3.1. Choice of Short Aliphatic Acids and Solvents
Four short aliphatic acids, including citric acid (CA), succinic acid, adipic acid, and malic acid (MA), were separately dispersed in high b.p. solvents, including glycerol, PEG 1000, PEI 600, and seven ionic liquids, which are listed in
Section 2.1. Then, the mixtures were treated with the oxygen-gas (10 sccm), RF-discharge (90 Watt) plasma produced between the capacitive-coupling, parallel-plate electrodes. If lower b.p. solvents, such as ethylene glycol (b.p. 197 °C) and 1,4-butanediol (b.p. 235 °C), were substituted for the high b.p. solvents, a large amount of vaporized solvent was rapidly produced just after the plasma initiation, and the vacuum pumping and RF power were quickly stopped. If only the acids were treated by the capacitively coupled plasma (CCP) without the addition of solvents, the white powder products looked like the native, untreated acids and showed no fluorescence. After dispersion in the high b.p. solvents and a 30 min treatment with O
2/CCP, dark brown products were obtained, re-dispersed in water, and measured by UV-Vis and fluorescence spectroscopies. The observed spectra are shown in
Table S1 in the Electronic supplementary information (ESI). Except for glycerol and PEG 1000, for which the CCP products did not show any fluorescence, most of the solvents had excitation-dependent emissions. Glycerol and PEG do not contain any nitrogen atoms in their chemical structures, but the other solvents do. The nitrogen involvement in O
2/CCP carbonization of the acids is crucial for obtaining fluorescent C-dots. Although some fluorescent C-dots were obtained from non-nitrogen-containing materials by solvothermal [
16], pyrolysis [
17], hydrothermal [
18], and microwave heating [
19], nitrogen gas in air can enter the heating apparatuses and encounter the reactants to introduce nitrogen atoms into the C-dot structures. Even heating glycerol or PEG solvents alone in a domestic microwave oven resulted in fluorescent C-dots [
20,
21]. In this study, the nitrogen in air could not enter the CCP vacuum chamber. The only nitrogen source was the high b.p. solvents. Currently, most fluorescent C-dots are synthesized from nitrogen-containing carbon sources, solvents, or additives. In these cases, nitrogen-containing solvents act both as a dispersant and a passivant.
As shown in
Table S1, the highest photoluminescence (PL) intensity (
Imax) observed at the corresponding emission (λ
em,max) and excitation (λ
ex,max) wavelengths varied with the acid and nitrogen-containing solvent used. Some of the solvents, including PEI 600, [BMIM]AlCl
4, [BMIM]BF
4, and [MOIM]Cl, possessed similar λ
em,max and λ
ex,max values for all the acids, but other solvents did not. Most of the CCP products possessed an obvious dependence of λ
em on λ
ex, but a few of them did not. Interestingly, the C-dots obtained from the O
2/CCP treatment of CA and MA in [EMIM]N(CN)
2 possessed the first and second highest photoluminescence (PL) intensities among the CCP products in
Table S1, but their dependence of λ
em on λ
ex was obscure. As shown in
Figure 1a and
Table S1, the emission redshifts were only 11 nm and 26 nm as an increase of 80 nm in λ
ex was applied to the CA-based C-dots (350 nm to 430 nm) and MA-based C-dots (300 nm to 380 nm), respectively. In contrast, as shown in
Figure 1(b), an obvious 58 nm redshift emission (λ
ex = 350~430 nm) was observed for the O
2/CCP of [EMIM]N(CN)
2 alone. Moreover, the λ
em,max position and the smaller PL intensity in
Figure 1b were different from those of the CA- and MA-based C-dots. There should be a synergistic effect of [EMIM]N(CN)
2 and the acids, CA and MA, on the formation of fluorescent C-dots in the O
2/CCP carbonization. Further discussion about the independence of λ
ex for the CA-based C-dots is addressed in
Section 3.2.
