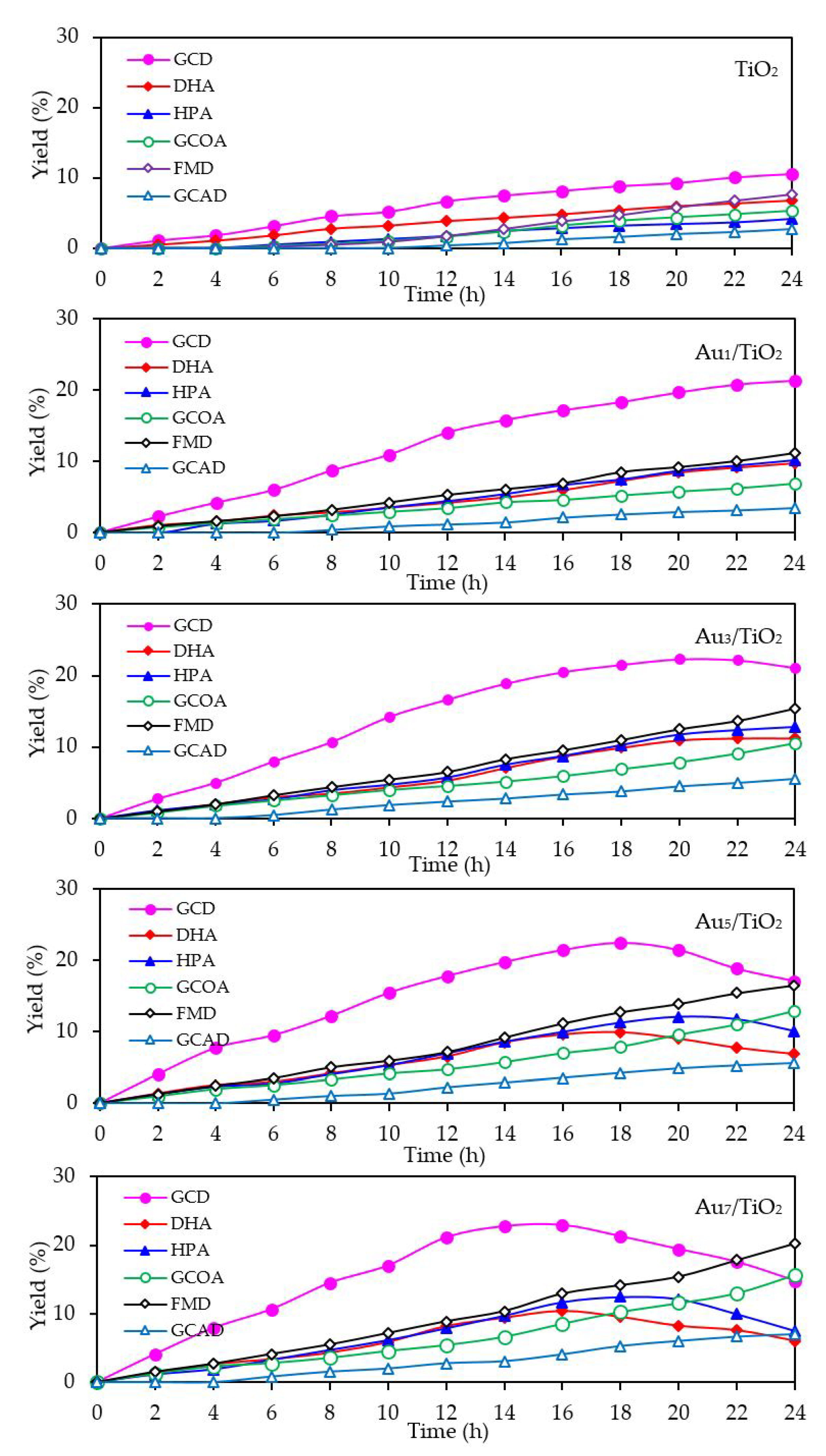
3.1. Au/TiO2 Photocatalysts
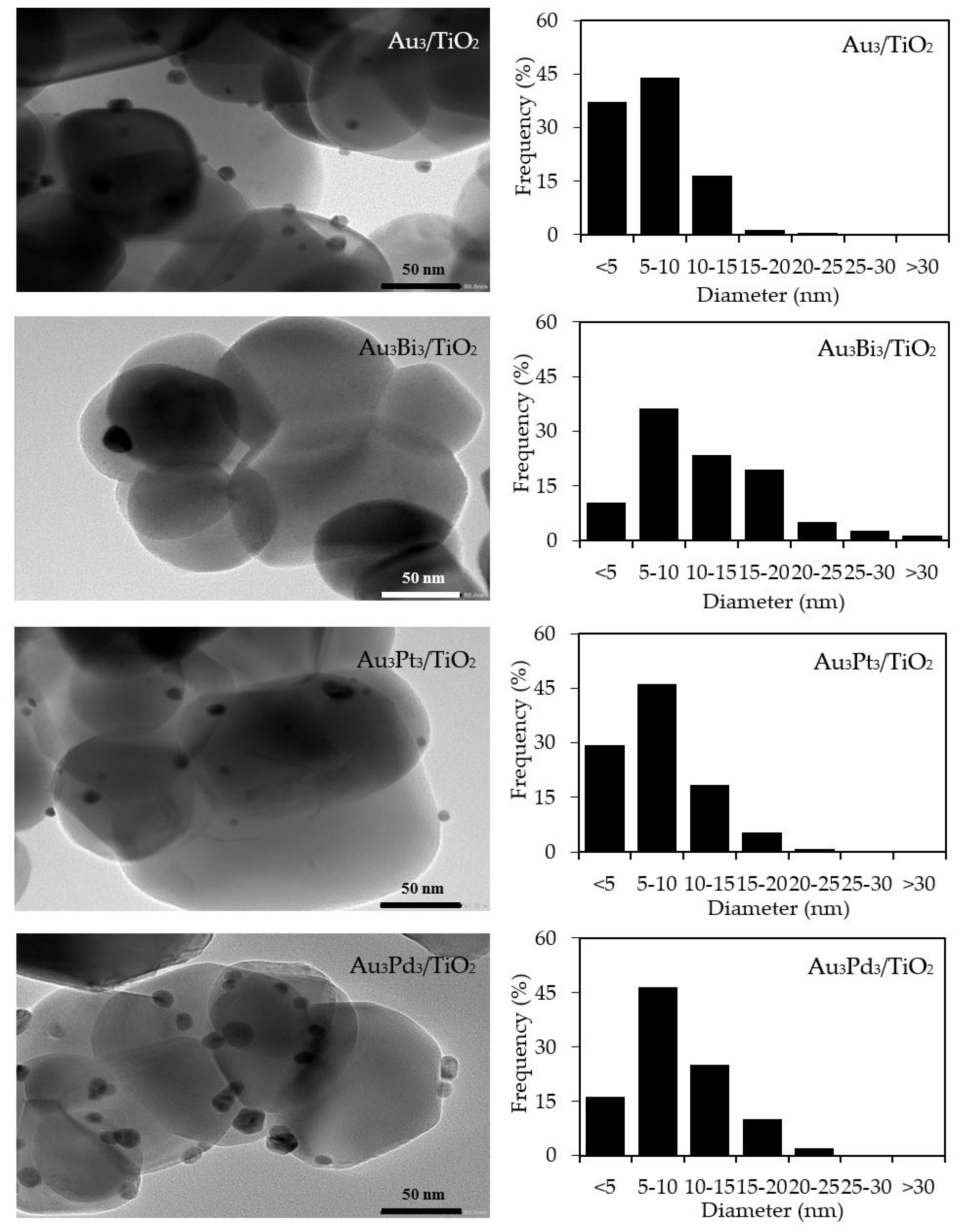
3.1.1. Photocatalyst Morphology
3.1.2. Photocatalytic Activity
3.2. Au3M3/TiO2 Photocatalysts
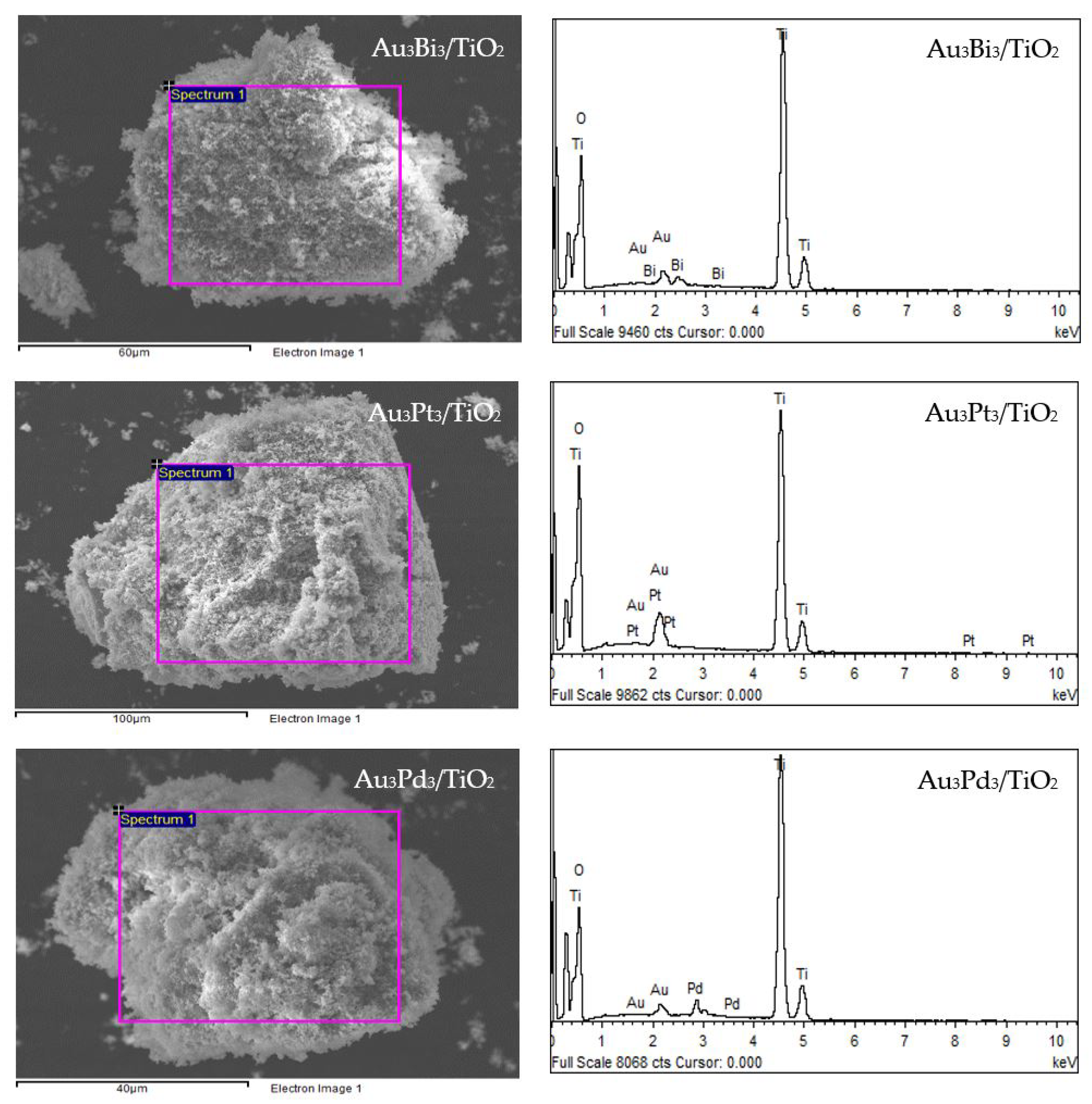
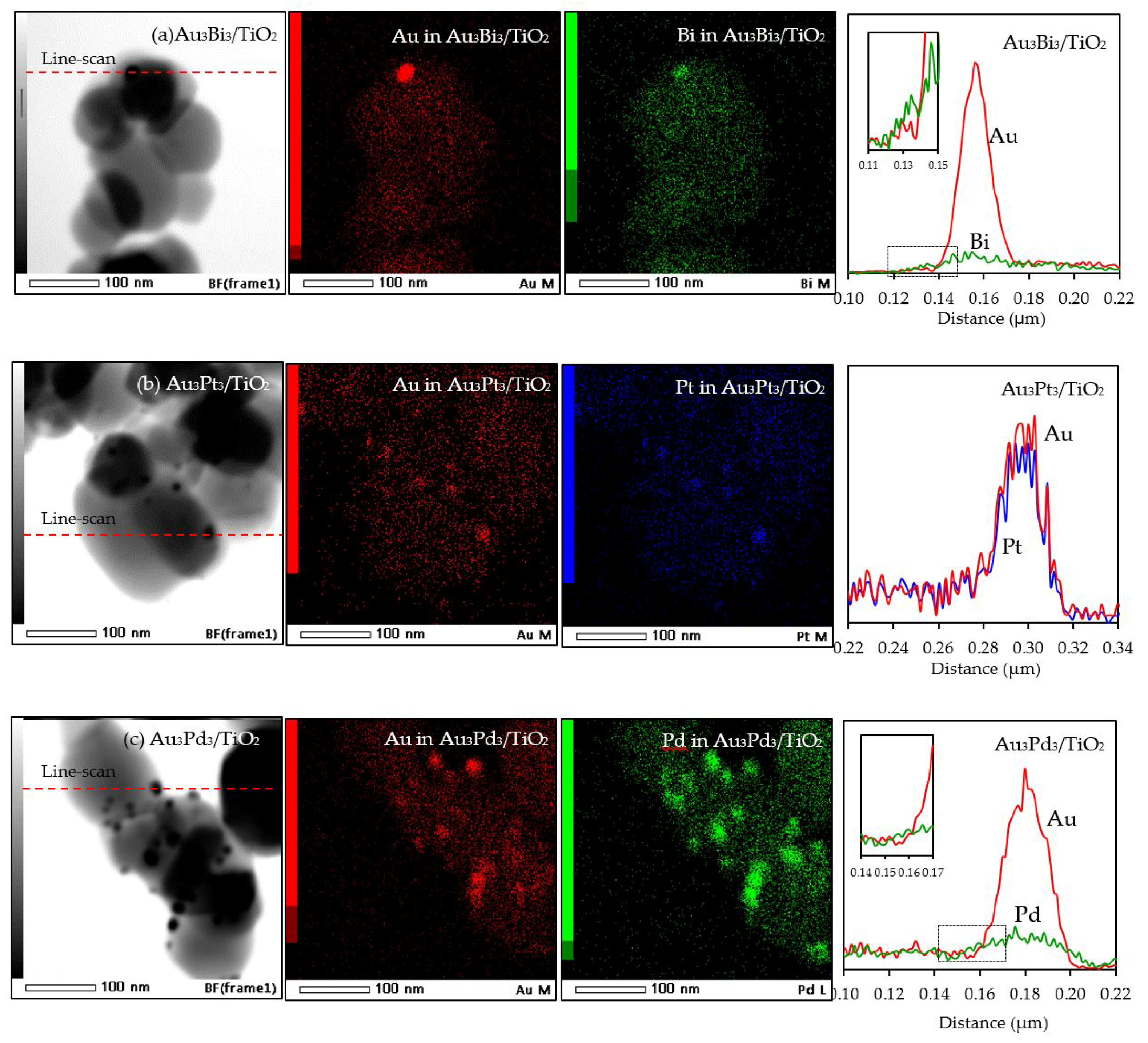
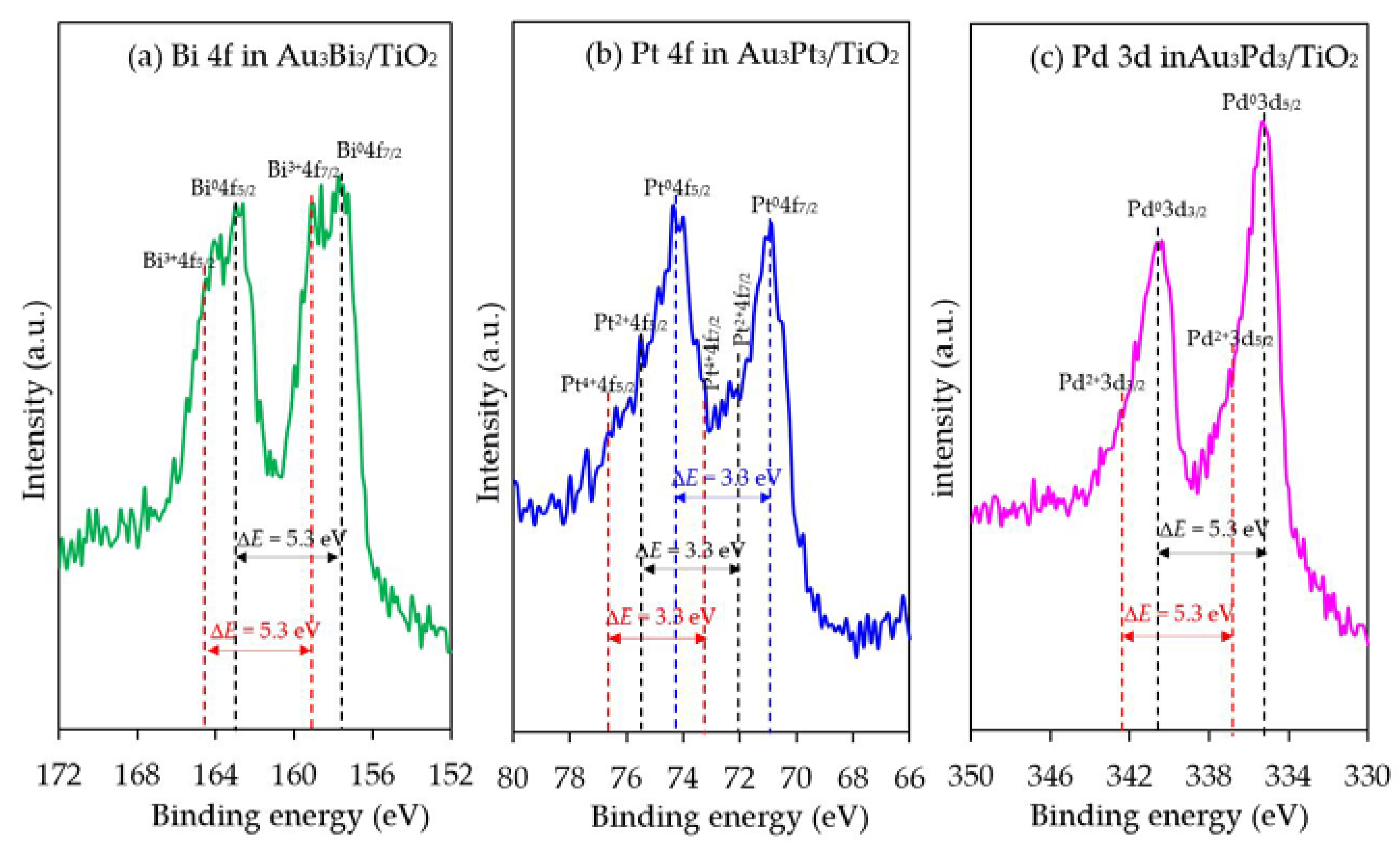
3.2.1. Photocatalyst Morphology
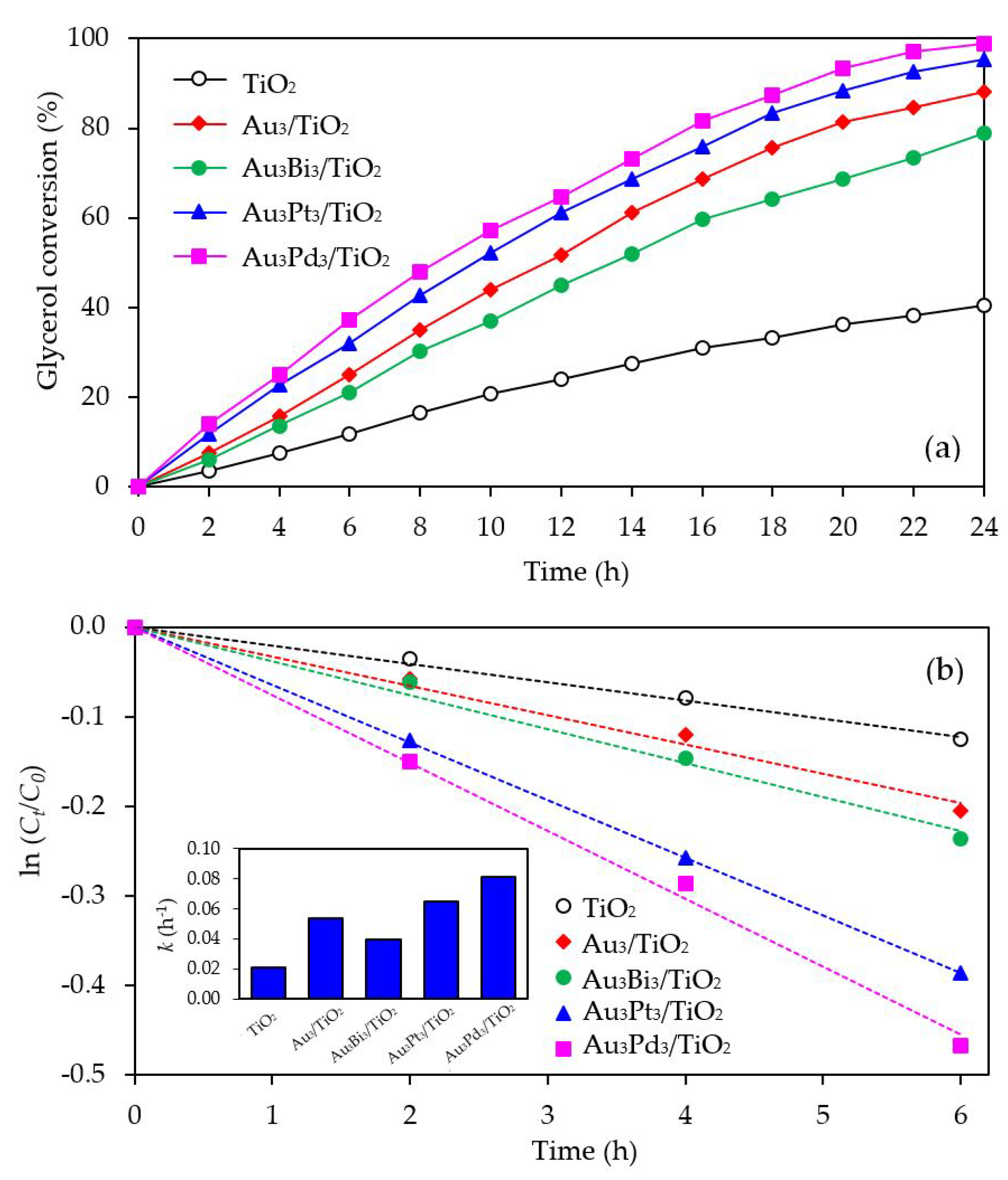
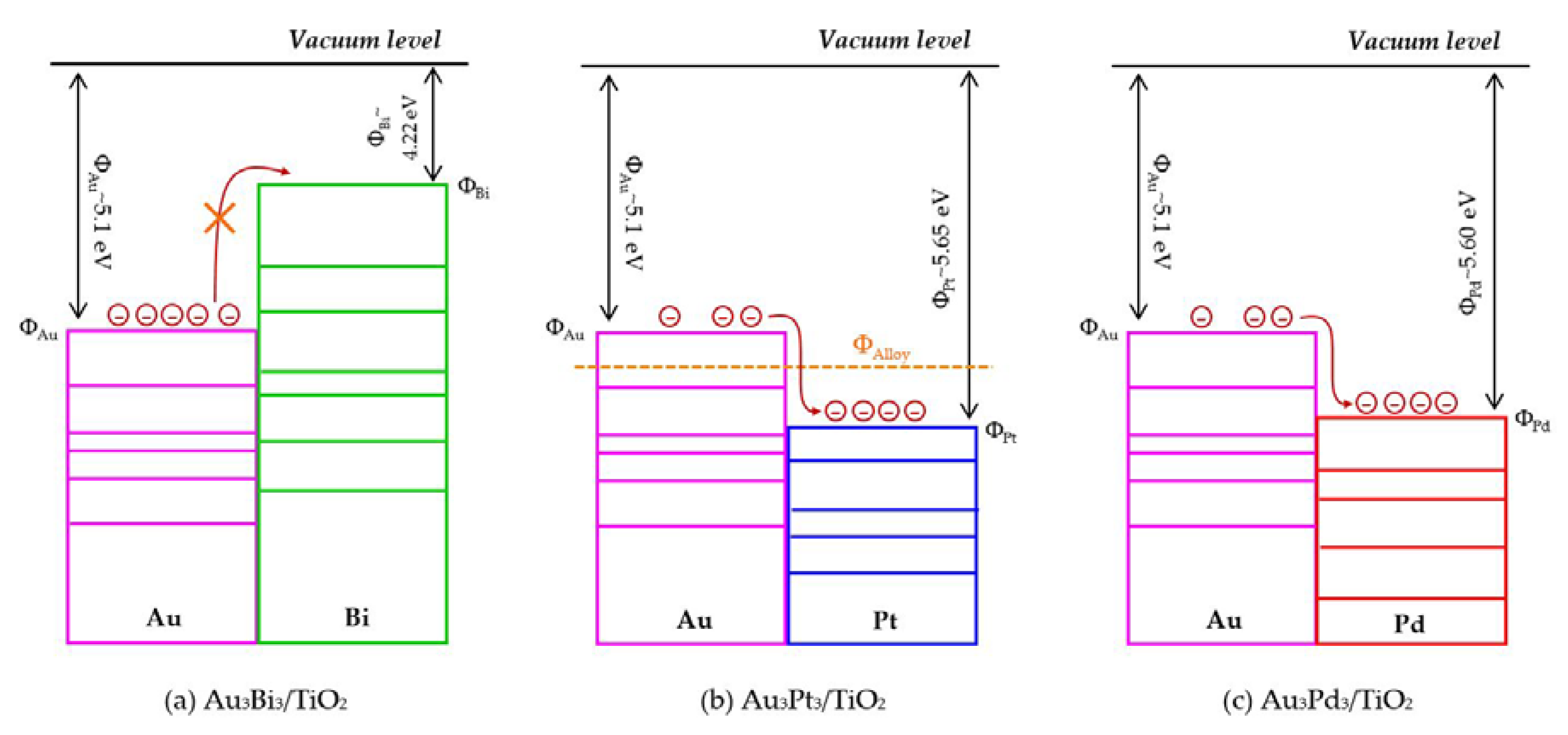
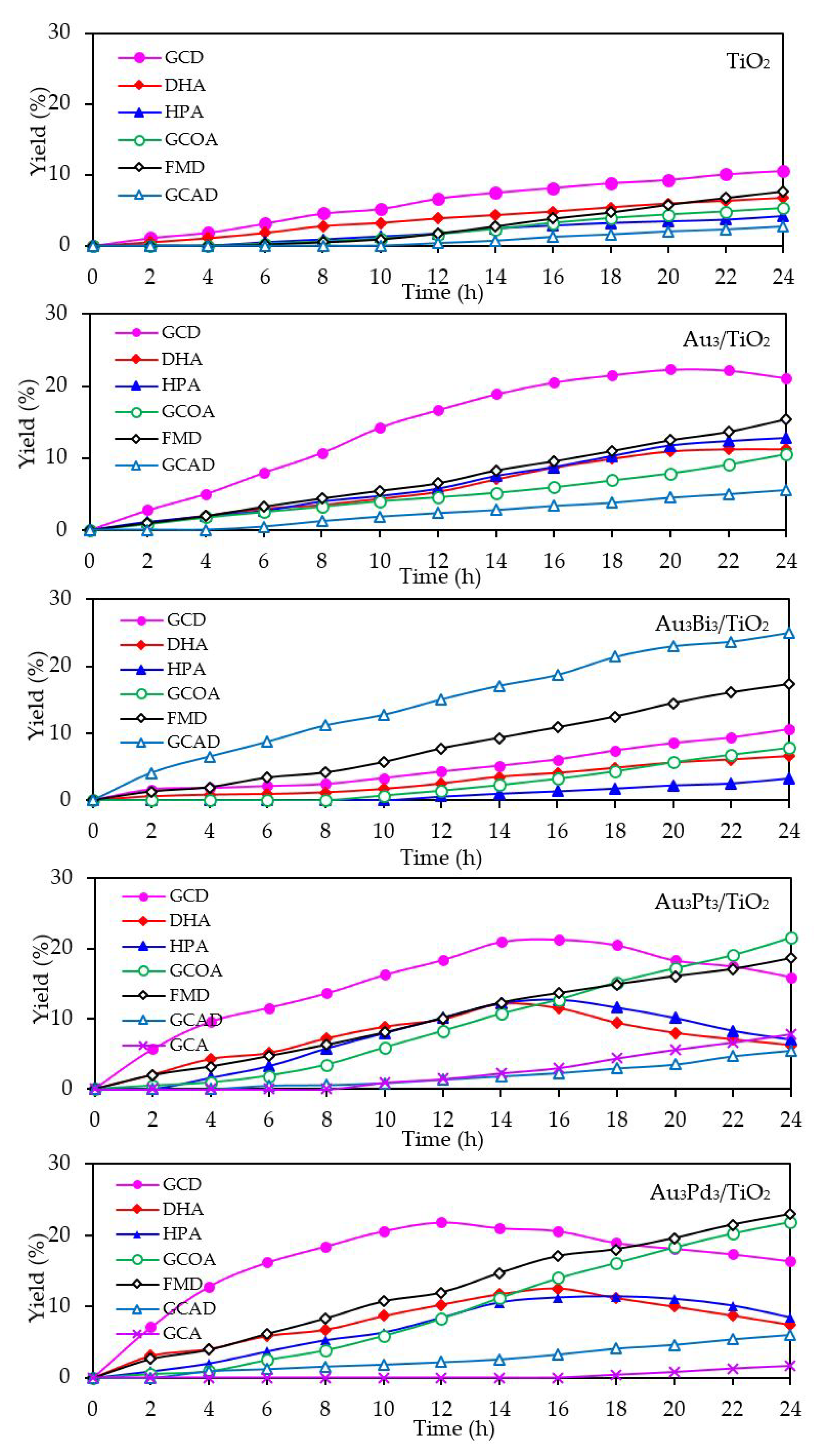
3.2.2. Photocatalytic Activity
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Clomburg, J.M.; Gonzalez, R. Anaerobic fermentation of glycerol: A platform for renewable fuels and chemicals. Trends Biotechnol. 2013, 31, 20–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotti, C.; Kašpar, J.; Farnetti, E. Dehydrogenation of glycerol to dihydroxyacetone catalyzed by iridium complexes with p–n ligands. Green Chem. 2010, 12, 1295–1300. [Google Scholar] [CrossRef]
- Zou, B.; Ren, S.; Ye, X.P. Glycerol dehydration to acrolein catalyzed by zsm-5 zeolite in supercritical carbon dioxide medium. ChemSusChem 2016, 9, 3268–3271. [Google Scholar] [CrossRef] [PubMed]
- Veiga, P.M.; Gomes, A.C.; Veloso, C.O.; Henriques, C.A. Acid zeolites for glycerol etherification with ethyl alcohol: Catalytic activity and catalyst properties. Appl. Catal. A Gen. 2017, 548, 2–15. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, Y.; Huang, Y.; Wang, J.; Gao, J.; Xu, J. Selective esterification of glycerol with acetic acid to diacetin using antimony pentoxide as reusable catalyst. J. Energy Chem. 2015, 24, 632–636. [Google Scholar] [CrossRef]
- Katryniok, B.; Kimura, H.; Skrzyńska, E.; Girardon, J.-S.; Fongarland, P.; Capron, M.; Ducoulombier, R.; Mimura, N.; Paul, S.; Dumeignil, F. Selective catalytic oxidation of glycerol: Perspectives for high value chemicals. Green Chem. 2011, 13, 1960–1979. [Google Scholar] [CrossRef]
- Villa, A.; Dimitratos, N.; Chan-Thaw, C.E.; Hammond, C.; Prati, L.; Hutchings, G.J. Glycerol oxidation using gold-containing catalysts. Acc. Chem. Res. 2015, 48, 1403–1412. [Google Scholar] [CrossRef] [PubMed]
- Puddu, V.; Choi, H.; Dionysiou, D.D.; Puma, G.L. TiO2 photocatalyst for indoor air remediation: Influence of crystallinity, crystal phase, and uv radiation intensity on trichloroethylene degradation. Appl. Catal. B Environ. 2010, 94, 211–218. [Google Scholar] [CrossRef]
- Pansa-Ngat, P.; Jedsukontorn, T.; Hunsom, M. Simultaneous H2 production and pollutant removal from biodiesel wastewater by photocatalytic oxidation with different crystal structure TiO2 photocatalysts. J. Taiwan Inst. Chem. Eng. 2017, 78, 386–394. [Google Scholar] [CrossRef]
