The Effect of Polymer Microstructure on Encapsulation Efficiency and Release Kinetics of Citropin 1.1 from the Poly(ε-caprolactone) Microparticles

 ,
, 


Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
Spectroscopic Data of PCLs Matrices
2.2. Single Emulsion Microspheres’ Preparation
2.3. Double Emulsion Microspheres’ Preparation
2.4. Determination of the Encapsulation Efficiency
2.5. In Vitro Studies of Citropin 1.1 Release From the Microspheres
2.6. Hemolytic Activity of the Peptide and Calculation of the Therapeutic Index
2.7. Measurements
3. Results and Discussion
3.1. Morphology, Size and Zeta Potential of MPs
3.2. Encapsulation Efficiency of CIT
3.3. The In Vitro Kinetics Release of CIP from PCL MPs
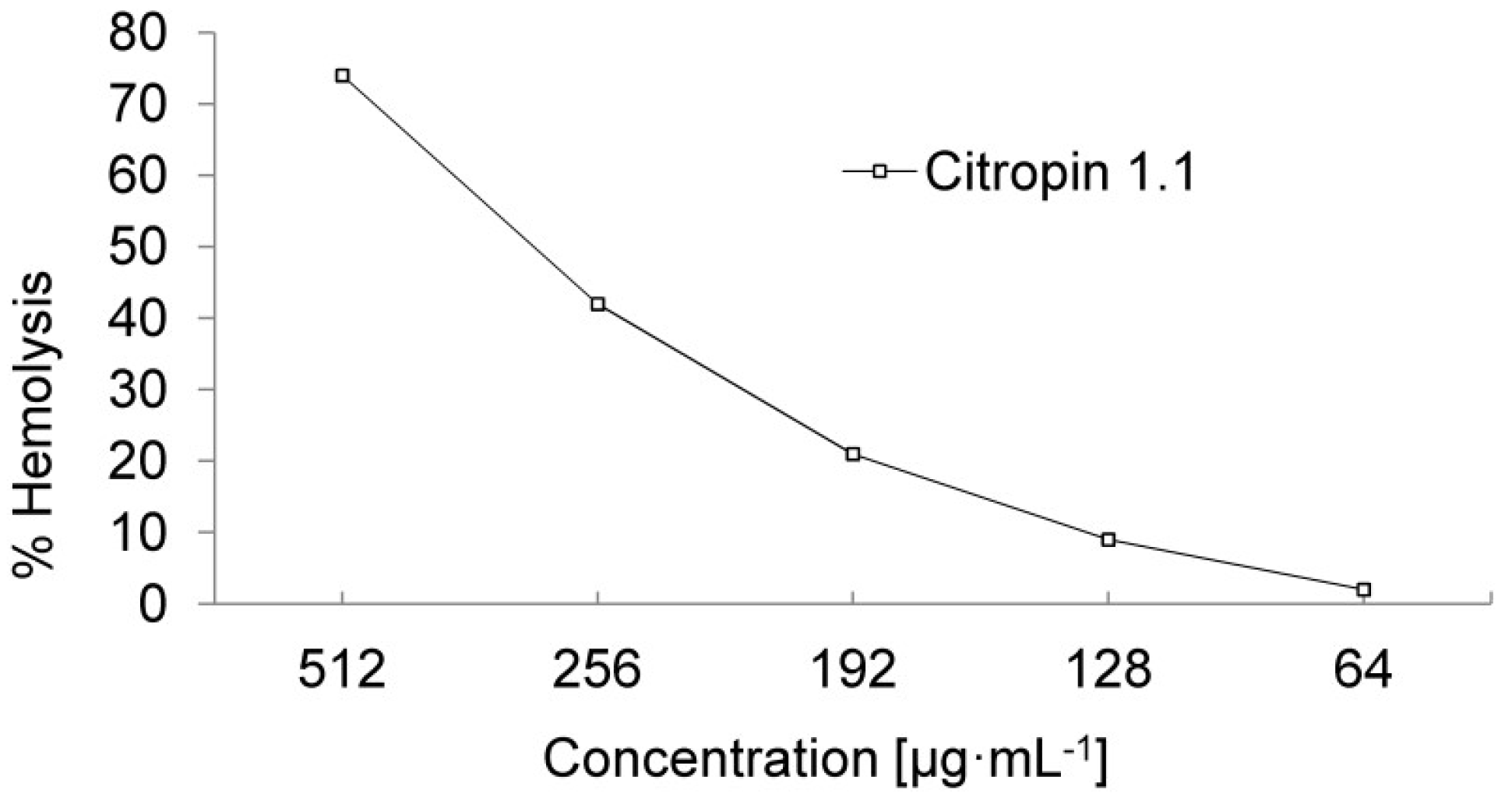
3.4. Hemolytic Activity of Peptides and Calculation of the Therapeutic Index
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kang, H.-K.; Kim, C.; Seo, C.H.; Park, Y. The therapeutic applications of antimicrobial peptides (AMPs): A patent review. J. Microbiol. 2017, 55, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, U.; Sobczak, M.; Oledzka, E. Current state of a dual behavior of antimicrobial peptides-therapeutic agents and promising delivery vectors. Chem. Biol. Drug Des. 2017, 90, 1079–1093. [Google Scholar] [CrossRef] [PubMed]
- Ron-Doitch, S.; Sawodny, B.; Kühbacher, A.; David, M.M.N.; Samanta, A.; Phopase, J.; Burger-Kentischer, A.; Griffith, M.; Golomb, G.; Rupp, S. Reduced cytotoxicity and enhanced bioactivity of cationic antimicrobial peptides liposomes in cell cultures and 3D epidermis model against HSV. J. Control. Release 2016, 229, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.M.; Edwards, M.A.; Li, J.; Yip, C.M.; Deber, C.M. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J. Biol. Chem. 2012, 28, 7738–7745. [Google Scholar] [CrossRef] [PubMed]
- Kamaly, N.; Yameen, B.; Wu, J.; Farokhzad, O.C. Degradable controlled-release polymers and polymeric nanoparticles: Mechanisms of controlling drug release. Chem. Rev. 2016, 116, 2602–2663. [Google Scholar] [CrossRef] [PubMed]
