In Situ X-ray Photoelectron Spectroscopic and Electrochemical Studies of the Bromide Anions Dissolved in 1-Ethyl-3-Methyl Imidazolium Tetrafluoroborate


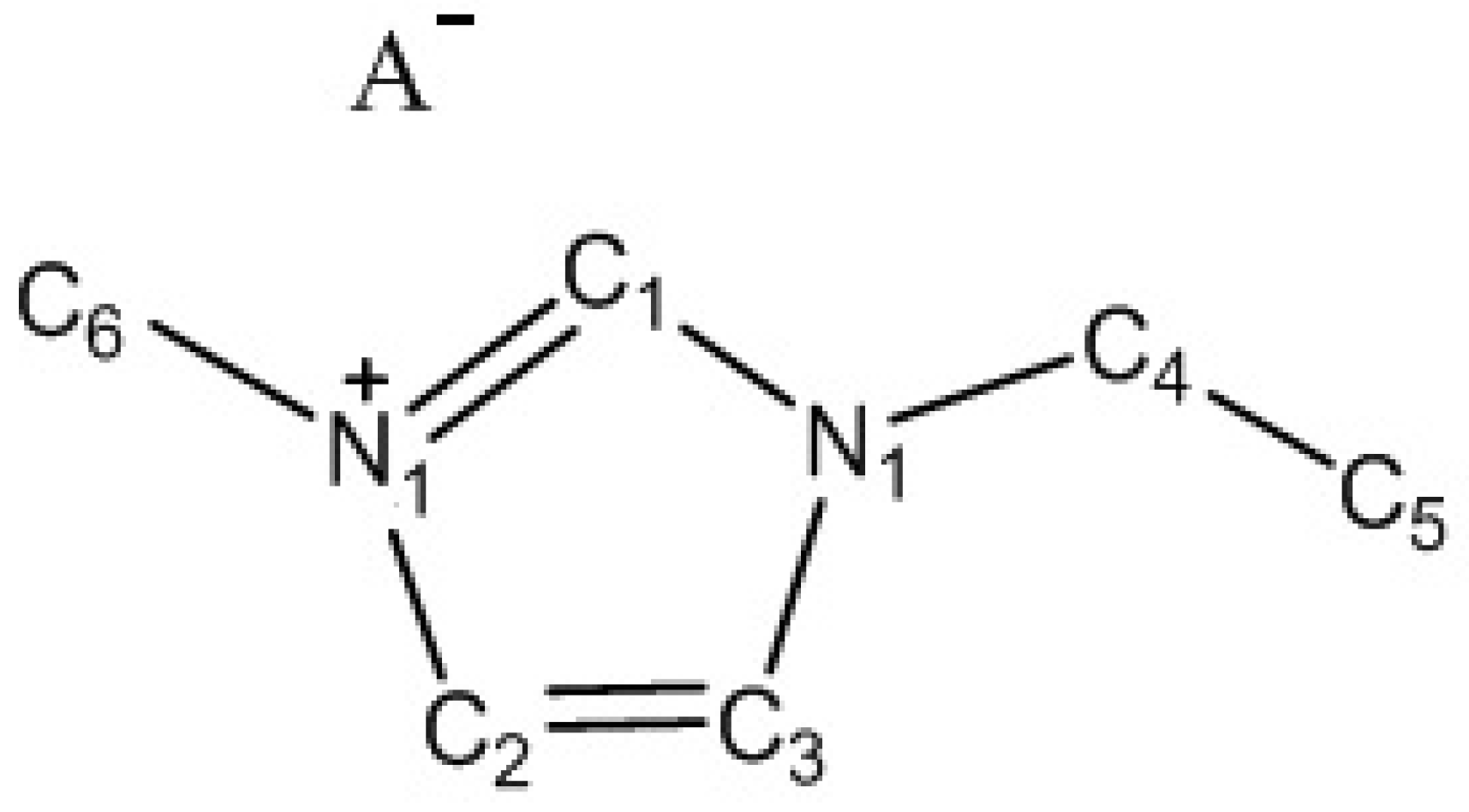{kind=link}
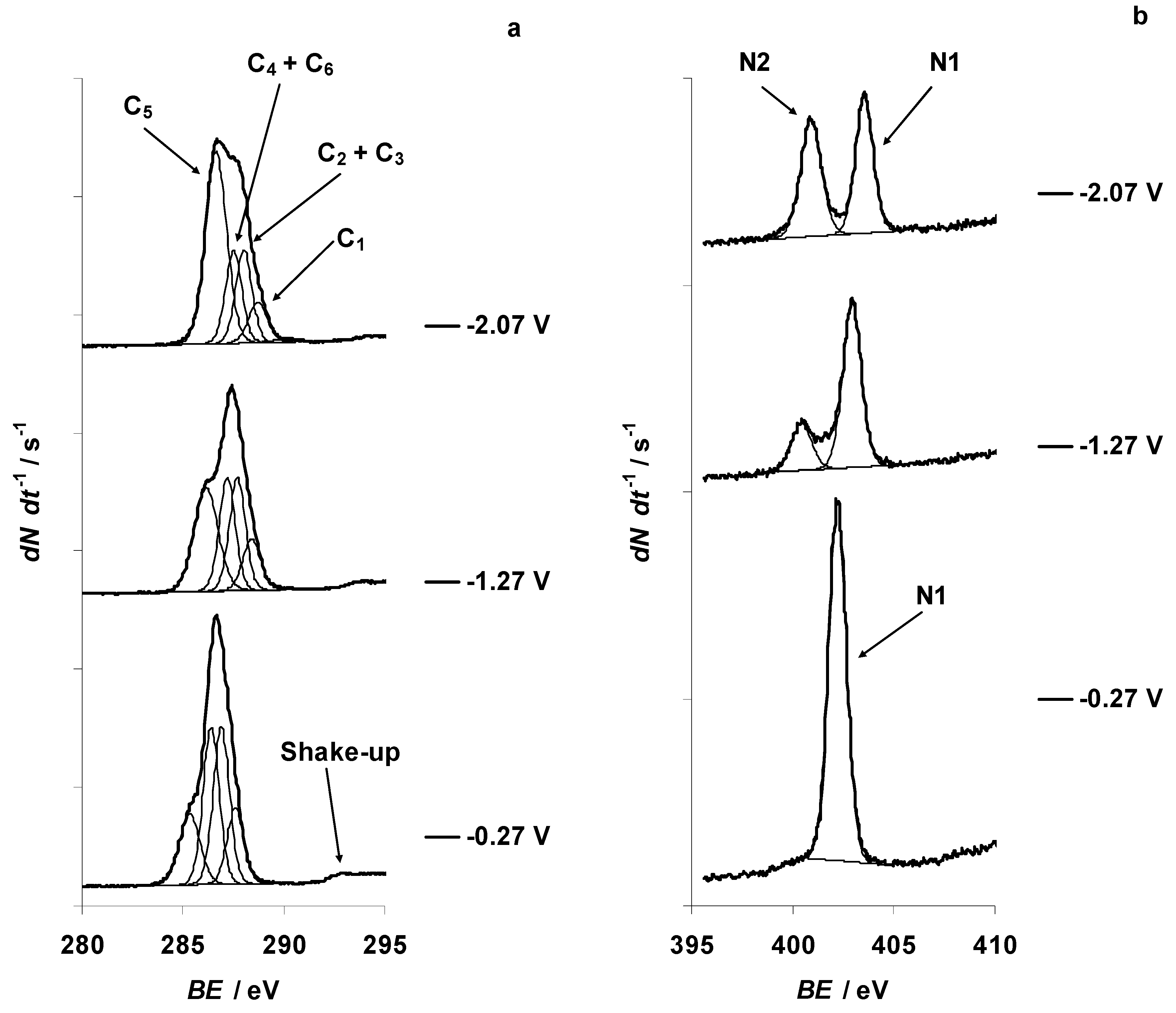{kind=link}
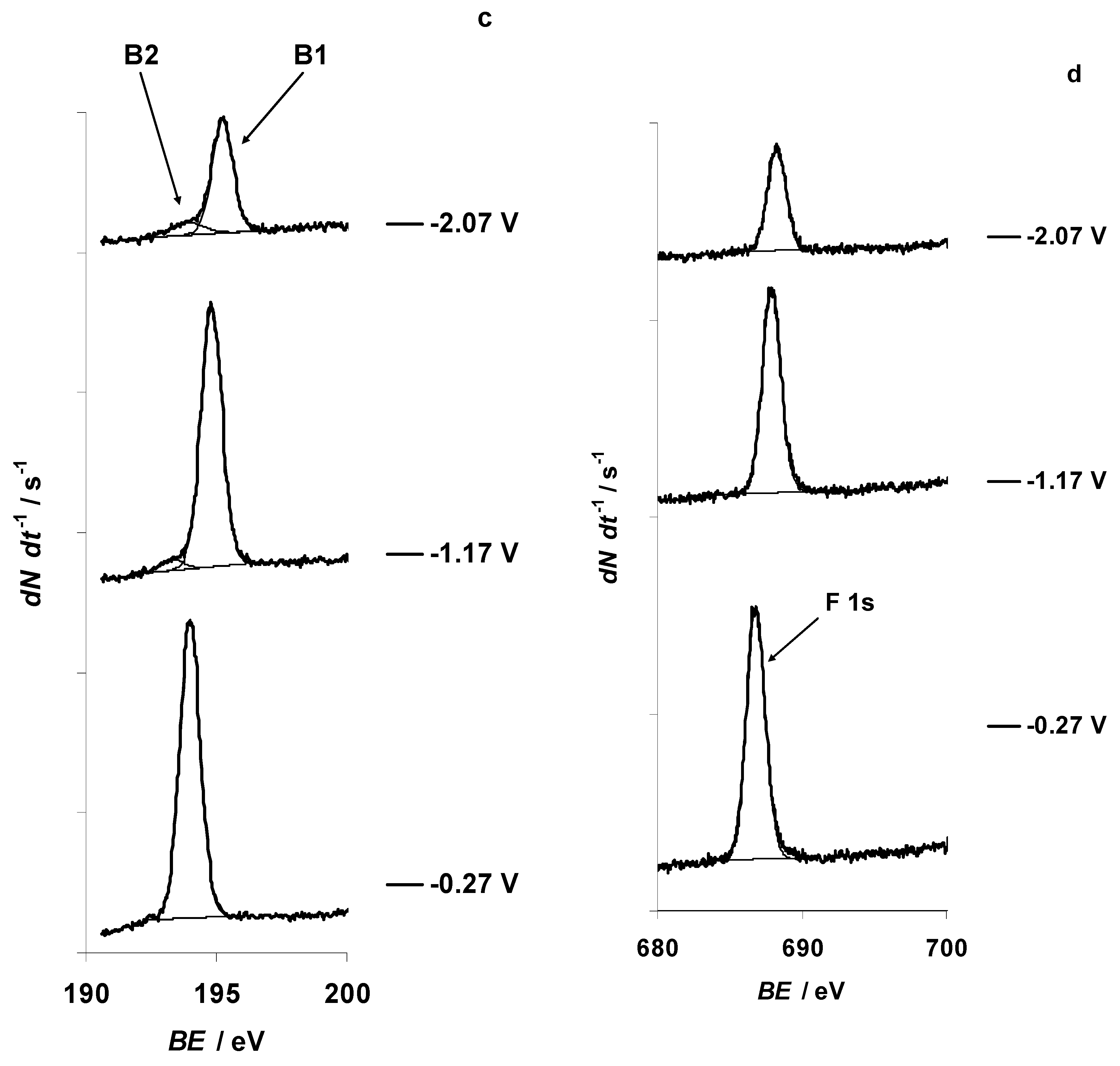{kind=link}
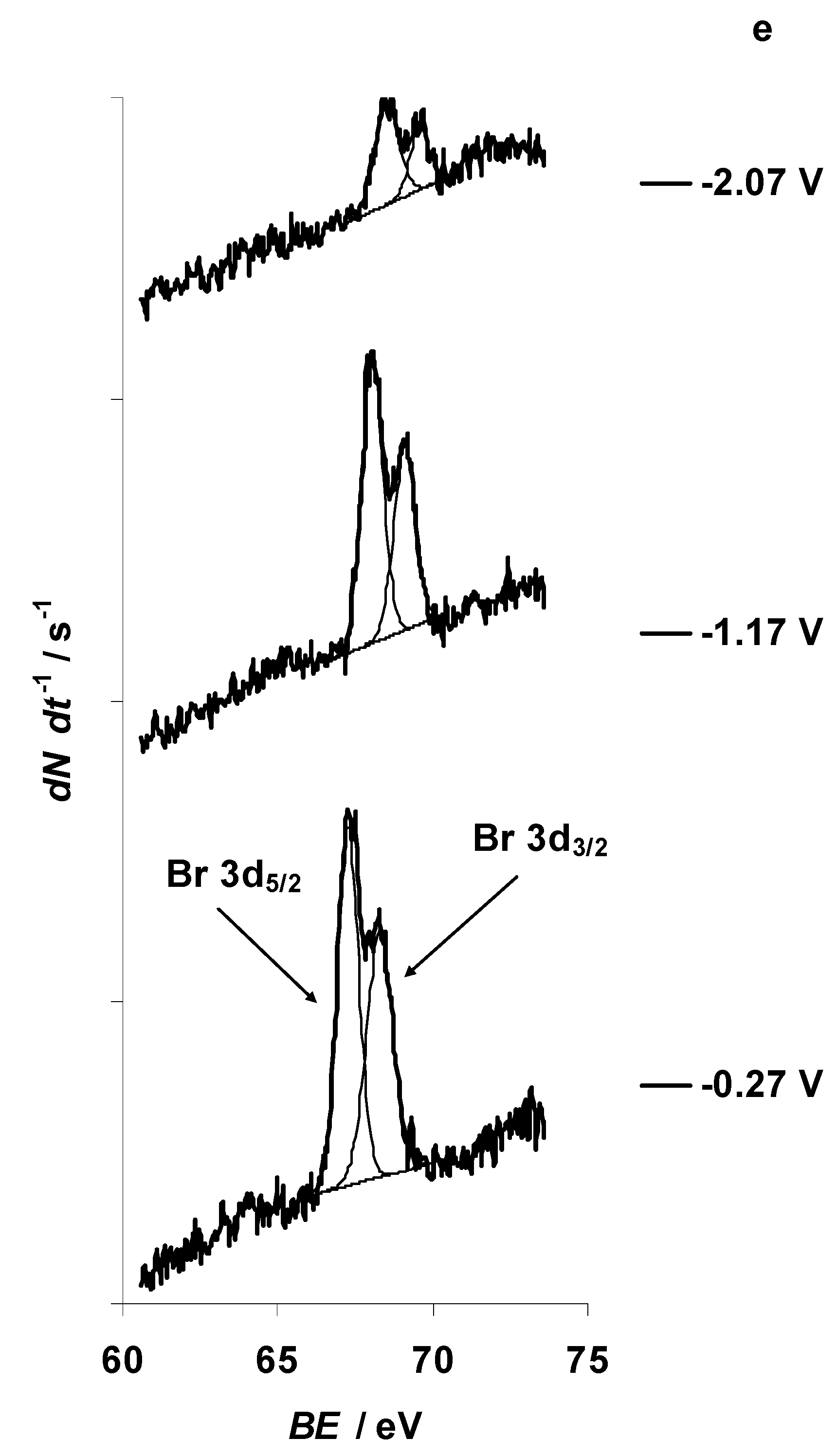{