- Low, J.; Cheng, B.; Yu, J. Surface modification and enhanced photocatalytic Co2 reduction performance of TiO2: A review. Appl. Surf. Sci. 2017, 392, 658–686. [Google Scholar] [CrossRef]
- Fujita, S.-I.; Kawamori, H.; Honda, D.; Yoshida, H.; Arai, M. Photocatalytic hydrogen production from aqueous glycerol solution using nio/tio2 catalysts: Effects of preparation and reaction conditions. Appl. Catal. B Environ. 2016, 181, 818–824. [Google Scholar] [CrossRef]
- Jedsukontorn, T.; Meeyoo, V.; Saito, N.; Hunsom, M. Route of glycerol conversion and product generation via TiO2-induced photocatalytic oxidation in the presence of H2O2. Chem. Eng. J. 2015, 281, 252–264. [Google Scholar] [CrossRef]
- Zhang, Z.; Wang, Z.; Cao, S.-W.; Xue, C. Au/pt nanoparticle-decorated TiO2 nanofibers with plasmon-enhanced photocatalytic activities for solar-to-fuel conversion. J. Phys. Chem. C 2013, 117, 25939–25947. [Google Scholar] [CrossRef]
- Bashir, S.; Idriss, H. Mechanistic study of the role of au, pd and au-pd in the surface reactions of ethanol over TiO2 in the dark and under photo-excitation. Catal. Sci. Technol. 2017, 7, 5301–5320. [Google Scholar] [CrossRef]
- Sellappan, R.; Nielsen, M.G.; González-Posada, F.; Vesborg, P.C.; Chorkendorff, I.; Chakarov, D. Effects of plasmon excitation on photocatalytic activity of ag/TiO2 and au/TiO2 nanocomposites. J. Catal. 2013, 307, 214–221. [Google Scholar] [CrossRef] [Green Version]
- Shuang, S.; Lv, R.; Xie, Z.; Zhang, Z. Surface plasmon enhanced photocatalysis of au/pt-decorated TiO2 nanopillar arrays. Sci. Rep. 2016, 6, 26670. [Google Scholar] [CrossRef] [PubMed]
- Leong, K.H.; Chu, H.Y.; Ibrahim, S.; Saravanan, P. Palladium nanoparticles anchored to anatase TiO2 for enhanced surface plasmon resonance-stimulated, visible-light-driven photocatalytic activity. Beilstein J. Nanotechnol. 2015, 6, 428. [Google Scholar] [CrossRef] [PubMed]
- Linic, S.; Christopher, P.; Ingram, D.B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 2011, 10, 911. [Google Scholar] [CrossRef] [PubMed]
- Ide, Y.; Matsuoka, M.; Ogawa, M. Efficient visible-light-induced photocatalytic activity on gold-nanoparticle-supported layered titanate. J. Am. Chem. Soc. 2010, 132, 16762–16764. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Teramura, K.; Hosokawa, S.; Kominami, H.; Tanaka, T. Visible light-induced water splitting in an aqueous suspension of a plasmonic au/TiO2 photocatalyst with metal co-catalysts. Chem. Sci. 2017, 8, 2574–2580. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Photocatalytic reactions under irradiation of visible light over gold nanoparticles supported on titanium (iv) oxide powder prepared by using a multi-step photodeposition method. Catal. Sci. Technol. 2014, 4, 1931–1938. [Google Scholar] [CrossRef]
