α-MoO3 Crystals with a Multilayer Stack Structure Obtained by Annealing from a Lamellar MoS2/g-C3N4 Nanohybrid



Abstract
:
1. Introduction
2. Materials and Methods
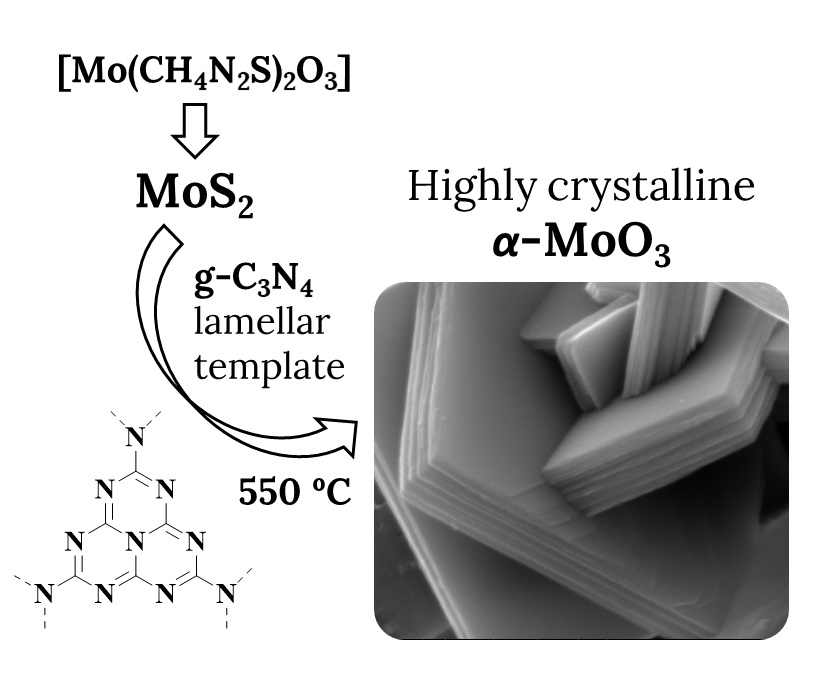
2.1. Reagents and Synthesis
2.1.1. Synthesis of MoS2
2.1.2. Synthesis of MoS2/g-C3N4
2.1.3. Synthesis of MoO3
2.2. Characterization
3. Results and Discussion
3.1. Vibrational Characterization
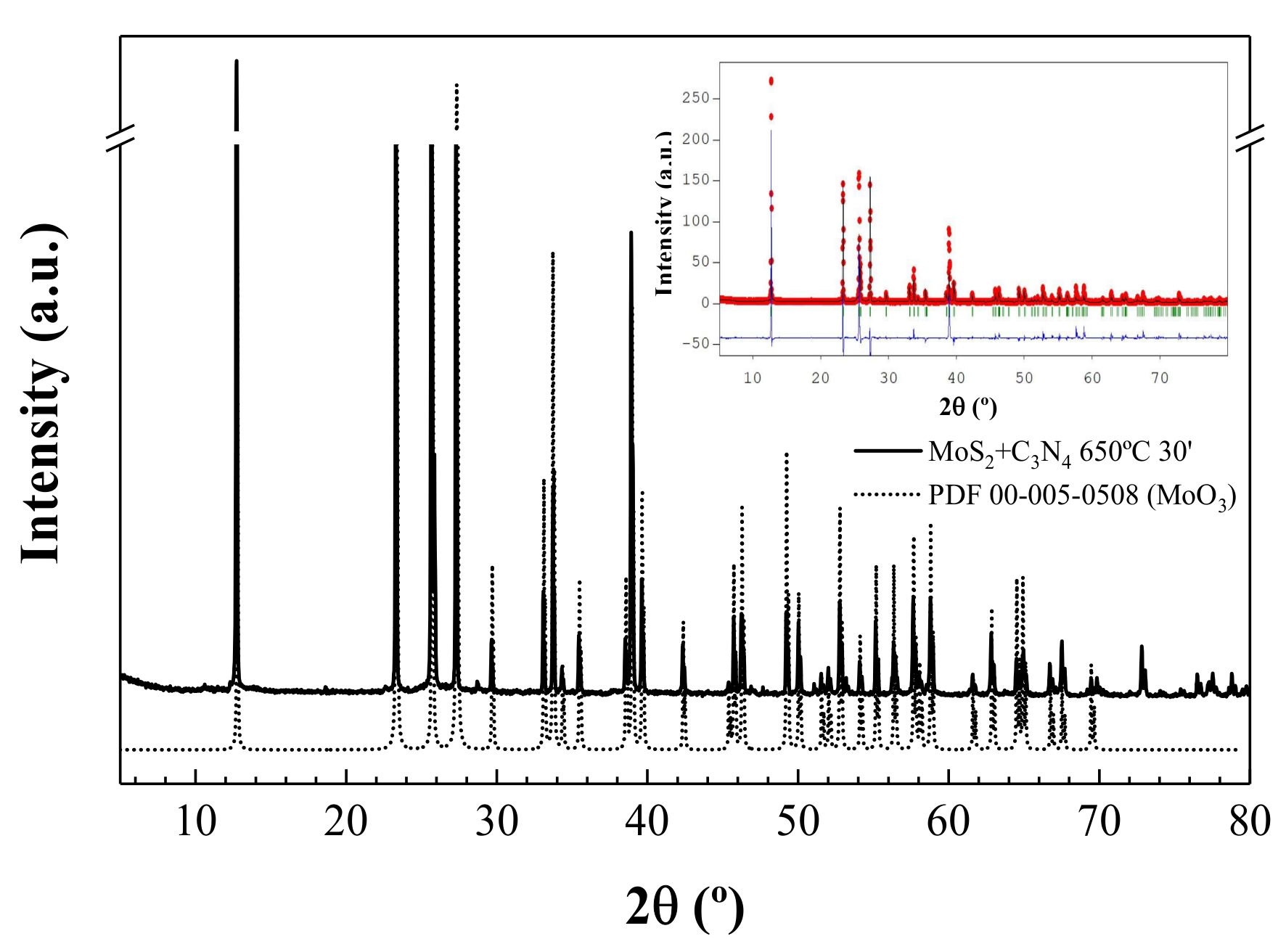
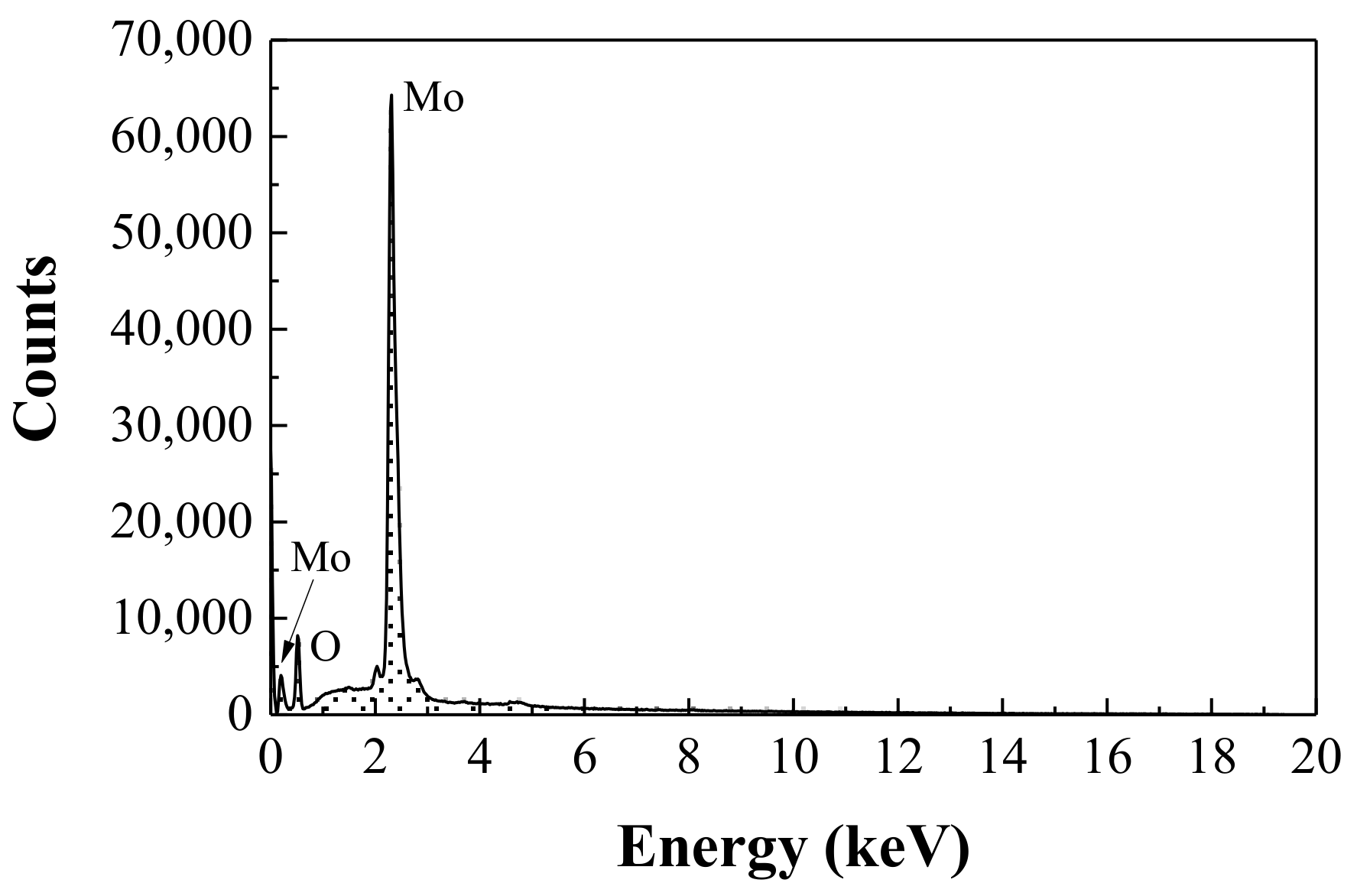
3.2. X-Ray Powder Diffraction and Energy-Dispersive X-Ray Spectroscopy Analyses
3.3. Scanning and Transmission Electron Microscopy Analyses
3.4. Surface Characterization
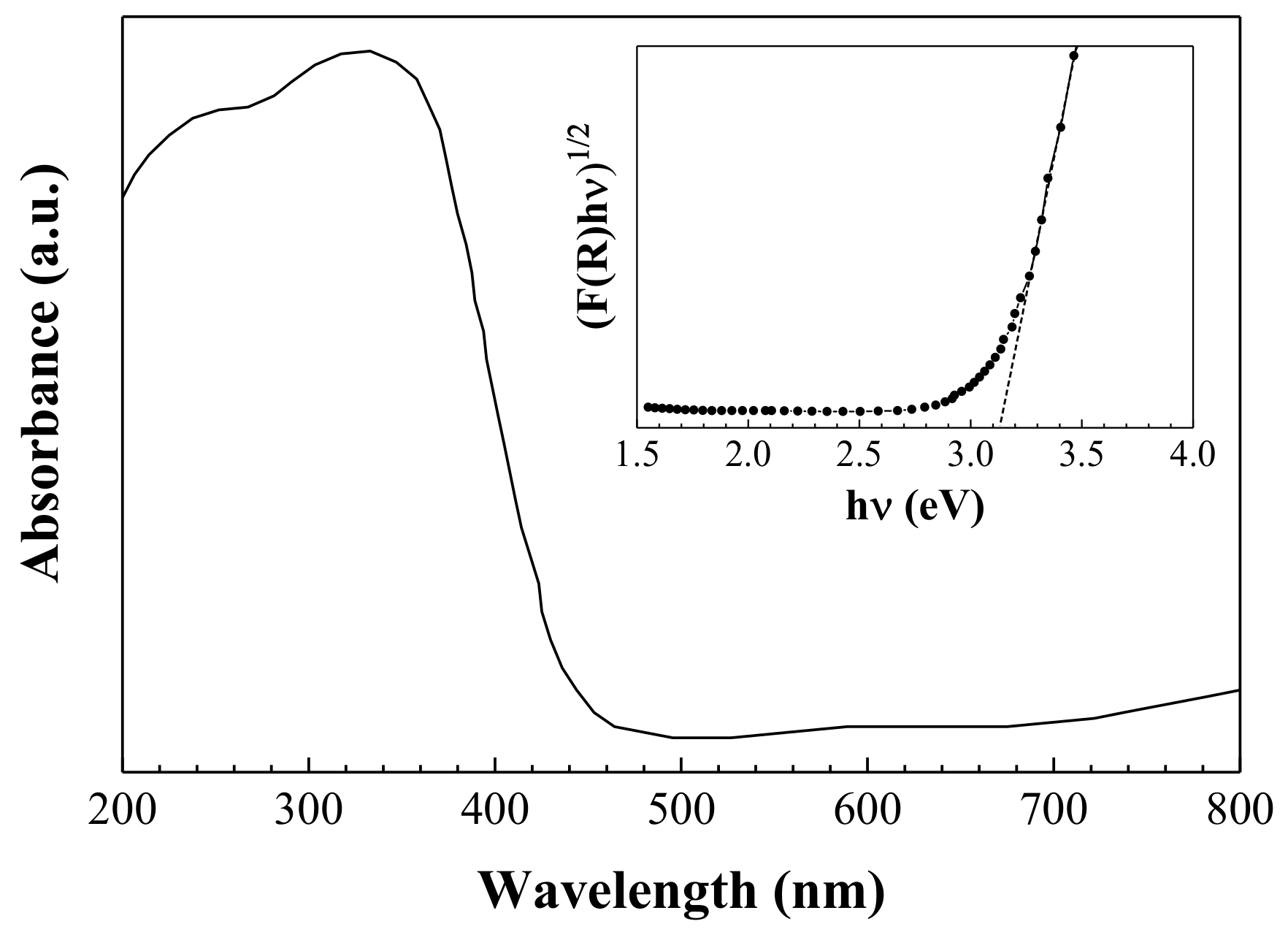
3.5. Optical Properties
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- de Castro, I.A.; Datta, R.S.; Ou, J.Z.; Castellanos-Gomez, A.; Sriram, S.; Daeneke, T.; Kalantar-zadeh, K. Molybdenum oxides—From fundamentals to functionality. Adv. Mater. 2017, 29. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.; Hamwi, S.; Kröger, M.; Kowalsky, W.; Riedl, T.; Kahn, A. Transition metal oxides for organic electronics: Energetics, device physics and applications. Adv. Mater. 2012, 24, 5408–5427. [Google Scholar] [CrossRef] [PubMed]
- Qu, Q.; Zhang, W.-B.; Huang, K.; Chen, H.-M. Electronic structure, optical properties and band edges of layered MoO3: A first-principles investigation. Comput. Mater. Sci. 2017, 130, 242–248. [Google Scholar] [CrossRef]