- Lassalle, V.; Ferreira, M.L. PLA nano-and microparticles for drug delivery: An overview of the methods of preparation. Macromol. Biosci. 2007, 7, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Na, R.; Wang, X.; Liu, H.; Zhao, L.; Sun, X.; Ma, G.; Cui, F. Fabrication of Antimicrobial Peptide-Loaded PLGA/Chitosan Composite Microspheres for Long-Acting Bacterial Resistance. Molecules 2017, 22, 1637. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Wang, S.; Li, H.; Wang, Y.; Chen, H.; Yuan, M. Characterization, Stability and Biological Activity In Vitro of Cathelicidin-BF-30 Loaded 4-Arm Star-Shaped PEG-PLGA Microspheres. Molecules 2018, 23, 497. [Google Scholar] [CrossRef] [PubMed]
- Piotrowska, U.; Oledzka, E.; Zgadzaj, A.; Bauer, M.; Sobczak, M. A Novel Delivery System for the Controlled Release of Antimicrobial Peptides: Citropin 1.1 and Temporin A. Polymers 2018, 10, 489. [Google Scholar] [CrossRef]
- Sobczak, M.; Kamysz, W.; Tyszkiewicz, W.; Dębek, C.; Kozłowski, R.; Olędzka, E.; Piotrowska, U.; Nałęcz-Jawecki, G.; Plichta, A.; Grzywacz, D. Biodegradable macromolecular conjugates of citropin: Synthesis, characterization and in vitro efficiency study. React. Funct. Polym. 2014, 83, 54–61. [Google Scholar] [CrossRef]
- Alexis, F. Factors affecting the degradation and drug-release mechanism of poly (lactic acid) and poly [(lactic acid)-co-(glycolic acid)]. Polym. Int. 2005, 54, 36–46. [Google Scholar] [CrossRef]
- Siepmann, J.; Göpferich, A. Mathematical modeling of bioerodible, polymeric drug delivery systems. Adv. Drug Deliv. Rev. 2001, 48, 229–247. [Google Scholar] [CrossRef]
- Dash, S.; Murthy, P.N.; Nath, L.; Chowdhury, P. Kinetic modeling on drug release from controlled drug delivery systems. Acta Pol. Pharm. 2010, 67, 217–223. [Google Scholar] [PubMed]
- Avrahami, D.; Shai, Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J. Biol. Chem. 2004, 279, 12277–12285. [Google Scholar] [CrossRef] [PubMed]
- De la Ossa, D.H.P.; Ligresti, A.; Gil-Alegre, M.; Aberturas, M.; Molpeceres, J.; Di Marzo, V.; Suárez, A.T. Poly-ε-caprolactone microspheres as a drug delivery system for cannabinoid administration: Development, characterization and in vitro evaluation of their antitumoral efficacy. J. Control. Release 2012, 161, 927–932. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, H.P.; Cesar, C.L.; Maria de Lourdes Barjas-Castro, M. Electrical properties of the red blood cell membrane and immunohematological investigation. Rev. Bras. Hematol. Hemoter. 2011, 33, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Badri, W.; Miladi, K.; Robin, S.; Viennet, C.; Nazari, Q.A.; Agusti, G.; Fessi, H.; Elaissari, A. Polycaprolactone Based Nanoparticles Loaded with Indomethacin for Anti-Inflammatory Therapy: From Preparation to Ex Vivo Study. Pharm. Res. 2017, 34, 1773–1783. [Google Scholar] [CrossRef] [PubMed]