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
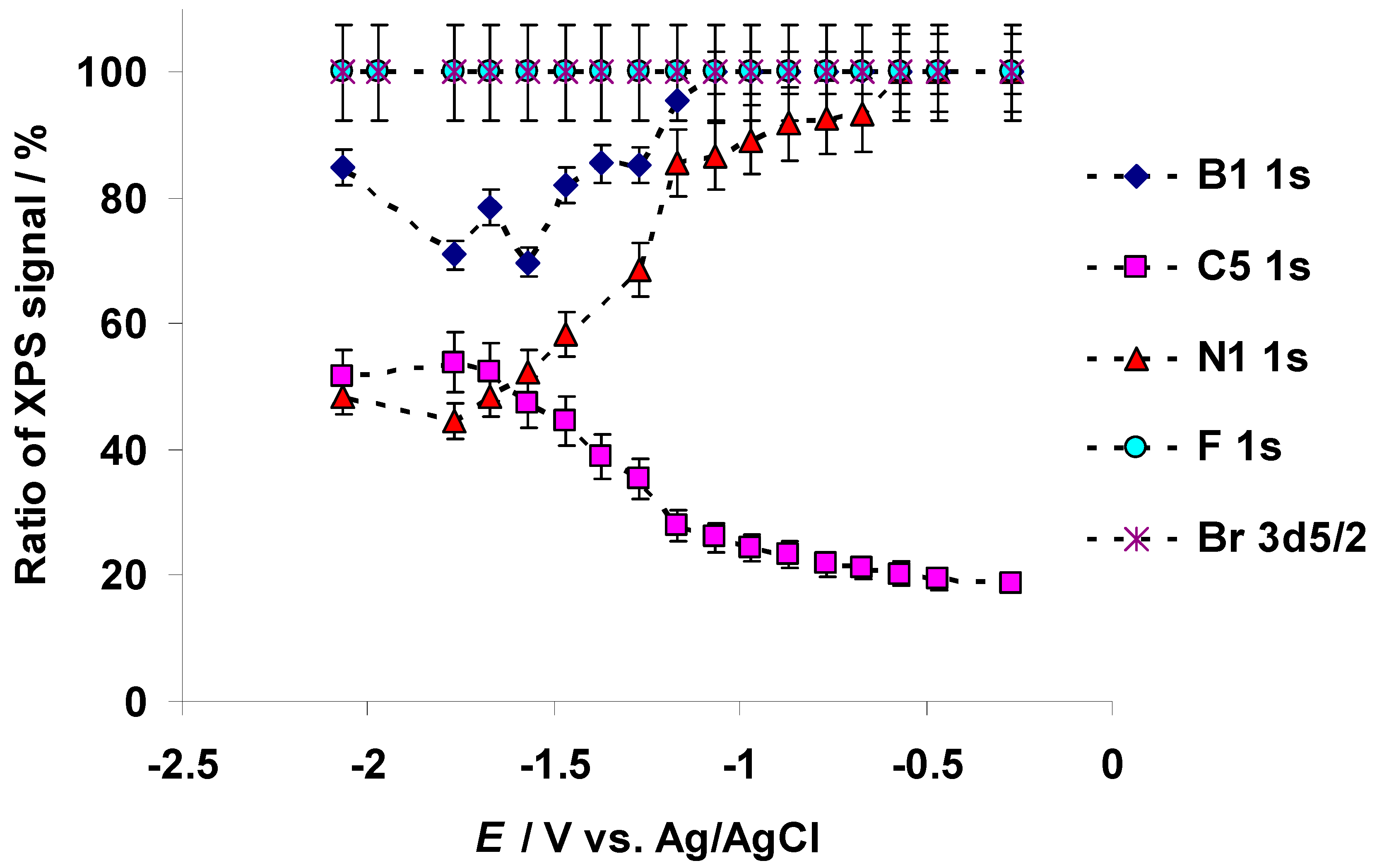
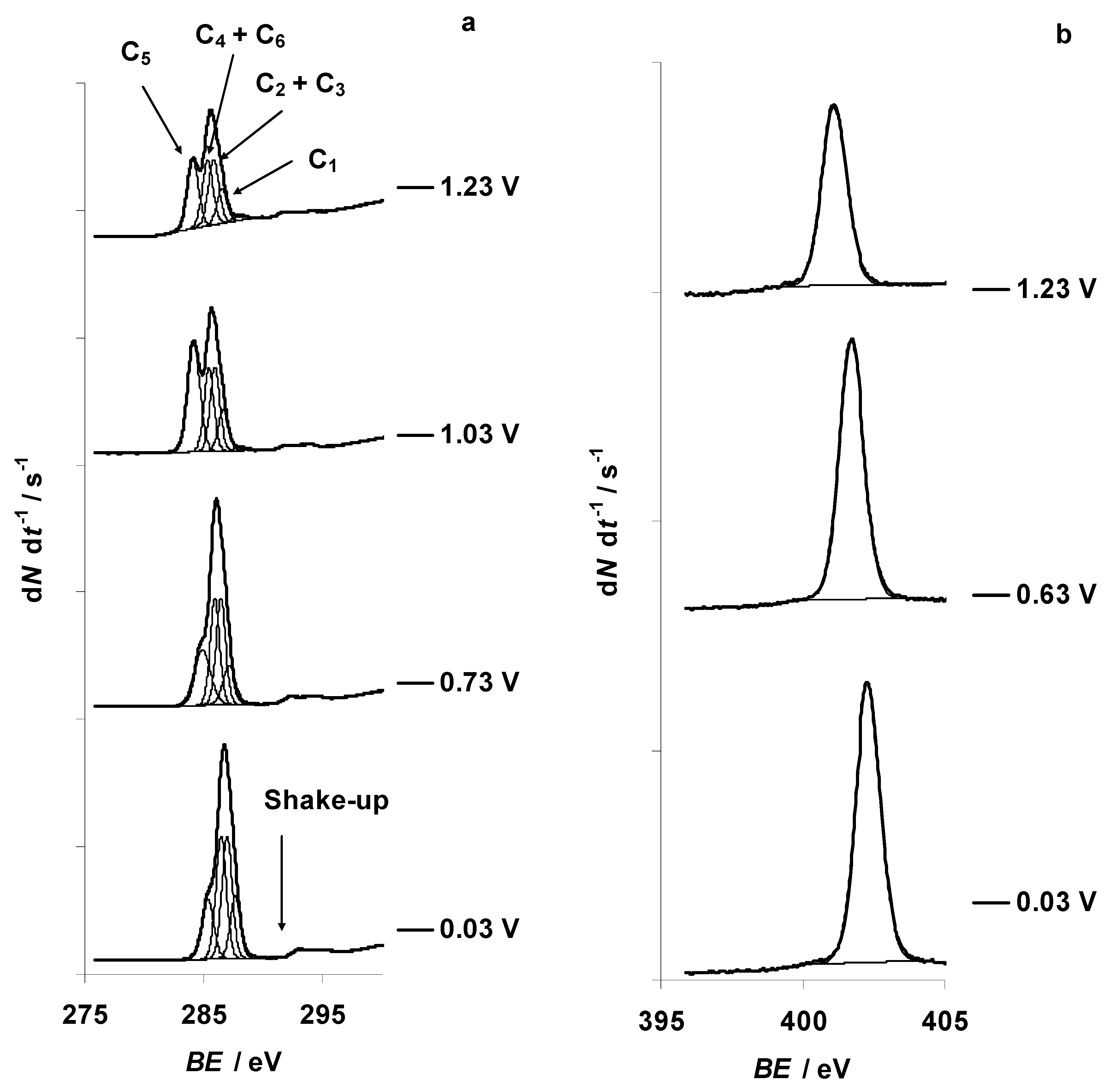
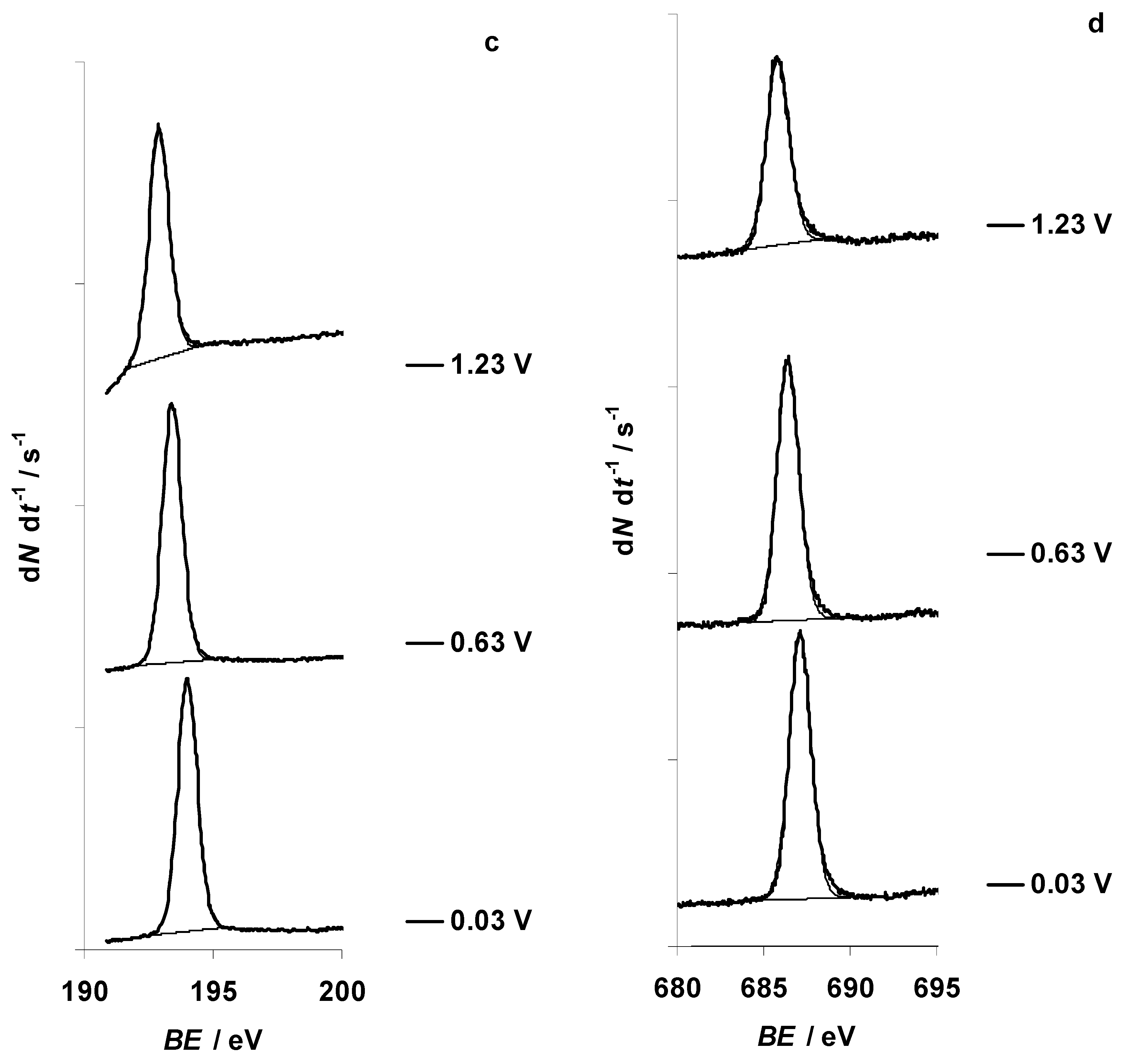