- Alsaif, M.M.Y.A.; Chrimes, A.F.; Daeneke, T.; Balendhran, S.; Bellisario, D.O.; Son, Y.; Field, M.R.; Zhang, W.; Nili, H.; Nguyen, E.P.; et al. High-performance field effect transistors using electronic inks of 2D molybdenum oxide nanoflakes. Adv. Funct. Mater. 2016, 26, 91–100. [Google Scholar] [CrossRef]
- Balendhran, S.; Deng, J.; Ou, J.Z.; Walia, S.; Scott, J.; Tang, J.; Wang, K.L.; Field, M.R.; Russo, S.; Zhuiykov, S.; et al. Enhanced charge carrier mobility in two-dimensional high dielectric molybdenum oxide. Adv. Mater. 2013, 25, 109–114. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-zadeh, K.; Tang, J.; Wang, M.; Wang, K.L.; Shailos, A.; Galatsis, K.; Kojima, R.; Strong, V.; Lech, A.; Wlodarski, W.; et al. Synthesis of nanometre-thick MoO3 sheets. Nanoscale 2010, 2, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Ji, F.; Ren, X.; Zheng, X.; Liu, Y.; Pang, L.; Jiang, J.; Liu, S. 2D-MoO3 nanosheets for superior gas sensors. Nanoscale 2016, 8, 8696–8703. [Google Scholar] [CrossRef] [PubMed]
- Rathnasamy, R.; Thangamuthu, R.; Alagan, V. Sheet-like orthorhombic MoO3 nanostructures prepared via hydrothermal approach for visible-light-driven photocatalytic application. Res. Chem. Intermed. 2017, 44, 1647–1660. [Google Scholar] [CrossRef]
- Shahab ud, D.; Ahmad, M.Z.; Qureshi, K.; Bhatti, I.A.; Zahid, M.; Nisar, J.; Iqbal, M.; Abbas, M. Hydrothermal synthesis of molybdenum trioxide, characterization and photocatalytic activity. Mater. Res. Bull. 2018, 100, 120–130. [Google Scholar] [CrossRef]
- Yao, D.D.; Ou, J.Z.; Latham, K.; Zhuiykov, S.; O’Mullane, A.P.; Kalantar-zadeh, K. Electrodeposited α- and β-Phase MoO3 Films and Investigation of Their Gasochromic Properties. Cryst. Growth Des. 2012, 12, 1865–1870. [Google Scholar] [CrossRef]