- Tanaka, A.; Hashimoto, K.; Ohtani, B.; Kominami, H. Non-linear photocatalytic reaction induced by visible-light surface-plasmon resonance absorption of gold nanoparticles loaded on titania particles. Chem. Commun. 2013, 49, 3419–3421. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; You, T.; Shi, W.; Li, J.; Guo, L. Au/TiO2/au as a plasmonic coupling photocatalyst. J. Phys. Chem. C 2012, 116, 6490–6494. [Google Scholar] [CrossRef]
- Hvolbæk, B.; Janssens, T.V.; Clausen, B.S.; Falsig, H.; Christensen, C.H.; Nørskov, J.K. Catalytic activity of au nanoparticles. Nano Today 2007, 2, 14–18. [Google Scholar] [CrossRef]
- Brust, M.; Walker, M.; Bethell, D.; Schiffrin, D.J.; Whyman, R. Synthesis of thiol-derivatised gold nanoparticles in a two-phase liquid–liquid system. J. Chem. Soc. Chem. Commun. 1994, 801–802. [Google Scholar] [CrossRef]
- Brust, M.; Fink, J.; Bethell, D.; Schiffrin, D.; Kiely, C. Synthesis and reactions of functionalised gold nanoparticles. J. Chem. Soc. Chem. Commun. 1995, 1655–1656. [Google Scholar] [CrossRef]
- Haruta, M.; Kobayashi, T.; Sano, H.; Yamada, N. Novel gold catalysts for the oxidation of carbon monoxide at a temperature far below 0 c. Chem. Lett. 1987, 16, 405–408. [Google Scholar] [CrossRef]
- Hutchings, G.J. Vapor phase hydrochlorination of acetylene: Correlation of catalytic activity of supported metal chloride catalysts. J. Catal. 1985, 96, 292–295. [Google Scholar] [CrossRef]
- Wang, J.; Gu, H. Novel metal nanomaterials and their catalytic applications. Molecules 2015, 20, 17070–17092. [Google Scholar] [CrossRef] [PubMed]
- Ayati, A.; Ahmadpour, A.; Bamoharram, F.F.; Tanhaei, B.; Mänttäri, M.; Sillanpää, M. A review on catalytic applications of au/TiO2 nanoparticles in the removal of water pollutant. Chemosphere 2014, 107, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Fiorenza, R.; Bellardita, M.; D’Urso, L.; Compagnini, G.; Palmisano, L.; Scirè, S. Au/ TiO2-CeO2 catalysts for photocatalytic water splitting and vocs oxidation reactions. Catalysts 2016, 6, 121. [Google Scholar] [CrossRef]
- Tanaka, A.; Sakaguchi, S.; Hashimoto, K.; Kominami, H. Preparation of au/TiO2 exhibiting strong surface plasmon resonance effective for photoinduced hydrogen formation from organic and inorganic compounds under irradiation of visible light. Catal. Sci. Technol. 2012, 2, 907–909. [Google Scholar] [CrossRef]