3.1. Characteristic Changes in C 1s, N 1s, B 1s, F 1s, and Br 3d In Situ X-ray Spectra Obtained at the Negatively Polarized C(Mo2C) Electrode
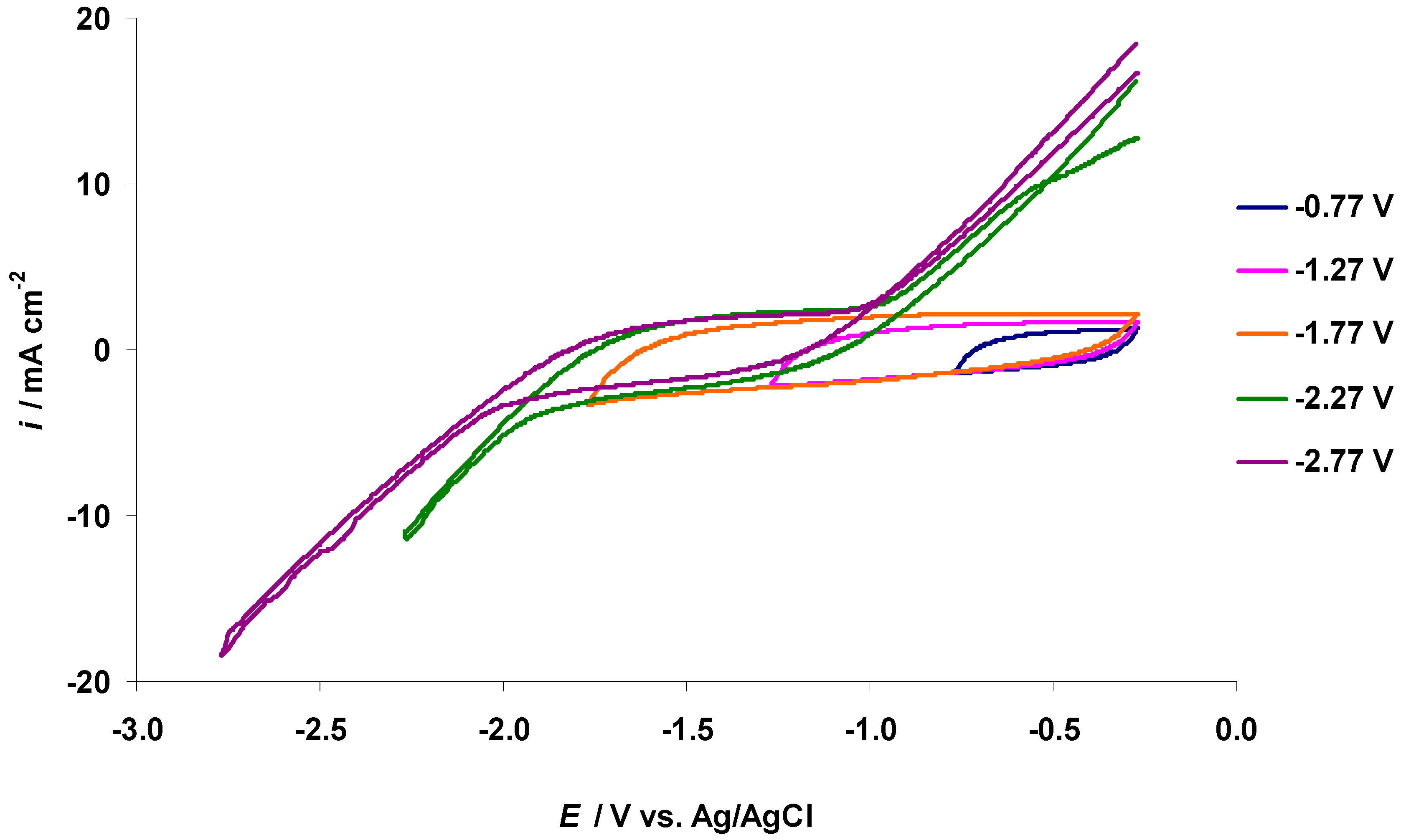