- Sinha, V.R.; Bansal, K.; Kaushik, R.; Kumria, R.; Trehan, A. Poly-epsilon-caprolactone microspheres and nanospheres: An overview. Int. J. Pharm. 2004, 278, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Ito, F.; Fujimori, H.; Makino, K. Incorporation of water-soluble drugs in PLGA microspheres. Colloids Surf. B BioInterf. 2007, 54, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, M.; Zafar, N.; Fessi, H.; Elaissari, A. Double emulsion solvent evaporation techniques used for drug encapsulation. Int. J. Pharm. 2015, 496, 173–190. [Google Scholar] [CrossRef] [PubMed]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer—Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Göpferich, A. Mechanisms of polymer degradation and erosion. In The Biomaterials: Silver Jubilee Compendium; Elsevier: New York, NY, USA, 2006; pp. 117–128. [Google Scholar]
- Uhrich, K.E.; Cannizzaro, S.M.; Langer, R.S.; Shakesheff, K.M. Polymeric systems for controlled drug release. Chem. Rev. 1999, 99, 3181–3198. [Google Scholar] [CrossRef] [PubMed]
- Bacalum, M.; Radu, M. Cationic antimicrobial peptides cytotoxicity on mammalian cells: An analysis using therapeutic index integrative concept. Int. J. Pept. Res. Ther. 2015, 21, 47–55. [Google Scholar] [CrossRef]
- Chen, Y.; Mant, C.T.; Farmer, S.W.; Hancock, R.E.; Vasil, M.L.; Hodges, R.S. Rational design of α-helical antimicrobial peptides with enhanced activities and specificity/therapeutic index. J. Biol. Chem. 2005, 280, 12316–12329. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, A.; Cirioni, O.; Kamysz, W.; Silvestri, C.; Del Prete, M.S.; Licci, A.; D’Amato, G.; Łukasiak, J.; Scalise, G. In vitro activity and killing effect of citropin 1.1 against gram-positive pathogens causing skin and soft tissue infections. Antimicrob. Agents Chemother. 2005, 49, 2507–2509. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mathematical Model | Equation |
|---|---|
| Zero-order model | F = kt |
| First-order model | log F = log F0 – kt·(2.303)−1 |
| Higuchi model | F= kt1/2 |
| Korsmeyer-Peppas model | F=ktn (F < 0.6) |
| Entry | Method 1 | Initial Contain of CIT in PCL (%, w/w) | Particle Size (µm) | Đ2 | Zeta Potential (mV) | EE3 (%) |
|---|---|---|---|---|---|---|