- Tanaka, A.; Hashimoto, K.; Kominami, H. A very simple method for the preparation of au/TiO2 plasmonic photocatalysts working under irradiation of visible light in the range of 600–700 nm. Chem. Commun. 2017, 53, 4759–4762. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Hashimoto, K.; Kominami, H. Selective photocatalytic oxidation of aromatic alcohols to aldehydes in an aqueous suspension of gold nanoparticles supported on cerium (iv) oxide under irradiation of green light. Chem. Commun. 2011, 47, 10446–10448. [Google Scholar] [CrossRef] [PubMed]
- Kominami, H.; Tanaka, A.; Hashimoto, K. Mineralization of organic acids in aqueous suspensions of gold nanoparticles supported on cerium (iv) oxide powder under visible light irradiation. Chem. Commun. 2010, 46, 1287–1289. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, K.; Tanaka, A.; Hashimoto, K.; Kominami, H. Photocatalytic hydrogenation of furan to tetrahydrofuran in alcoholic suspensions of metal-loaded titanium (iv) oxide without addition of hydrogen gas. Phys. Chem. Chem. Phys. 2017, 19, 20206–20212. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Nishino, Y.; Sakaguchi, S.; Yoshikawa, T.; Imamura, K.; Hashimoto, K.; Kominami, H. Functionalization of a plasmonic au/TiO2 photocatalyst with an ag co-catalyst for quantitative reduction of nitrobenzene to aniline in 2-propanol suspensions under irradiation of visible light. Chem. Commun. 2013, 49, 2551–2553. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Han, Y.; Fu, H.; Qu, X.; Xu, Z.; Zheng, S. Au@ pd/TiO2 with atomically dispersed pd as highly active catalyst for solvent-free aerobic oxidation of benzyl alcohol. Chem. Eng. J. 2017, 313, 1–9. [Google Scholar] [CrossRef]
- Colmenares, J.C.; Lisowski, P.; Łomot, D.; Chernyayeva, O.; Lisovytskiy, D. Sonophotodeposition of bimetallic photocatalysts pd–au/TiO2: Application to selective oxidation of methanol to methyl formate. ChemSusChem 2015, 8, 1676–1685. [Google Scholar] [CrossRef] [PubMed]
- Jones, W.; Su, R.; Wells, P.P.; Shen, Y.; Dimitratos, N.; Bowker, M.; Morgan, D.; Iversen, B.B.; Chutia, A.; Besenbacher, F. Optimised photocatalytic hydrogen production using core–shell aupd promoters with controlled shell thickness. Phys. Chem. Chem. Phys. 2014, 16, 26638–26644. [Google Scholar] [CrossRef] [PubMed]
- Gallo, A.; Marelli, M.; Psaro, R.; Gombac, V.; Montini, T.; Fornasiero, P.; Pievo, R.; Santo, V.D. Bimetallic au-pt/TiO2 photocatalysts active under uv-a and simulated sunlight for H2 production from ethanol. Green Chem. 2012, 14, 330–333. [Google Scholar] [CrossRef]
- Douthwaite, M.; Huang, X.; Iqbal, S.; Miedziak, P.J.; Brett, G.L.; Kondrat, S.A.; Edwards, J.K.; Sankar, M.; Knight, D.W.; Bethell, D. The controlled catalytic oxidation of furfural to furoic acid using aupd/mg (oh) 2. Catal. Sci. Technol. 2017, 7, 5284–5293. [Google Scholar] [CrossRef]