- Chang, W.-C.; Qi, X.; Kuo, J.-C.; Lee, S.-C.; Ng, S.-K.; Chen, D. Post-deposition annealing control of phase and texture for the sputtered MoO3 films. CrystEngComm 2011, 13, 5125–5132. [Google Scholar] [CrossRef]
- Datta, R.S.; Haque, F.; Mohiuddin, M.; Carey, B.J.; Syed, N.; Zavabeti, A.; Zhang, B.; Khan, H.; Berean, K.J.; Ou, J.Z.; et al. Highly active two dimensional α-MoO3−x for the electrocatalytic hydrogen evolution reaction. J. Mater. Chem. A 2017, 5, 24223–24231. [Google Scholar] [CrossRef]
- Zhang, B.Y.; Zavabeti, A.; Chrimes, A.F.; Haque, F.; O’Dell, L.A.; Khan, H.; Syed, N.; Datta, R.; Wang, Y.; Chesman, A.S.R.; et al. Degenerately hydrogen doped molybdenum oxide nanodisks for ultrasensitive plasmonic biosensing. Adv. Funct. Mater. 2018, 28, 1706006. [Google Scholar] [CrossRef]
- Dante, R.C.; Martín-Ramos, P.; Correa-Guimaraes, A.; Martín-Gil, J. Synthesis of graphitic carbon nitride by reaction of melamine and uric acid. Mater. Chem. Phys. 2011, 130, 1094–1102. [Google Scholar] [CrossRef]
- Vikraman, D.; Akbar, K.; Hussain, S.; Yoo, G.; Jang, J.-Y.; Chun, S.-H.; Jung, J.; Park, H.J. Direct synthesis of thickness-tunable MoS2 quantum dot thin layers: Optical, structural and electrical properties and their application to hydrogen evolution. Nano Energy 2017, 35, 101–114. [Google Scholar] [CrossRef]
- Vattikuti, S.V.P.; Byon, C. Synthesis and Characterization of Molybdenum Disulfide Nanoflowers and Nanosheets: Nanotribology. J. Nanomater. 2015, 2015, 1–11. [Google Scholar] [CrossRef]