Instead of O
2/CCP, the N
2/CCP treatment of CA in glycerol under the same CCP conditions converted non-fluorescent C-dots to fluorescent ones, as shown in
Figure 1c, and they present a λ
em,max of 400 nm with a λ
ex,max of 330 nm and a 19 nm redshift emission with an excitation from 290 nm to 370 nm. The definite difference in the fluorescence and UV-Vis spectra between
Figure 1a,c implies the different routes of C-dot formation. Although the N
2/CCP treatment aided the use of non-nitrogen-containing glycerol, its PL intensity was much lower than that of the product from the O
2/CCP treatment of CA in [EMIM]N(CN)
2 at the same dilution factor. Because a fluorophore with a high PL intensity is advantageous for analytical uses, CA dispersed in [EMIM]N(CN)
2 was selected to be the carbon source for the O
2/CCP carbonization in the following discussion.
3.2. Characterization of CCP-Treated C-Dots
The high-resolution TEM (HRTEM) image of the C-dots obtained in the conditions of
Figure 1a is shown in
Figure S1, and the average size of the 55 particles analyzed by the ImageJ freeware was 8.6 nm (σ = 1.1 nm). Most of the C-dots did not have any clear lattice fringes in the HRTEM image, but a few indicated spaces between the graphene layers, as shown in
Figure 2a and
Figure S1. This was further supported by the X-ray diffraction (XRD) pattern in
Figure 2b, which displayed a broad diffraction peak due to the amorphous nature of the sample and a distinct peak centered at 2θ = 27.3° for the (002) facet of graphite. The prepared C-dots would be classified as carbon quantum dots rather than carbon nanodots according to the classification in a recent paper [
22]. Capillary zone electrophoresis was used to analyze the changes in the surface charge of the C-dots via the electrophoretic mobility (μ
ep) at the pH of the running buffers. As shown in
Figure 2c, the μ
ep values, which were determined by subtracting the electroosmotic mobilities (μ
eof) from the apparent mobilities (μ
app) and inferred net charge on the C-dot surface, were positive below pH 5.0 and negative above pH 8.9. The dissociation of carboxylic acid and protonated amine or 1.3-diketones would occur on the C-dot surface as their pKa values are near to 5.0 and 8.9, respectively. The full scan of the XPS spectrum presents the main peaks of C 1s, N 1s, and O 1s in
Figure 2d. The carbon content (49.5%) in the O
2/CCP product was reasonable and in the range between the reactants CA (C: 37.5%) and [EMIM]N(CN)
2 (C: 58.9%). However, a large decrease in the oxygen percentage from 58.3% in CA to 13.2% in the product was distinct from the slight increase in the nitrogen percentage from 34.4% in [EMIM]N(CN)
2 to 37.4% in the product. This indicates that the oxygen atoms in CA were more easily subtracted by the oxygen plasma than the nitrogen atoms bound in the imidazole ring in [EMIM]N(CN)
2. The deconvolution of the main peaks is shown in
Figure S2. In detail, four peaks related to the C–C (284.3 eV), C–N (284.9 eV), C–O (285.9 eV), and C=O/C=N (287.6 eV) functional groups were deconvoluted from the C 1s spectrum. The N 1s spectrum contained three nitrogen bonding groups, including C–N–C (399.5 eV), N–(C)
3 (400.2 eV), and N–H (401.9 eV). The O 1s spectrum contained two characteristic peaks corresponding to the C=O (530.7 ev) and C–OH/C–O–C (532.3 eV) groups. Although the peaks of C=N and N–(C)
3 may support the existence of the imidazole group, the peaks for C–N, C–N–C, and N–H imply that parts of the imidazole rings were pyrolyzed. Further evidence of the decomposition of imidazole rings was given by the FTIR spectra in
Figure 2e, which show that the characteristic absorption bands of the aromatic CN heterocycles observed at 1465 to 1600 cm
−1 in [EMIM]N(CN)
2 disappeared after the O
2/CCP treatment. Even the dicyanamide anion decomposed, and its C≡N group absorption at 2145 cm
−1 also vanished after the plasma action. The other typical peaks of the plasma product were recognized as specific chemical bonding in
Figure 2e.