- Redina, E.; Greish, A.; Novikov, R.; Strelkova, A.; Kirichenko, O.; Tkachenko, O.; Kapustin, G.; Sinev, I.; Kustov, L. Au/pt/TiO2 catalysts prepared by redox method for the chemoselective 1,2-propanediol oxidation to lactic acid and an nmr spectroscopy approach for analyzing the product mixture. Appl. Catal. A Gen. 2015, 491, 170–183. [Google Scholar] [CrossRef]
- Tan, T.H.; Scott, J.A.; Ng, Y.H.; Taylor, R.A.; Aguey-Zinsou, K.-F.; Amal, R. Plasmon enhanced selective electronic pathways in TiO2 supported atomically ordered bimetallic au-cu alloys. J. Catal. 2017, 352, 638–648. [Google Scholar] [CrossRef]
- Durán-Álvarez, J.C.; Avella, E.; Ramírez-Zamora, R.M.; Zanella, R. Photocatalytic degradation of ciprofloxacin using mono-(au, ag and cu) and bi-(au–ag and au–cu) metallic nanoparticles supported on TiO2 under uv-c and simulated sunlight. Catal. Today 2016, 266, 175–187. [Google Scholar] [CrossRef]
- Villa, A.; Jouve, A.; Sanchez Trujillo, F.J.; Motta, D.; Prati, L.; Dimitratos, N. Exploring the effect of au/pt ratio on glycerol oxidation in presence and absence of a base. Catalysts 2018, 8, 54. [Google Scholar] [CrossRef]
- Dimitratos, N.; Porta, F.; Prati, L. Au, pd (mono and bimetallic) catalysts supported on graphite using the immobilisation method: Synthesis and catalytic testing for liquid phase oxidation of glycerol. Appl. Catal. A Gen. 2005, 291, 210–214. [Google Scholar] [CrossRef]
- Evans, C.D.; Kondrat, S.A.; Smith, P.J.; Manning, T.D.; Miedziak, P.J.; Brett, G.L.; Armstrong, R.D.; Bartley, J.K.; Taylor, S.H.; Rosseinsky, M.J. The preparation of large surface area lanthanum based perovskite supports for aupt nanoparticles: Tuning the glycerol oxidation reaction pathway by switching the perovskite b site. Faraday Discuss. 2016, 188, 427–450. [Google Scholar] [CrossRef] [PubMed]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Anthonykutty, J.M.; Brett, G.; Carley, A.F.; Tiruvalam, R.C.; Herzing, A.A.; Kiely, C.J.; Knight, D.W.; Hutchings, G.J. Oxidation of glycerol using gold–palladium alloy-supported nanocrystals. Phys. Chem. Chem. Phys. 2009, 11, 4952–4961. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, C.L.; Canton, P.; Dimitratos, N.; Porta, F.; Prati, L. Selective oxidation of glycerol with oxygen using mono and bimetallic catalysts based on au, pd and pt metals. Catal. Today 2005, 102, 203–212. [Google Scholar] [CrossRef]
- Dimitratos, N.; Lopez-Sanchez, J.A.; Lennon, D.; Porta, F.; Prati, L.; Villa, A. Effect of particle size on monometallic and bimetallic (au, pd)/c on the liquid phase oxidation of glycerol. Catal. Lett. 2006, 108, 147–153. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Karamerou, E.E.; Kondarides, D.I. Kinetics and mechanism of glycerol photo-oxidation and photo-reforming reactions in aqueous TiO2 and pt/TiO2 suspensions. Catal. Today 2013, 209, 91–98. [Google Scholar] [CrossRef]