- Wang, J.; Guan, Z.; Huang, J.; Li, Q.; Yang, J. Enhanced photocatalytic mechanism for the hybrid g-C3N4/MoS2 nanocomposite. J. Mater. Chem. A 2014, 2, 7960–7966. [Google Scholar] [CrossRef]
- Li, J.; Liu, E.; Ma, Y.; Hu, X.; Wan, J.; Sun, L.; Fan, J. Synthesis of MoS2/g-C3N4 nanosheets as 2D heterojunction photocatalysts with enhanced visible light activity. Appl. Surf. Sci. 2016, 364, 694–702. [Google Scholar] [CrossRef]
- Ge, L.; Han, C.; Xiao, X.; Guo, L. Synthesis and characterization of composite visible light active photocatalysts MoS2–g-C3N4 with enhanced hydrogen evolution activity. Int. J. Hydrogen Energy 2013, 38, 6960–6969. [Google Scholar] [CrossRef]
- Kharlamov, A.; Bondarenko, M.; Kharlamova, G.; Gubareni, N. Features of the synthesis of carbon nitride oxide (g-C3N4)O at urea pyrolysis. Diamond Relat. Mater. 2016, 66, 16–22. [Google Scholar] [CrossRef]
- Nagaraju, G.; Tharamani, C.N.; Chandrappa, G.T.; Livage, J. Hydrothermal synthesis of amorphous MoS2 nanofiber bundles via acidification of ammonium heptamolybdate tetrahydrate. Nanoscale Res. Lett. 2007, 2, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Siciliano, T.; Tepore, A.; Filippo, E.; Micocci, G.; Tepore, M. Characteristics of molybdenum trioxide nanobelts prepared by thermal evaporation technique. Mater. Chem. Phys. 2009, 114, 687–691. [Google Scholar] [CrossRef]
- Rodríguez-Carvajal, J. Recent developments of the program FULLPROF. Comm. Powder Diffr. (IUCr) Newslett. 2001, 26, 12–19. [Google Scholar]