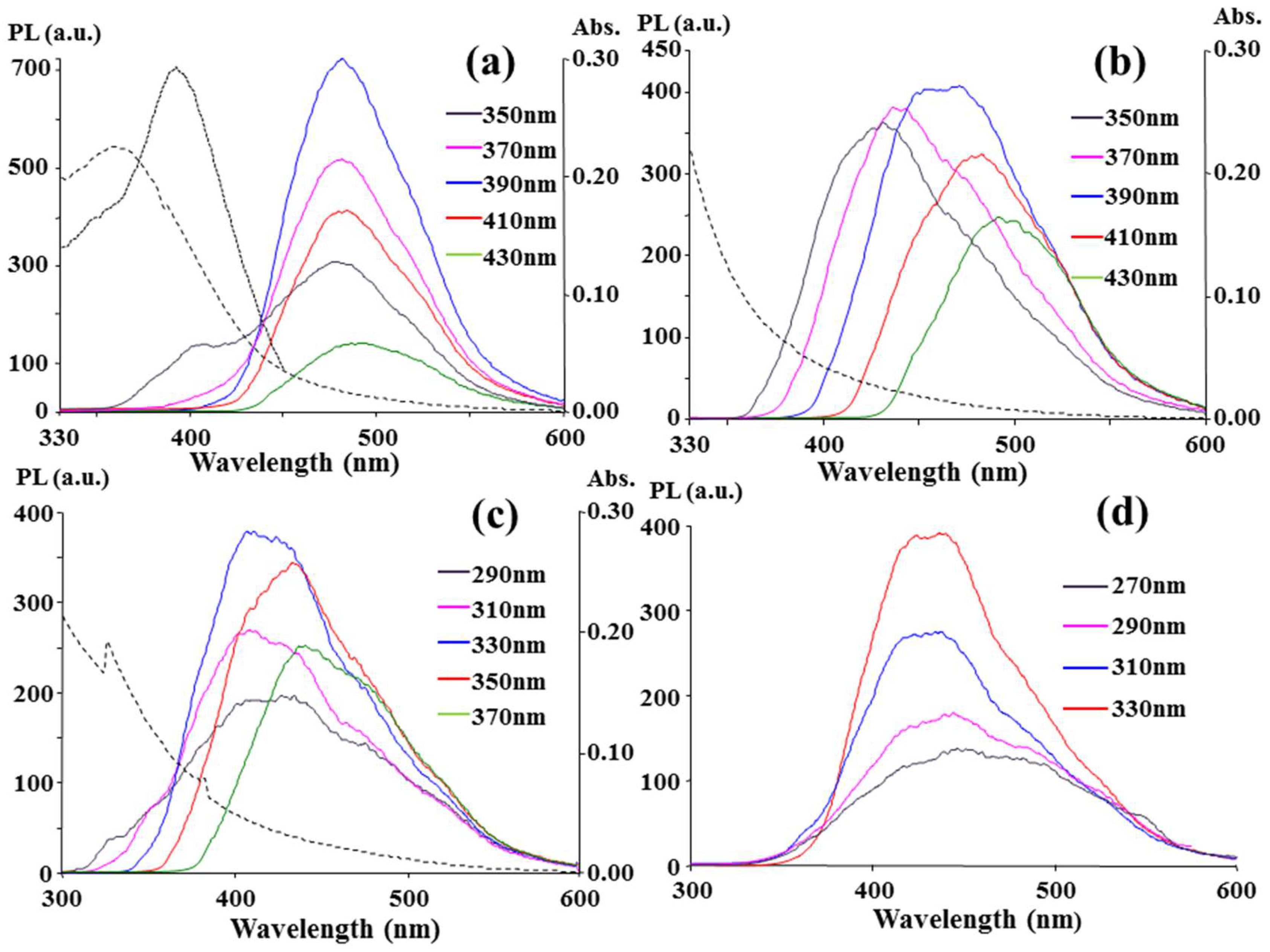
The PL, PLE (PL excitation), and UV-Vis absorption spectra of the as-prepared C-dots are shown in
Figure 1a, and an excitation-independent PL and an absorption peak at 360 nm, which was different from λ
ex,max = 390 nm, were observed. The emission at approximately 480 nm is independent of the excited light beam from 350 nm to 430 nm. At λ
ex,max = 390 nm, the quantum yield of the C-dots was 12.4% based on a calibration against the reference quinine sulfate in 0.5 M H
2SO
4, as shown in
Figure S3. The same phenomenon occurred in
Figure 1d, and the excited light beam from 270 to 330 nm produced another independent emission at approximately 430 nm for the C-dots. At λ
ex,max = 330 nm, the quantum yield was calculated to be 7.2% with reference to 2-aminopyridine in 0.5 M H
2SO
4, as shown in
Figure S3. The two λ
ex-independent emissions at 430 nm and 480 nm indicate two emissive states for each uniform energy distribution on the C-dots. Neither of the two emissions could be categorized as an intrinsic emission (π*→π) from the carbon core because the quantum confinement effect determines that 8.6 nm in diameter C-dots would have a longer-wavelength emission than near infrared but not visible emissions [
23]. The molecule states, which are determined solely by the fluorescent molecules connected on the surface or interior of the C-dots, may be emissive states because λ