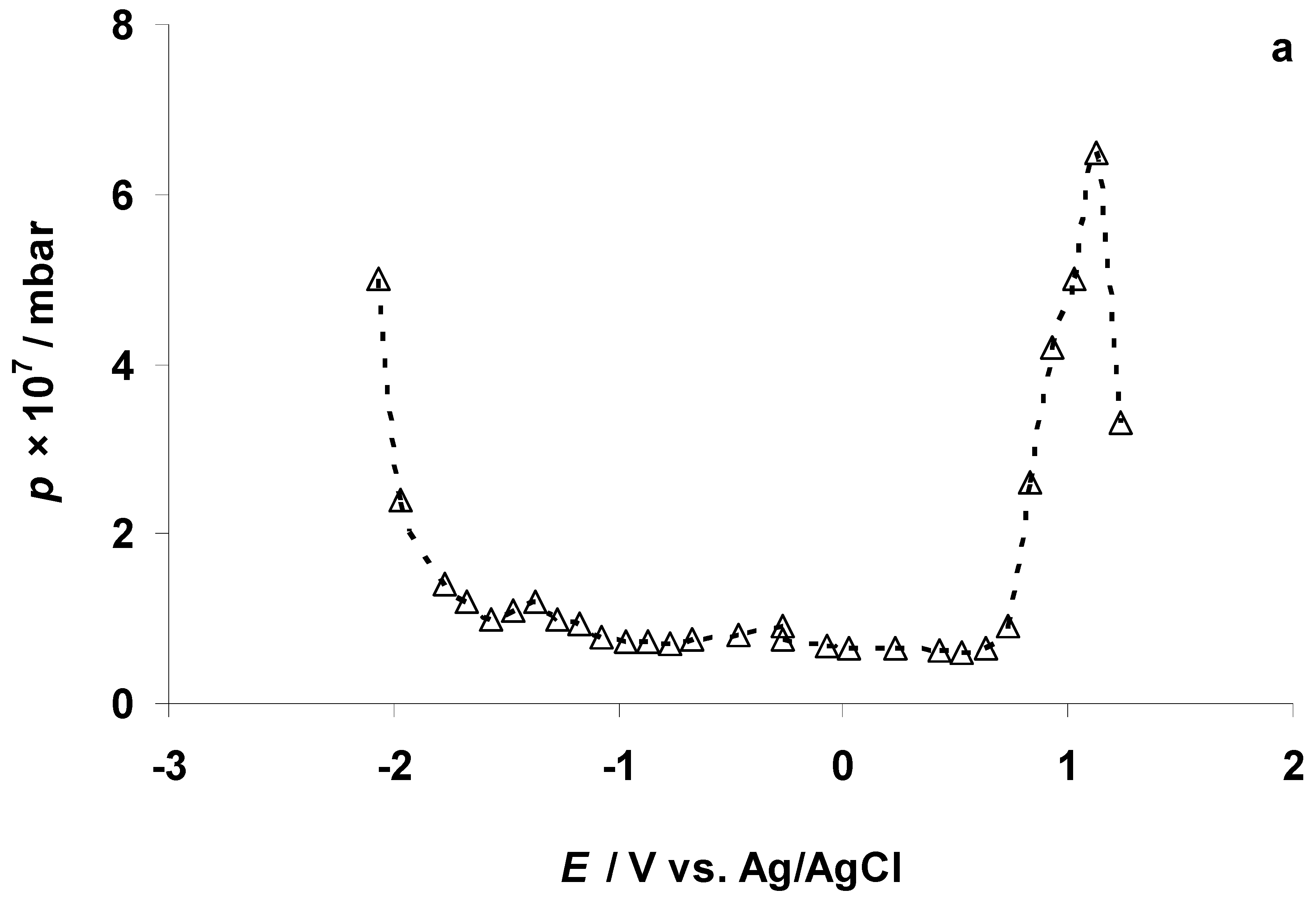
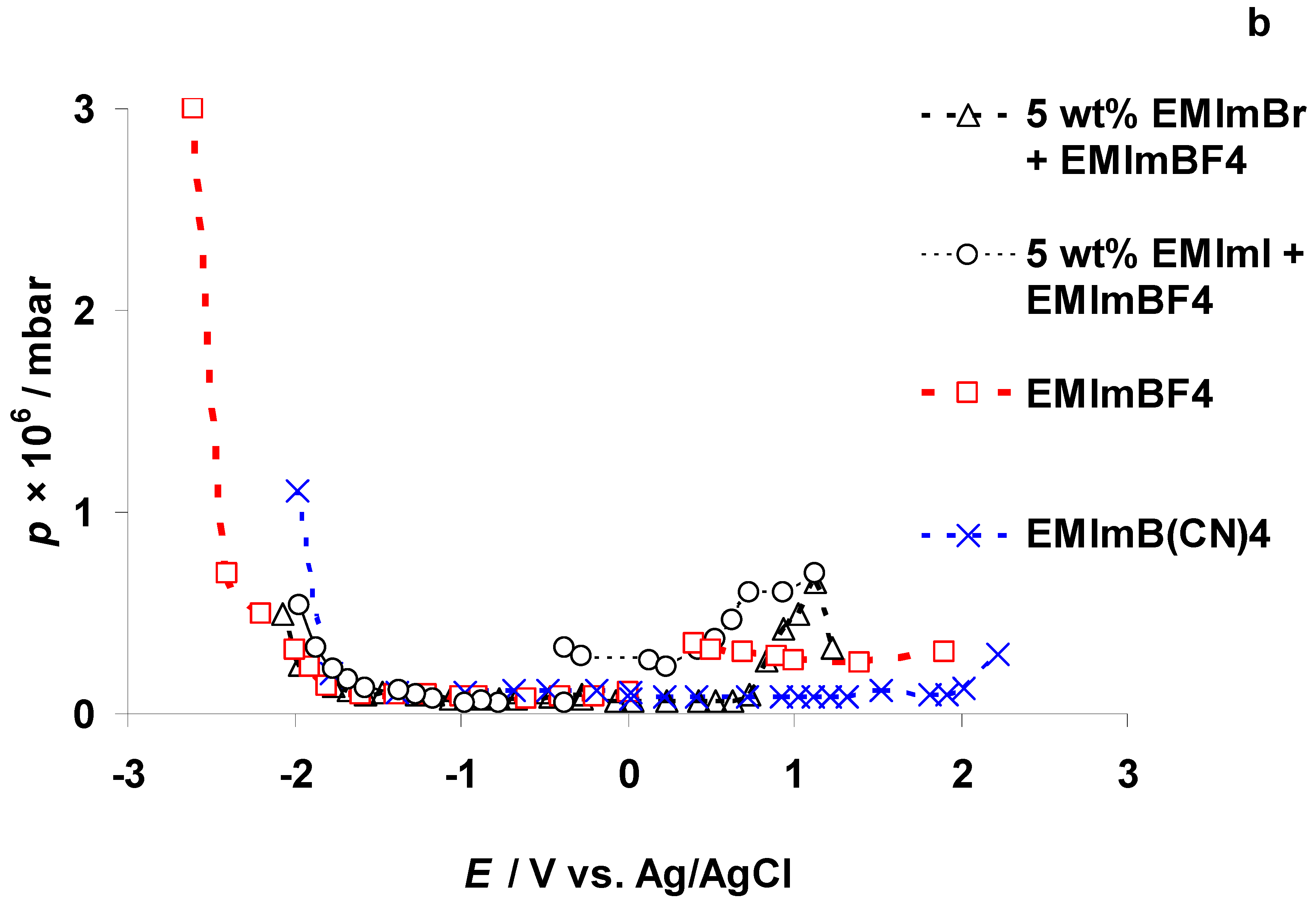
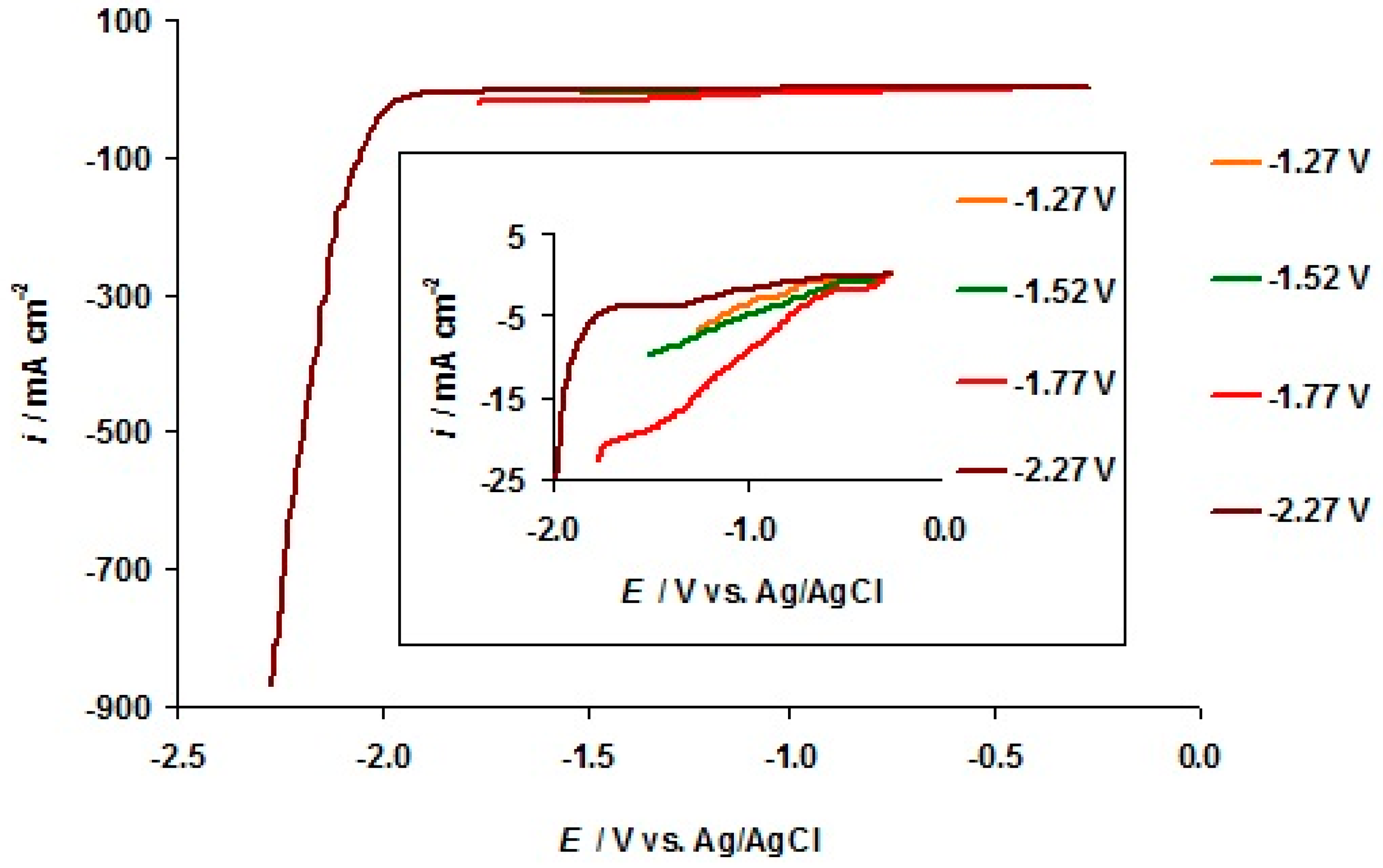
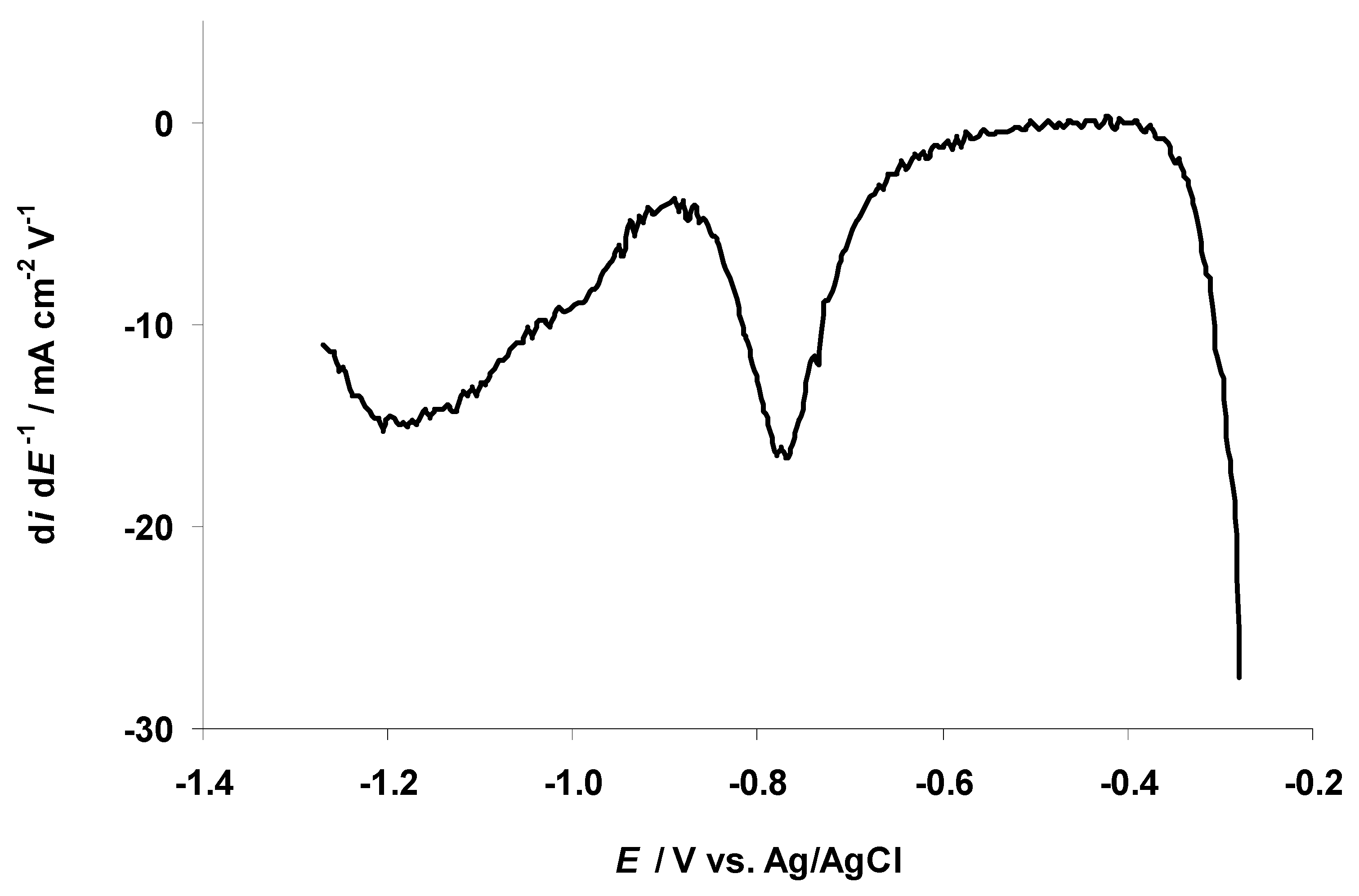
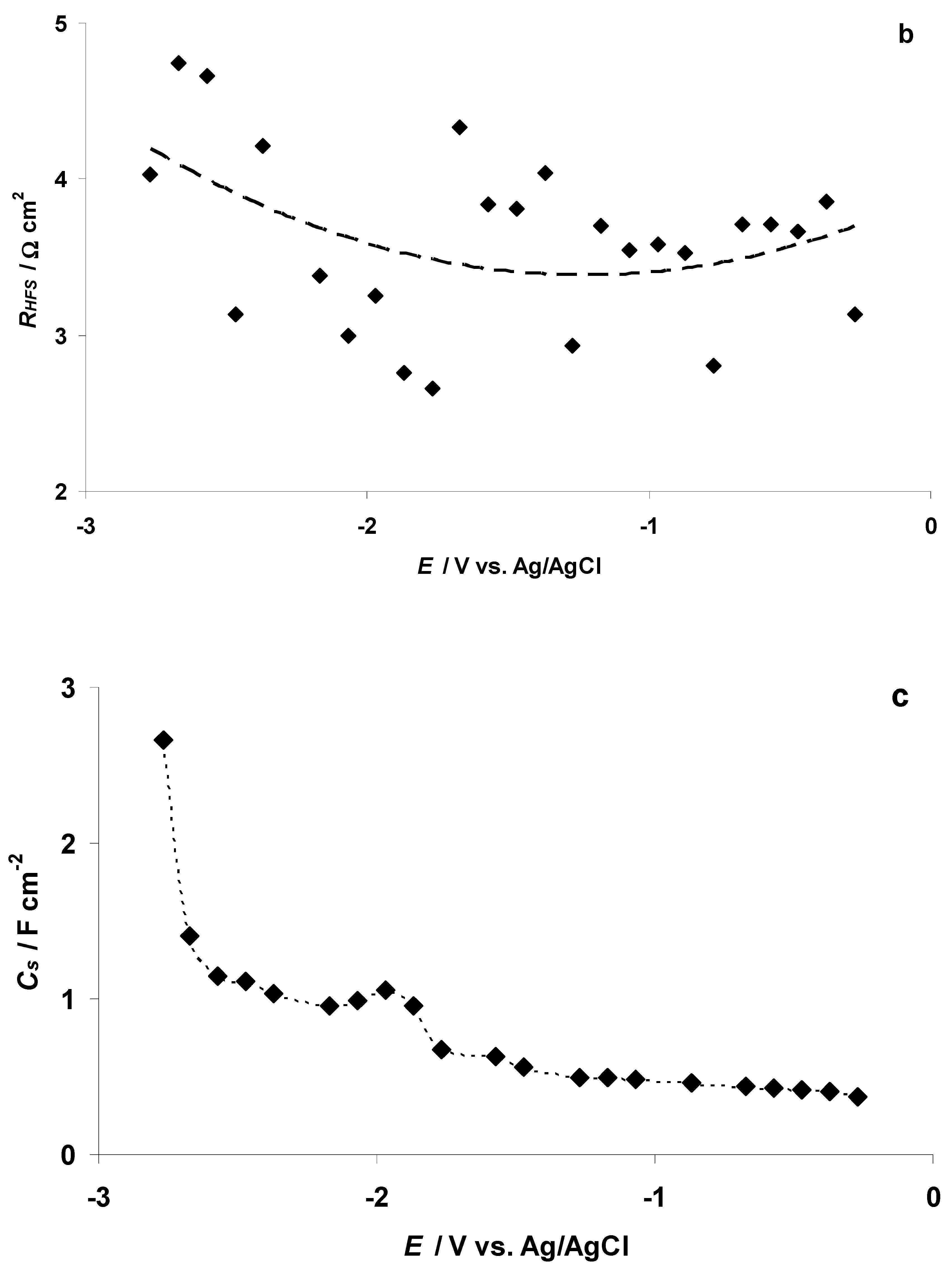