- Wang, T.; Li, J.; Zhao, G. Synthesis of MoS2 and MoO3 hierarchical nanostructures using a single-source molecular precursor. Powder Technol. 2014, 253, 347–351. [Google Scholar] [CrossRef]
- Lou, S.N.; Yap, N.; Scott, J.; Amal, R.; Ng, Y.H. Influence of MoO3 (110) crystalline plane on its self-charging photoelectrochemical properties. Sci. Rep. 2014, 4, 7428. [Google Scholar] [CrossRef] [PubMed]
- Vila, M.; Díaz-Guerra, C.; Jerez, D.; Lorenz, K.; Piqueras, J.; Alves, E. Intense luminescence emission from rare-earth-doped MoO3 nanoplates and lamellar crystals for optoelectronic applications. J. Phys. D Appl. Phys. 2014, 47, 35. [Google Scholar] [CrossRef]
- Wongkrua, P.; Thongtem, T.; Thongtem, S. Synthesis of h- and α-MoO3 by refluxing and calcination combination: Phase and morphology transformation, photocatalysis, and photosensitization. J. Nanomater. 2013, 2013, 1–8. [Google Scholar] [CrossRef]
- Klinbumrung, A.; Thongtem, T.; Thongtem, S. Characterization of orthorhombic α-MoO3 microplates produced by a microwave plasma process. J. Nanomater. 2012, 2012, 1–5. [Google Scholar] [CrossRef]
- Xia, T.; Li, Q.; Liu, X.; Meng, J.; Cao, X. Morphology-controllable synthesis and characterization of single-crystal molybdenum trioxide. J. Phys. Chem. B 2006, 110, 2006–2012. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.K.S.; Dewangan, K.; Gajbhiye, N.S. Synthesis and room temperature d0 ferromagnetic properties of α-MoO3 nanofibers. J. Mater. Sci. Technol. 2015, 31, 453–457. [Google Scholar] [CrossRef]