| MP-PCL-1-1 | A | 0 | 3.69 ± 1.88 | 0.22 | −19.3 ± 5.3 | - |
| MP-PCL-1-2 | B | 0 | 5.50 ± 1.70 | 0.25 | −13.1 ± 3.9 | - |
| MP-PCL-1-CIT-0.5 | B | 0.50 | 5.46 ± 0.71 | 0.38 | −16.0 ± 4.3 | 46.8 ± 1.8 |
| MP-PCL-1-3 | B | 0 | 3.90 ± 1.81 | 0.18 | −17.5 ± 4.9 | - |
| MP-PCL-1-CIT-1.0 | B | 1.02 | 4.67 ± 2.10 | 0.23 | −16.8 ± 5.2 | 42.2 ± 1.2 |
| MP-PCL-2-1 | A | 0 | 3.92 ± 2.13 | 0.24 | −15.6 ± 5.7 | - |
| MP-PCL-2-2 | B | 0 | 4.67 ± 2.00 | 0.21 | −12.2 ± 3.5 | - |
| MP-PCL-2-CIT-0.5 | B | 0.51 | 5.06 ± 1.77 | 0.13 | −17.9 ± 5.7 | 51.5 ± 1.4 |
| MP-PCL-2-3 | B | 0 | 4.52 ± 0.89 | 0.13 | −19.7 ± 5.4 | - |
| MP-PCL-2-CIT-1.0 | B | 1.04 | 5.56 ± 1.61 | 0.12 | −19.5 ± 6.4 | 50.5 ± 1.7 |
| No. | Zero Order Model | First Order Model | Higuchi Model | Korsmeyer-Peppas Model | |
|---|---|---|---|---|---|
| R2 | R2 | R2 | R2 | n | |
| MP-PCL-2-CIT-0.5 | - | - | - | - | - |
| Phase I | 0.9708 | 0.9670 | 0.9822 | 0.9848 | 0.79 |
| Phase II | 0.9835 | 0.9689 | 0.9866 | 0.9818 | > 0.85 |
| MP-PCL-2-CIT-1.0 | 0.9878 | 0.8173 | 0.9714 | 0.9893 | > 0.85 |
| MP-PCL-1-CIT-0.5 | 0.9562 | 0.9854 | 0.9887 | 0.9855 | > 0.85 |
| MP-PCL-1-CIT-1.0 | 0.9289 | 0.9814 | 0.9831 | 0.9836 | > 0.85 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piotrowska, U.; Oledzka, E.; Kamysz, W.; Białek, S.; Sobczak, M. The Effect of Polymer Microstructure on Encapsulation Efficiency and Release Kinetics of Citropin 1.1 from the Poly(ε-caprolactone) Microparticles. Nanomaterials 2018, 8, 482. https://doi.org/10.3390/nano8070482
Piotrowska U, Oledzka E, Kamysz W, Białek S, Sobczak M. The Effect of Polymer Microstructure on Encapsulation Efficiency and Release Kinetics of Citropin 1.1 from the Poly(ε-caprolactone) Microparticles. Nanomaterials. 2018; 8(7):482. https://doi.org/10.3390/nano8070482
Chicago/Turabian StylePiotrowska, Urszula, Ewa Oledzka, Wojciech Kamysz, Sławomir Białek, and Marcin Sobczak. 2018. "The Effect of Polymer Microstructure on Encapsulation Efficiency and Release Kinetics of Citropin 1.1 from the Poly(ε-caprolactone) Microparticles" Nanomaterials 8, no. 7: 482. https://doi.org/10.3390/nano8070482
APA StylePiotrowska, U., Oledzka, E., Kamysz, W., Białek, S., & Sobczak, M. (2018). The Effect of Polymer Microstructure on Encapsulation Efficiency and Release Kinetics of Citropin 1.1 from the Poly(ε-caprolactone) Microparticles. Nanomaterials, 8(7), 482. https://doi.org/10.3390/nano8070482