- Jedsukontorn, T.; Saito, N.; Hunsom, M. Photocatalytic behavior of metal-decorated TiO2 and their catalytic activity for transformation of glycerol to value added compounds. Mol. Catal. 2017, 432, 160–171. [Google Scholar] [CrossRef]
- Ning, X.; Li, Y.; Yu, H.; Peng, F.; Wang, H.; Yang, Y. Promoting role of bismuth and antimony on pt catalysts for the selective oxidation of glycerol to dihydroxyacetone. J. Catal. 2016, 335, 95–104. [Google Scholar] [CrossRef]
- Brandner, A.; Lehnert, K.; Bienholz, A.; Lucas, M.; Claus, P. Production of biomass-derived chemicals and energy: Chemocatalytic conversions of glycerol. Top. Catal. 2009, 52, 278–287. [Google Scholar] [CrossRef]
- Xue, W.; Wang, Z.; Liang, Y.; Xu, H.; Liu, L.; Dong, J. Promoting role of bismuth on hydrotalcite-supported platinum catalysts in aqueous phase oxidation of glycerol to dihydroxyacetone. Catalysts 2018, 8, 20. [Google Scholar] [CrossRef]
- Porta, F.; Prati, L. Selective oxidation of glycerol to sodium glycerate with gold-on-carbon catalyst: An insight into reaction selectivity. J. Catal. 2004, 224, 397–403. [Google Scholar] [CrossRef]
- Naldoni, A.; Riboni, F.; Marelli, M.; Bossola, F.; Ulisse, G.; Di Carlo, A.; Píš, I.; Nappini, S.; Malvestuto, M.; Dozzi, M.V. Influence of TiO2 electronic structure and strong metal–support interaction on plasmonic au photocatalytic oxidations. Catal.Sci. Technol. 2016, 6, 3220–3229. [Google Scholar] [CrossRef]
- Link, S.; El-Sayed, M.A. Size and temperature dependence of the plasmon absorption of colloidal gold nanoparticles. J. Phys. Chem. B 1999, 103, 4212–4217. [Google Scholar] [CrossRef]
- De, G.; Rao, C. Au–pt alloy nanocrystals incorporated in silica films. J. Mater. Chem. 2005, 15, 891–894. [Google Scholar] [CrossRef]
- Biesinger, M.C.; Lau, L.W.; Gerson, A.R.; Smart, R.S.C. Resolving surface chemical states in xps analysis of first row transition metals, oxides and hydroxides: Sc, ti, v, cu and zn. Appl. Surf. Sci. 2010, 257, 887–898. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, C.; Zhang, L.; Zhang, X.; Yao, H.; Shi, J. Pd-catalyzed instant hydrogenation of TiO2 with enhanced photocatalytic performance. Energy Environ. Sci. 2016, 9, 2410–2417. [Google Scholar] [CrossRef]