- Chithambararaj, A.; Bose, A.C. Hydrothermal synthesis of hexagonal and orthorhombic MoO3 nanoparticles. J. Alloys Compd. 2011, 509, 8105–8110. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Element | Series | [wt.%] | [norm. wt.%] | [norm. at.%] | Error in wt.% (3σ) |
|---|---|---|---|---|---|
| Oxygen | K-series | 25.473 | 36.624 | 77.403 | 9.409 |
| Aluminum | K-series | 0.070 | 0.100 | 0.126 | 0.089 |
| Sulfur * | K-series | 0.172 | 0.247 | 0.124 | 0.097 |
| Molybdenum | L-series | 43.839 | 63.029 | 22.210 | 4.732 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martín-Ramos, P.; Fernández-Coppel, I.A.; Avella, M.; Martín-Gil, J. α-MoO3 Crystals with a Multilayer Stack Structure Obtained by Annealing from a Lamellar MoS2/g-C3N4 Nanohybrid. Nanomaterials 2018, 8, 559. https://doi.org/10.3390/nano8070559
Martín-Ramos P, Fernández-Coppel IA, Avella M, Martín-Gil J. α-MoO3 Crystals with a Multilayer Stack Structure Obtained by Annealing from a Lamellar MoS2/g-C3N4 Nanohybrid. Nanomaterials. 2018; 8(7):559. https://doi.org/10.3390/nano8070559
Chicago/Turabian StyleMartín-Ramos, Pablo, Ignacio A. Fernández-Coppel, Manuel Avella, and Jesús Martín-Gil. 2018. "α-MoO3 Crystals with a Multilayer Stack Structure Obtained by Annealing from a Lamellar MoS2/g-C3N4 Nanohybrid" Nanomaterials 8, no. 7: 559. https://doi.org/10.3390/nano8070559
APA StyleMartín-Ramos, P., Fernández-Coppel, I. A., Avella, M., & Martín-Gil, J. (2018). α-MoO3 Crystals with a Multilayer Stack Structure Obtained by Annealing from a Lamellar MoS2/g-C3N4 Nanohybrid. Nanomaterials, 8(7), 559. https://doi.org/10.3390/nano8070559