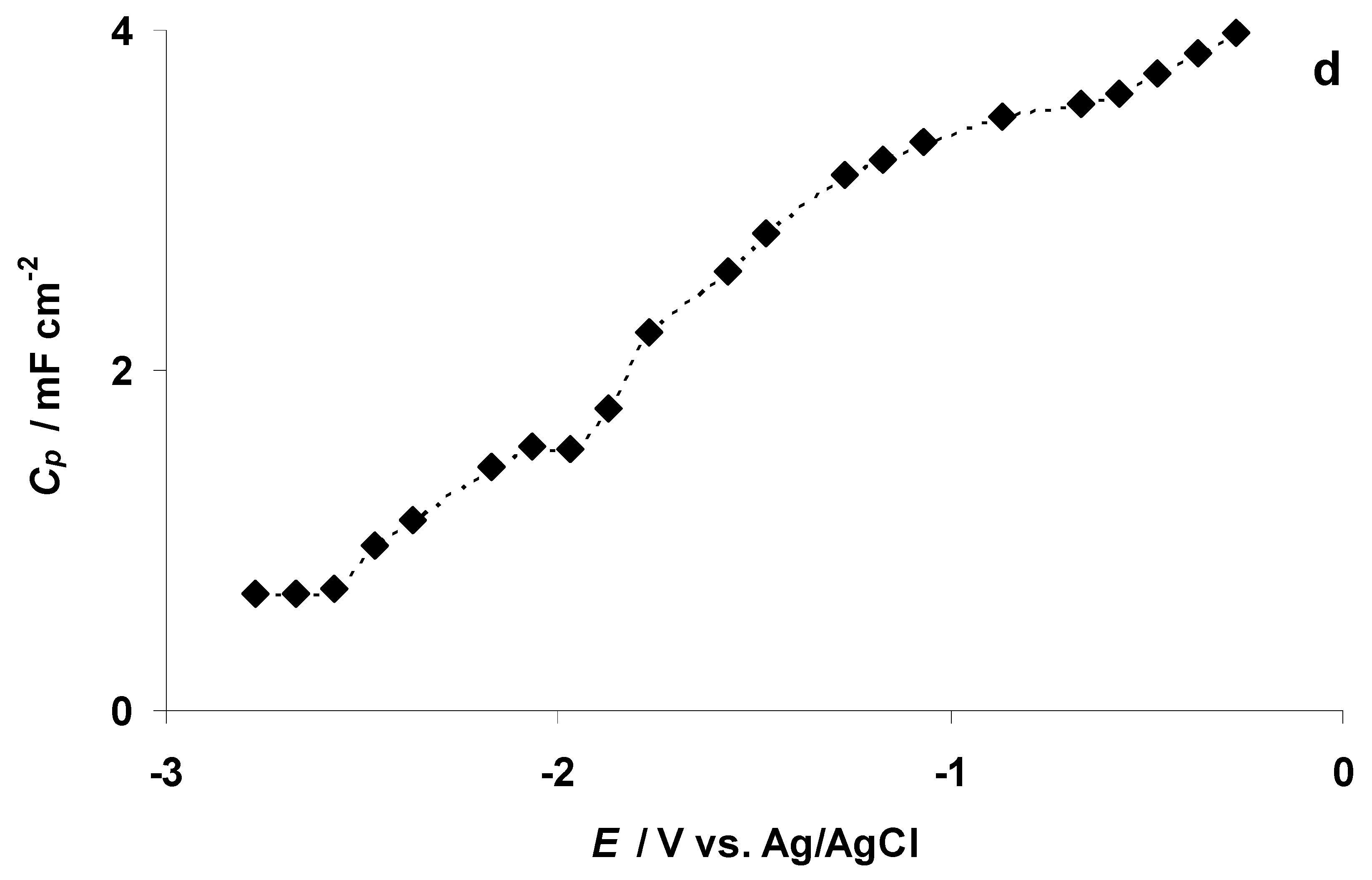
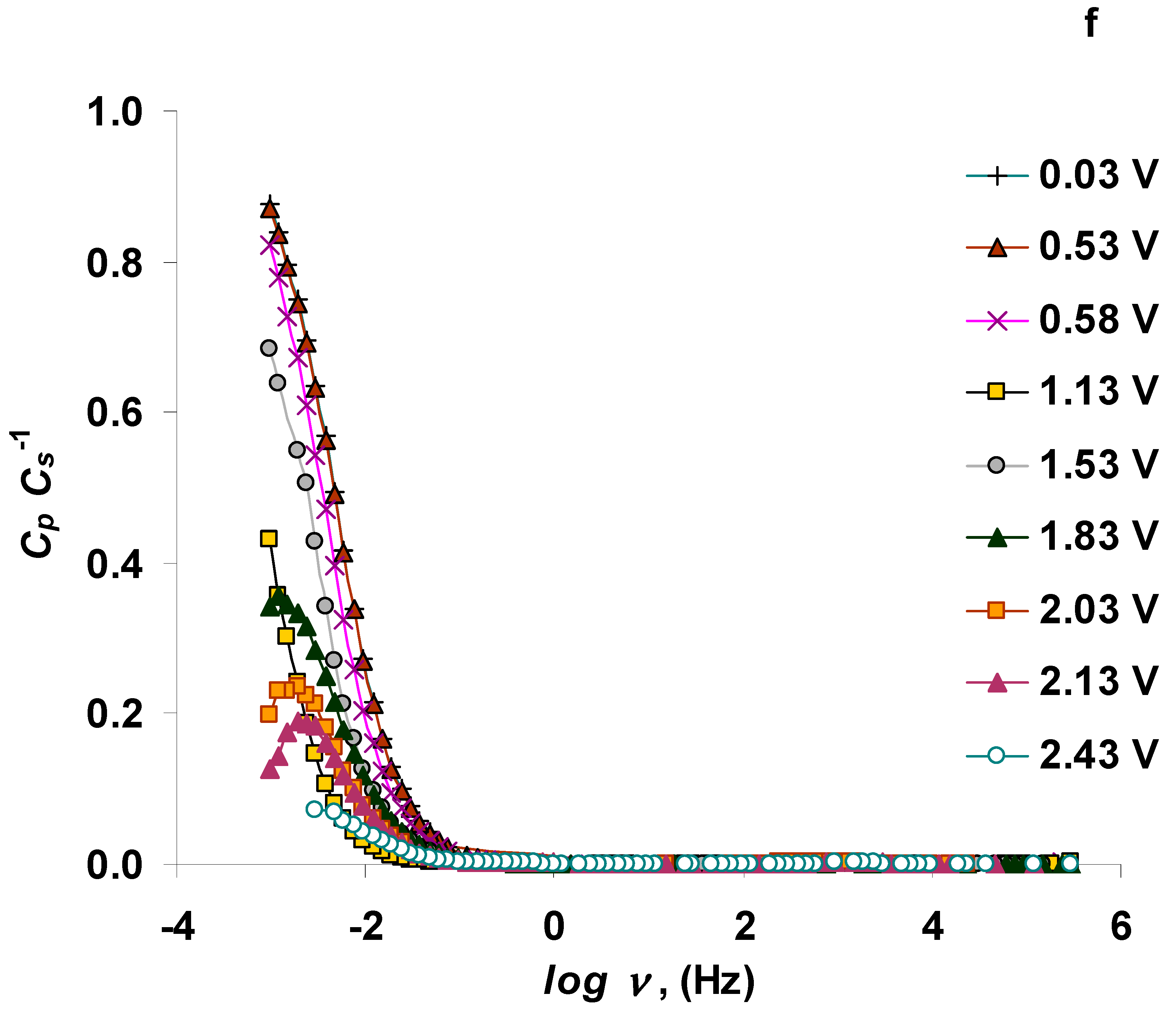
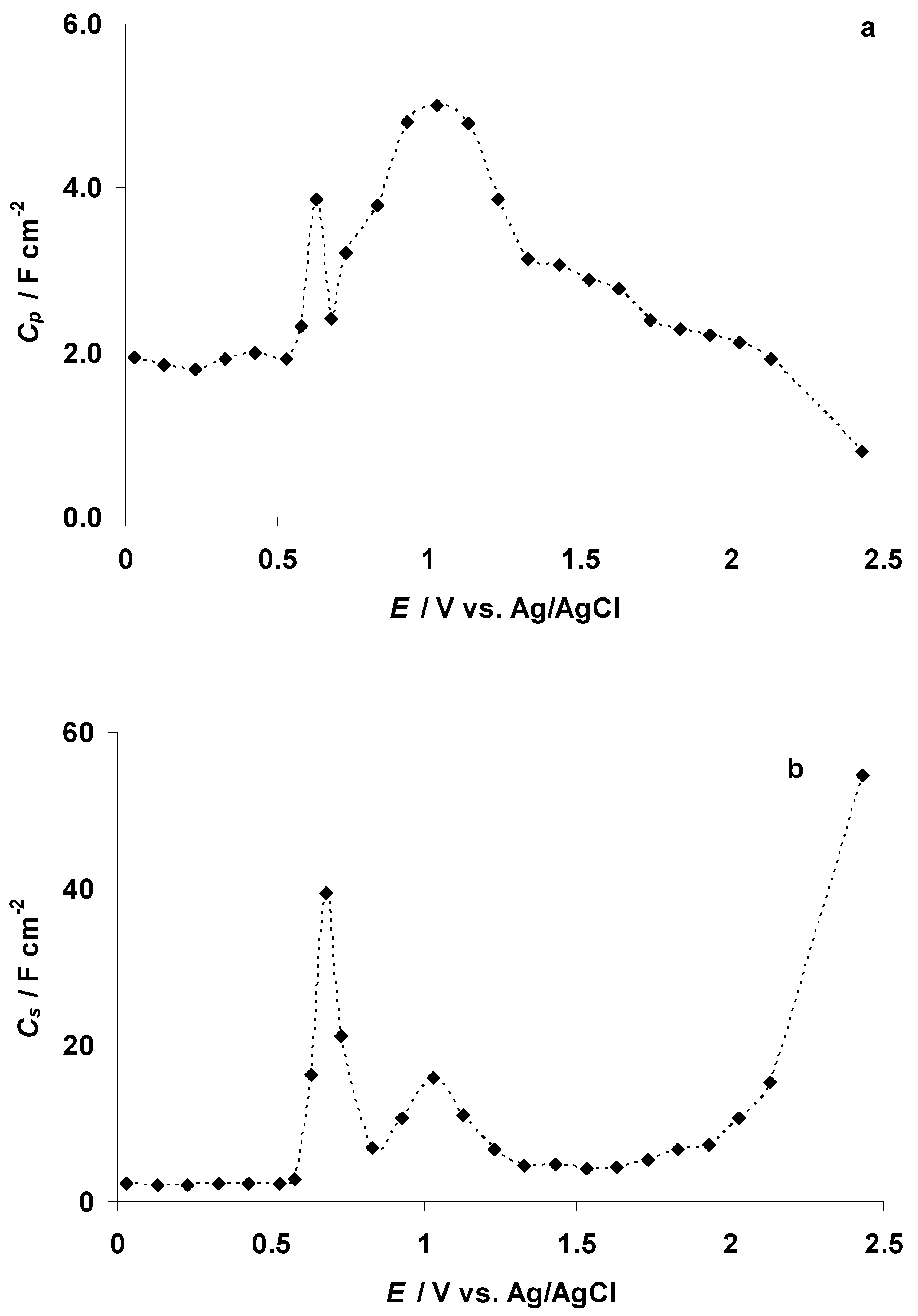
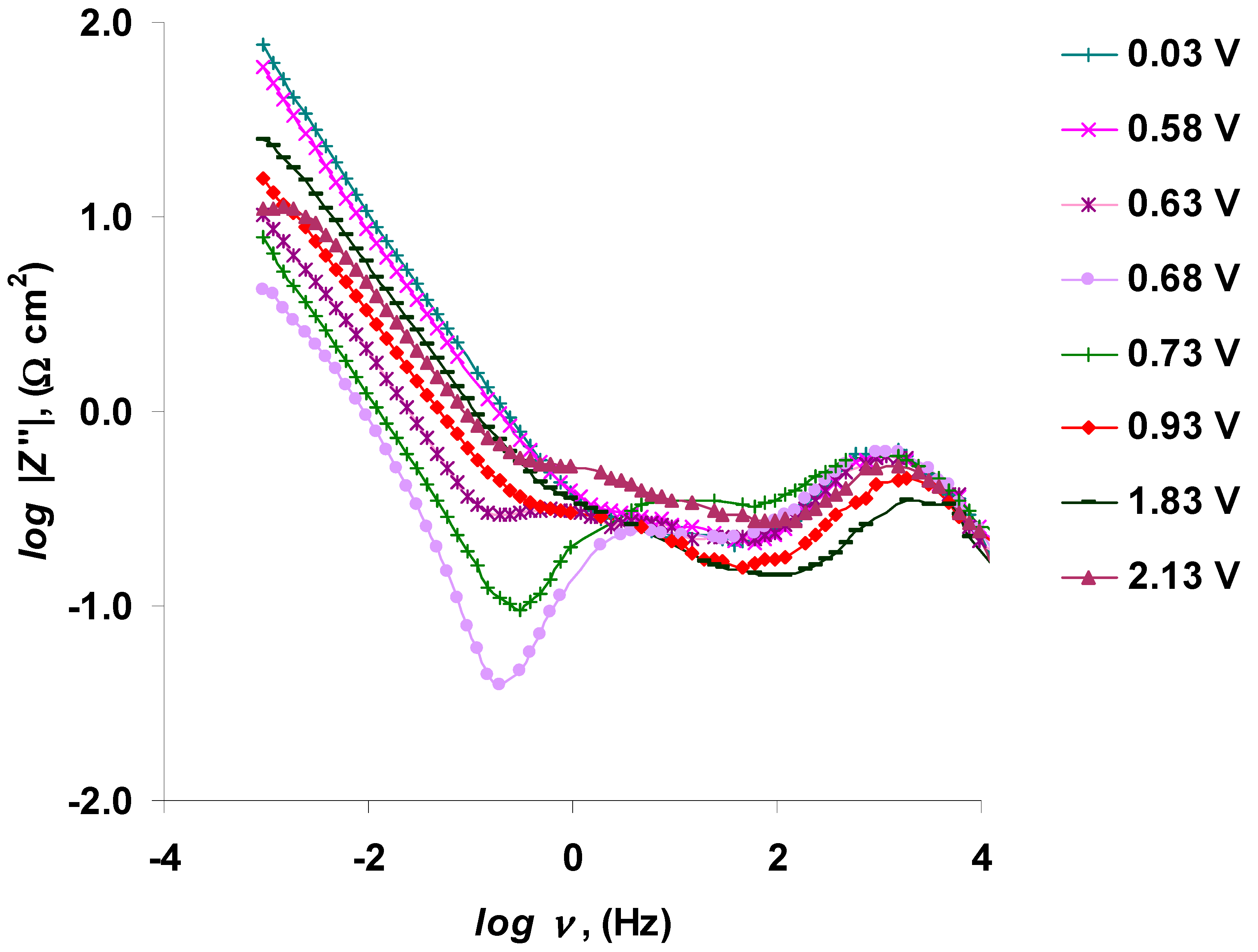