ex independence is a characteristic of a molecule state [
24,
25,
26,
27]. However, the possibility of forming molecule states on the C-dots was excluded because no reasonable route to form a fluorophore molecule via the reaction of CA and [EMIM]N(CN)
2 could be inferred, whereas fluorophores were reasonably formed by the reaction of CA and a primary amine, such as ethanolamine and ethylene diamine [
24,
27]. Therefore, the two emissions should come from the surface states, which consisted of hybridization of the carbon backbone and the connected chemical groups because the π* and molecule states were excluded as the emissive states [
28].
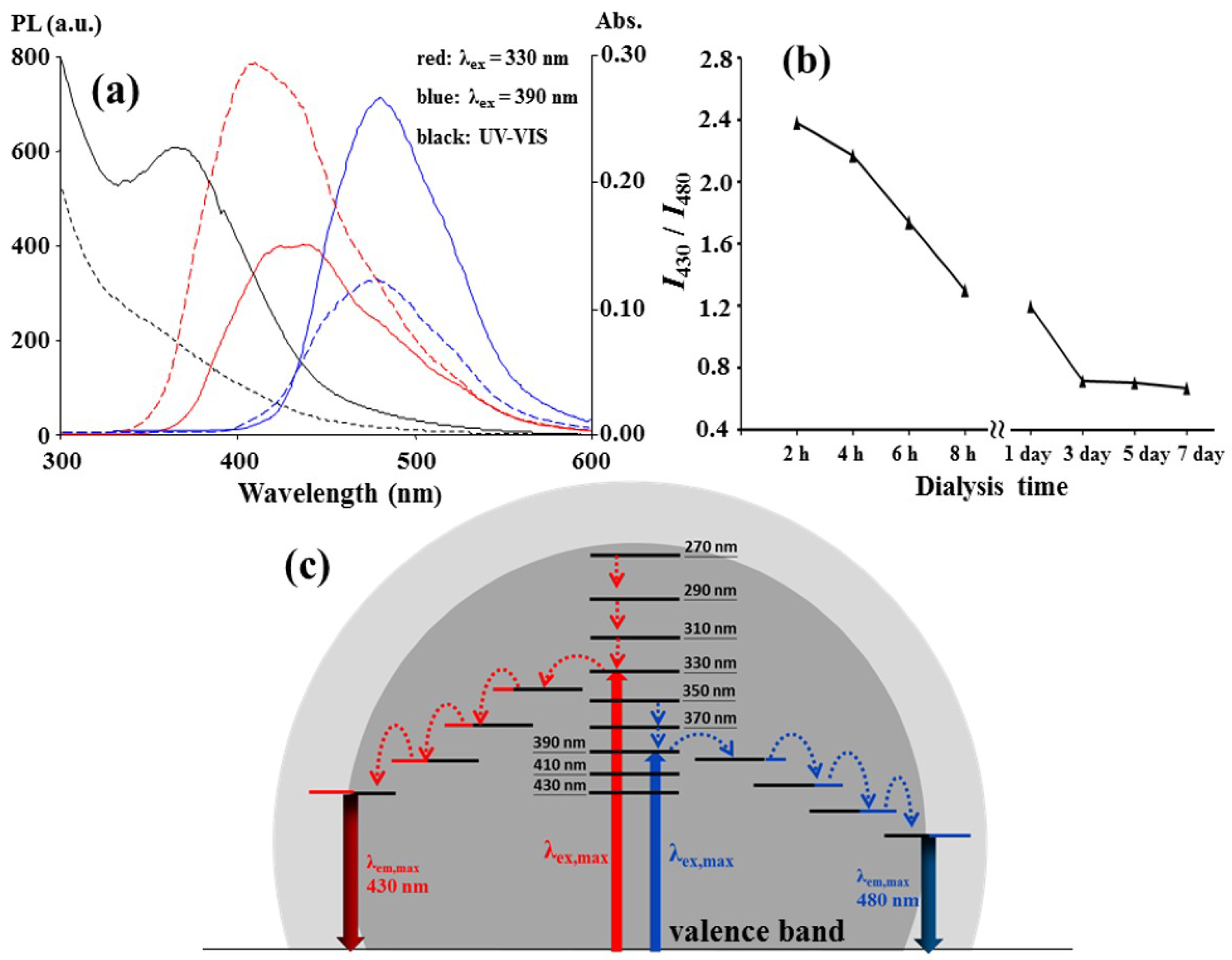
A dialysis membrane (cellulose ester, 500–1000 Da molecular weight cut-off) was used as an ultrafilter for the as-prepared C-dots to separate the two surface states. As shown in
Figure 3a, the 430 nm emission of the dialyzed C-dots that collected outside the membrane grew stronger after the first 2 h dialysis, but the 480 nm emission was weaker than that inside the membrane. Moreover, the UV absorption peak at approximately 360 nm for the dialyzed C-dots vanished, and this could be related to the weakness of the 480 nm emission. The fluorescence spectra of the C-dots dialyzed at various times are assembled in
Figure S4, and the changes in the ratio of the PL intensity at 430 nm to that at 480 nm with the dialysis time are plotted in
Figure 3b. The plot shows that the C-dot particles with higher ratio values penetrate through membrane faster than those with lower ratios, and each as-prepared C-dot particle did not have a uniform surface composition with an identical PL intensity ratio. The faster penetration was apparently not due to the smaller particle size because the average diameter (3.1 nm, σ = 1.0 nm) of the C-dots dialyzed for the first two hours was close to that (3.6 nm, σ = 1.1 nm) for those dialyzed for 8 h, as shown in
Figure S5. The main reason for different penetration rate is that some of the functional groups or surface states leading to the 430 nm emission favored penetration, but other surface states that lead to the 480 nm emission did not favor penetration. These two groups simultaneously collected on a C-dot particle, but the buildup of the groups on the particles was in different mole ratios.
Figure 3c shows a possible energy level diagram for a C-dot particle. For the two emissions, the excitation radiation beams actuated the valence π electrons in the core of the C-dot particle to the π* conduction band. Then, the excited electrons transitioned to the emissive surface states via radiationless relaxation between the lower π* levels and/or their hybrid states, which were hybridized with core carbon and heteroatoms, such as nitrogen and oxygen atoms, during the nucleation step in the carbonation.