- Gallo, A.; Montini, T.; Marelli, M.; Minguzzi, A.; Gombac, V.; Psaro, R.; Fornasiero, P.; Dal Santo, V. H2 production by renewables photoreforming on pt–au/TiO2 catalysts activated by reduction. ChemSusChem 2012, 5, 1800–1811. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Pang, G.; He, G.; Li, Y.; He, C.; Hao, Z. Layered sphere-shaped TiO2 capped with gold nanoparticles on structural defects and their catalysis of formaldehyde oxidation. J. Environ. Sci. 2016, 39, 77–85. [Google Scholar] [CrossRef] [PubMed]
- Boyen, H.-G.; Kästle, G.; Weigl, F.; Koslowski, B.; Dietrich, C.; Ziemann, P.; Spatz, J.P.; Riethmüller, S.; Hartmann, C.; Möller, M. Oxidation-resistant gold-55 clusters. Science 2002, 297, 1533–1536. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Liu, X.; Zhu, Z.; Gao, Y.; Wang, Q.; Zhu, F.; Xie, Z. Enhanced visible light photocatalytic performance of cds sensitized TiO2 nanorod arrays decorated with au nanoparticles as electron sinks. Sci. Rep. 2017, 7, 973. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-Q.; Liu, Y.; Zhang, M.-X.; Min, F.-F. Electronic structure and visible-light absorption of transition metals (tm= cr, mn, fe, co) and zn-codoped srtio3: A first-principles study. Chin. Phys. Lett. 2018, 35, 017101. [Google Scholar] [CrossRef]
- Minero, C.; Bedini, A.; Maurino, V. Glycerol as a probe molecule to uncover oxidation mechanism in photocatalysis. Appl. Catal. B Environ. 2012, 128, 135–143. [Google Scholar] [CrossRef]
- Jedsukontorn, T.; Meeyoo, V.; Saito, N.; Hunsom, M. Effect of electron acceptors H2O2 and O2 on the generated reactive oxygen species 1O2 and OH in TiO2-catalyzed photocatalytic oxidation of glycerol. Chin. J. Catal. 2016, 37, 1975–1981. [Google Scholar] [CrossRef]
- Devarajan, S.; Bera, P.; Sampath, S. Bimetallic nanoparticles: A single step synthesis, stabilization, and characterization of au–ag, au–pd, and au–pt in sol–gel derived silicates. J. Colloid Interface Sci. 2005, 290, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Liz-Marzan, L.M.; Philipse, A.P. Stable hydrosols of metallic and bimetallic nanoparticles immobilized on imogolite fibers. J. Phys. Chem. 1995, 99, 15120–15128. [Google Scholar] [CrossRef]
- Feng, N.; Liu, F.; Huang, M.; Zheng, A.; Wang, Q.; Chen, T.; Cao, G.; Xu, J.; Fan, J.; Deng, F. Unravelling the efficient photocatalytic activity of boron-induced Ti3+ species in the surface layer of TiO2. Sci. Rep. 2016, 6, 34765. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Pan, F.; Li, Y. A review on the effects of TiO2 surface point defects on CO2 photoreduction with H2O. J. Materiomics 2017, 3, 17–32. [Google Scholar] [CrossRef]