3.2. The Electrochemical Measurements Data at Negatively Polarized C(Mo2C) Electrode
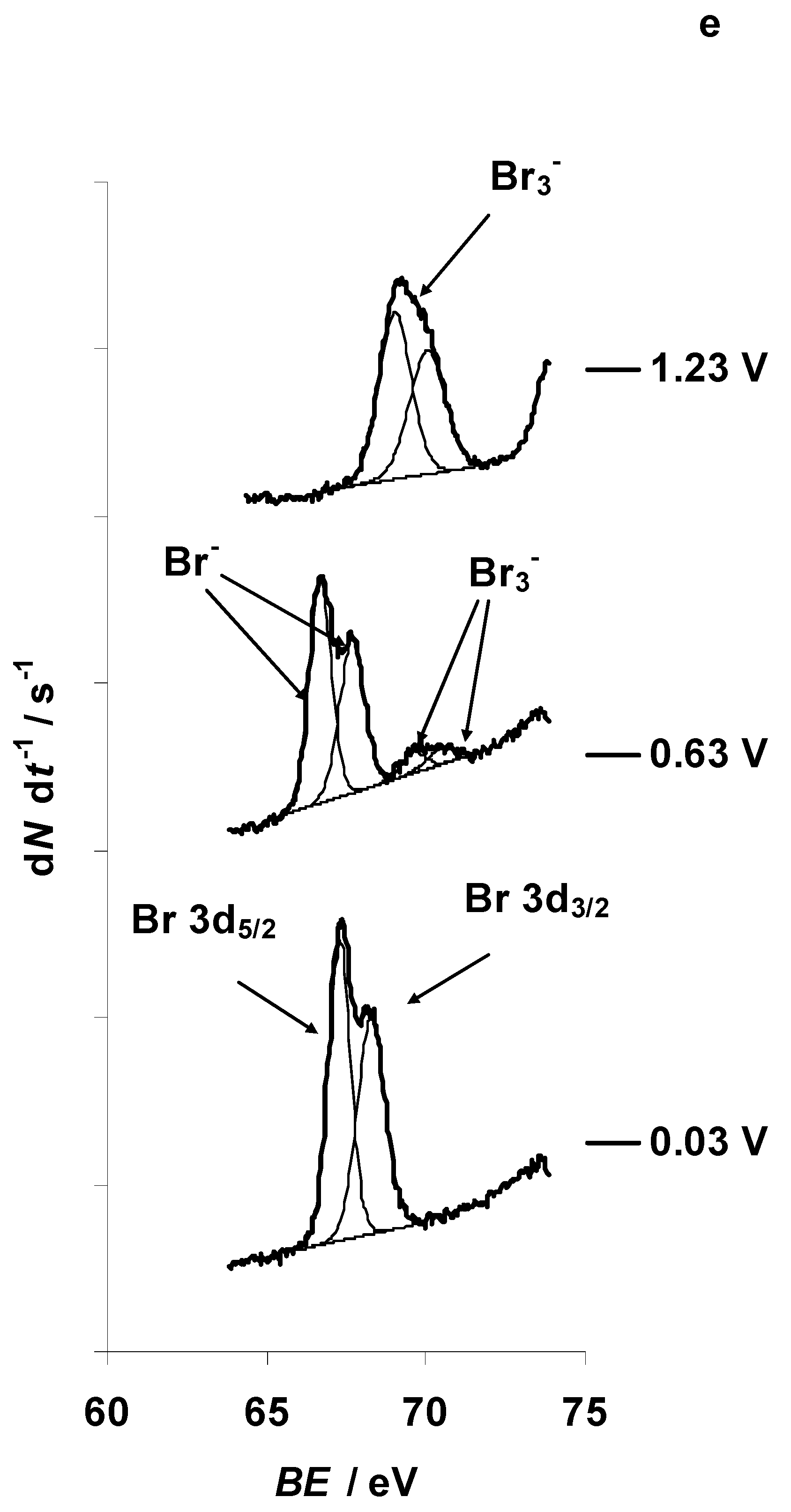
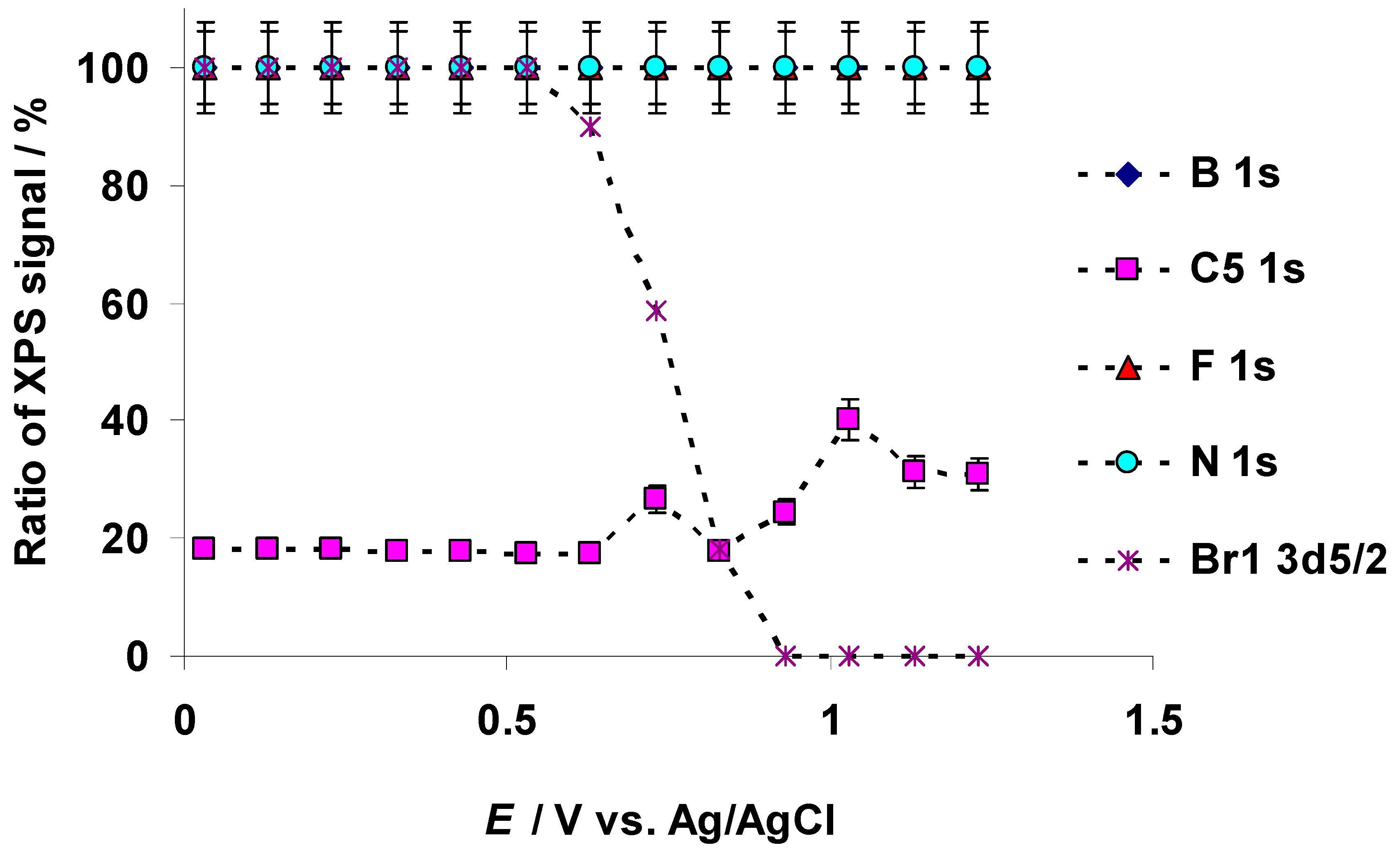