The upconversion emission at λ
em,max = 480 nm was observed for the C-dots with 710–790 nm excitation (λ
ex,max = 790 nm), as shown in
Figure S6. The emission intensity increased with the excitation wavelength. A longer-wavelength light than 800 nm was not available due to the limitations of the fluorospectrophotometer used in this study, but a longer wavelength source, such as a near-IR laser, might induce a higher PL intensity. A 790 nm light stimulated the 480 nm up-conversion emission, and a 390 nm light also stimulated the 480 nm emission, as shown in
Figure 1a. This was expected because 790 nm is nearly double 390 nm and is suitable for the sequential absorption of two long wavelength photons. However, a 430 nm up-conversion emission was not observed in the excitation range from 430 to 790 nm. The π* transit states (2λ = 660 nm) that correlated with the 430 nm emission at λ
ex,max = 330 nm, as shown in
Figure 1d, would be less stable for the sequential absorption than those (2λ = 780 nm) that correlated with the 480 nm and underwent radiationless relaxation.
3.3. Comparison between CCP, Microwave, and Hydrothermal Carbonation
For a comparison, CA and [EMIM]N(CN)
2 were mixed in a crucible and heated in a domestic microwave oven at 90 Watt. The heating was stopped after 4.5 min because a severe burning flame appeared on the crucible during the microwave irradiation. As shown in
Figure 4a, the microwave-treated product emission at 430 nm is stronger than its emission at 480 nm. Therefore, the number of surface states corresponding to the 430 nm emission should be larger than the number corresponding to the 480 nm emission. Furthermore, the chemical composition corresponding to the two surface states caused by microwave radiation with a sufficient oxygen supply should be different from that caused by the CCP ion bombardment under a limited oxygen supply. The stronger 430 nm emission obtained with a sufficient oxygen supply implied that the formation of the 430 nm emissive surface states involved oxygen atoms.
Figure 4a shows the λ
ex,max was 370 nm or 390 nm for the maximum emission at λ
em,max = 430 nm, and the λ
ex,max was 430 nm for that at λ
em,max = 480 nm. The two λ
ex,max values are larger (approximately 40 nm) than those in
Figure 1d (λ
ex,max = 330 nm) and
Figure 1a (λ
ex,max = 390 nm), respectively. In addition to the changes in the surface composition, the severe carbonation in a microwave oven might extend the domain of the double bond and shrink the energy gap between π and π* to cause the 40 nm redshift compared with that for the CCP treatment. Without the addition of CA, heating [EMIM]N(CN)
2 alone in a microwave could not provide the considerable 480 nm emission and only resulted in a strong 430 nm emission (λ
em,max = 400 nm) at a shorter λ
ex (290~350 nm), as shown in
Figure 4b. In comparison with
Figure 4a, the participation of CA in the carbonation could mainly contribute the 480 nm emissive states and the extension of the conjugated double bonds to the C-dot structure.
The mixture of CA and [EMIM]N(CN)
2 was also placed in a Teflon-lined autoclave and heated in a hot-air oven at 200 °C for 30 min. The PL and UV-Vis absorption spectra of the hydrothermal products are plotted in
Figure 4c and are similar to those in
Figure 1a, which were obtained from O
2/CCP at 10 sccm, except the PL intensity was smaller than that in
Figure 1a. Furthermore, the spectrum of the [EMIM]N(CN)
2 alone sample treated by a hydrothermal method was similar to that from the CCP method, i.e., comparison of
Figure 4d with
Figure 1b. It is believed that fine tuning the temperature and duration of the hydrothermal reaction in an autoclave could enhance the similarity of the reaction to the CCP carbonation in a vacuum chamber with a low O
2 supply, i.e., 10 sccm in our system.
Figure 4d shows the results from 5.0 h of heating [EMIM]N(CN)
2 alone in an autoclave. Only 30 min of heating by the hydrothermal method could not obtain a PL spectrum, but
Figure 1b spectrum by the O
2/CCP method was obtained.