- Yi, C.-W.; Luo, K.; Wei, T.; Goodman, D. The composition and structure of pd−au surfaces. J. Phys. Chem. B 2005, 109, 18535–18540. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Wang, X.; Sun, H.; Huo, M. Photocatalytic degradation of malathion in aqueous solution using an au–pd–TiO2 nanotube film. J. Hazard. Mater. 2010, 184, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Tripković, A.; Popović, K.D.; Stevanović, R.; Socha, R.; Kowal, A. Activity of a ptbi alloy in the electrochemical oxidation of formic acid. Electrochem. Commun. 2006, 8, 1492–1498. [Google Scholar] [CrossRef]
- Zhang, L.-M.; Wang, Z.-B.; Sui, X.-L.; Li, C.-Z.; Zhao, L.; Gu, D.-M. Nitrogen-doped carbon with mesoporous structure as high surface area catalyst support for methanol oxidation reaction. RSC Adv. 2016, 6, 39310–39316. [Google Scholar] [CrossRef]
- Cheng, H.; Huang, B.; Dai, Y.; Qin, X.; Zhang, X.; Wang, Z.; Jiang, M. Visible-light photocatalytic activity of the metastable Bi20TiO32 synthesized by a high-temperature quenching method. J. Solid State Chem. 2009, 182, 2274–2278. [Google Scholar] [CrossRef]
- Wong, R.J.; Scott, J.; Kappen, P.; Low, G.K.-C.; Hart, J.N.; Amal, R. Enhancing bimetallic synergy with light: The effect of uv light pre-treatment on catalytic oxygen activation by bimetallic au–pt nanoparticles on a TiO2 support. Catal. Sci. Technol. 2017, 7, 4792–4805. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Photocatalyst | Au Loading (wt. %) a | M Loading (wt. %) a | Decorated Metal Particle Size (nm) b | Bandgap Energy (eV) c | Ti3+/Ti4+ d |
|---|---|---|---|---|---|
| Au1/TiO2 | 0.98 ± 0.10 | n.d. | 6.36 ± 0.75 | 3.00 | 0.1220 |
| Au3/TiO2 | 3.02 ± 0.21 | n.d. | 6.69 ± 0.86 | 3.00 | 0.1478 |
| Au5/TiO2 | 4.96 ± 0.19 | n.d. | 10.04 ± 1.11 | 2.94 | 0.2198 |
| Au7/TiO2 | 7.08 ± 0.35 | n.d. | 10.84 ± 1.01 | 2.80 | 0.2412 |
| Au3Bi3/TiO2 | 2.96 ± 0.12 | 3.11 ± 0.28 | 11.84 ± 1.11 | 2.85 | 0.1960 |
| Au3Pt3/TiO2 | 3.06 ± 0.10 | 3.01 ± 0.20 | 7.61 ± 0.91 | 2.85 | 0.1941 |
| Au3Pd3/TiO2 | 3.09 ± 0.14 | 3.05 ± 0.22 | 9.66 ± 1.06 | 2.79 | 0.3561 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jedsukontorn, T.; Saito, N.; Hunsom, M. Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts. Nanomaterials 2018, 8, 269. https://doi.org/10.3390/nano8040269
Jedsukontorn T, Saito N, Hunsom M. Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts. Nanomaterials. 2018; 8(4):269. https://doi.org/10.3390/nano8040269
Chicago/Turabian StyleJedsukontorn, Trin, Nagahiro Saito, and Mali Hunsom. 2018. "Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts" Nanomaterials 8, no. 4: 269. https://doi.org/10.3390/nano8040269
APA StyleJedsukontorn, T., Saito, N., & Hunsom, M. (2018). Photoinduced Glycerol Oxidation over Plasmonic Au and AuM (M = Pt, Pd and Bi) Nanoparticle-Decorated TiO2 Photocatalysts. Nanomaterials, 8(4), 269. https://doi.org/10.3390/nano8040269