3.4. Fluorescent Sensing by the O2/CCP-Treated C-Dots
The fluorescent C-dots prepared by O
2/CCP of a mixture of CA and [EMIM]N(CN)
2 in the conditions shown in the legend of
Figure 1a were used to separately probe fifteen metal ions and nine flavonoids. At first, the λ
ex-independent emissions at 430 nm (λ
ex,max = 330 nm) and 480 nm (λ
ex,max = 390 nm) of the C-dots were used to evaluate their responses to the analytes. The 480 nm emission was quenched by Cu
2+, Ag
+, and Hg
2+, while the 430 nm emission was further quenched by Fe
3+ in addition to those ions, as shown in
Figure 5a, where
I and
I0 are the emission intensities in the absence and presence of the sample, respectively. The ions, Fe
3+, Cu
2+, and Ag
+, quenched 10~15% of each emission, and nearly 30% of the 430 nm emission and 65% of the 480 nm emission could be heavily quenched by Hg
2+. The quenching was sensitive to Hg
2+, especially the 480 nm emission, but the coexistence of the Fe
3+, Cu
2+, and Ag
+ ions in a real sample would interfere with the detection of Hg
2+. However, the synthetic C-dots are good starting fluorescent materials for further ligand attachment on them to improve a sensor’s specificity for Hg
2+ in the presence of interfering ions.
For the flavonoid samples, quercetin, hesperetin, and naringenin could quench nearly half of the 430 nm emission, but daidzein and 5-methoxyflavone passivated some unknown surface traps and enhanced the 430 nm PL intensity, as shown in
Figure 5b. In contrast to the 430 nm emission, the 480 nm emission was only selectively quenched by quercetin, and the other flavonoids did not affect the 480 nm emission. Therefore, the C-dots would directly sense quercetin in a flavonoid-rich sample, such as
Citrus reticulata cv. Chachiensis, which is a sun-dried peel used as a traditional Chinese medicine, called “Guang-Chen-Pi” in Chinese. For quantification, the 480 nm PL intensities were measured (
Figure 5c) after equilibrium with quercetin standards in a phosphate buffer, pH 7.0, 50 mM, and they were plotted against the quercetin concentrations ranging from 2.4 μM to 119 μM in
Figure 5d. As shown in
Figure 5d, the Stern–Volmer plot,
I0/
I v.s. [quercetin], is not linear. The deviation from linearity is frequently attributed to a combination of dynamic and static quenching and can be corrected using a modified Stern–Volmer plot, i.e., the Perrin model:
where α =
NAV, where
NA is Avogadro’s number and
V is the volume of the active sphere of quenching. Based on the good linearity (
R2 = 0.9992) and high slope (1.96 × 10
4 M
−1) of the Perrin relationship, the radius of the effective quenching sphere was calculated to be 19.8 nm, which is 2.3 times the C-dot radius (8.6 nm) and allowed an efficient, photoinduced electron-transfer process of the encounter pair between the C-dot (as an electron donor) and quercetin (as an electron acceptor) without coupling reagents. The relative standard deviation for seven replicate measurements of 12.1 μM quercetin solutions was 3.3%. Based on the 3σ of the blank response (σ = 3.1%,
n = 10), the detection limit was calculated to be 0.5 μM. As shown in the inset of
Figure 5d, the quercetin in the ethanol extract of Guang-Chen-Pi was determined by a standard addition method to be 4.20 ± 0.15 mg/g, which was nearly nine times higher than 0.47 mg/g found in the air-dried peel of
Citrus reticulate Blanco [
29]. Some factors, such as the citrus species used, growth environment, and method of drying the peel, accounted for the difference. The matrix effect was evaluated by the ratio defined as (
S1 −
S2) × 100%/
S2, where
S1 and
S2 are the slopes of the calibration curves obtained by standard addition (
S1 = 2.12 × 10
4 M
−1,
R2 = 0.9990) and external standard (
S2 = 1.96 × 10
4 M
−1) methods, respectively [
30]. The calculated ratio, 8.16%, was lower than 10%, which suggested that the matrix effect could be ignored by the standard addition method. The selectivity of the 480 nm emission from the C-dots for quercetin helped reduce the matrix effect.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