3.3. Characteristic Changes in the C 1s, N 1s, B 1s, F 1s, and Br 3d In Situ X-ray Spectra Collected at the Positively Polarized C(Mo2C) Electrode
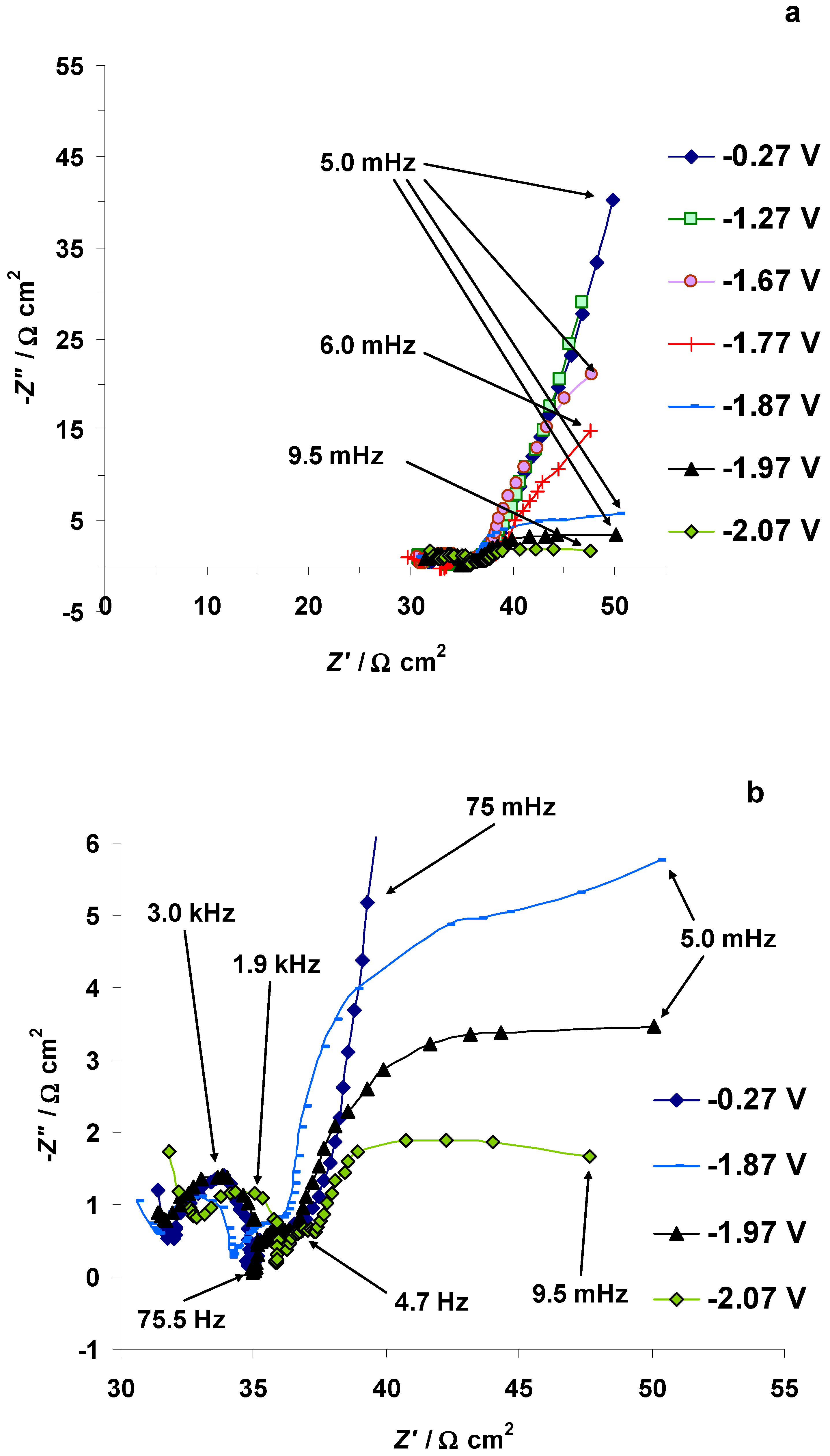
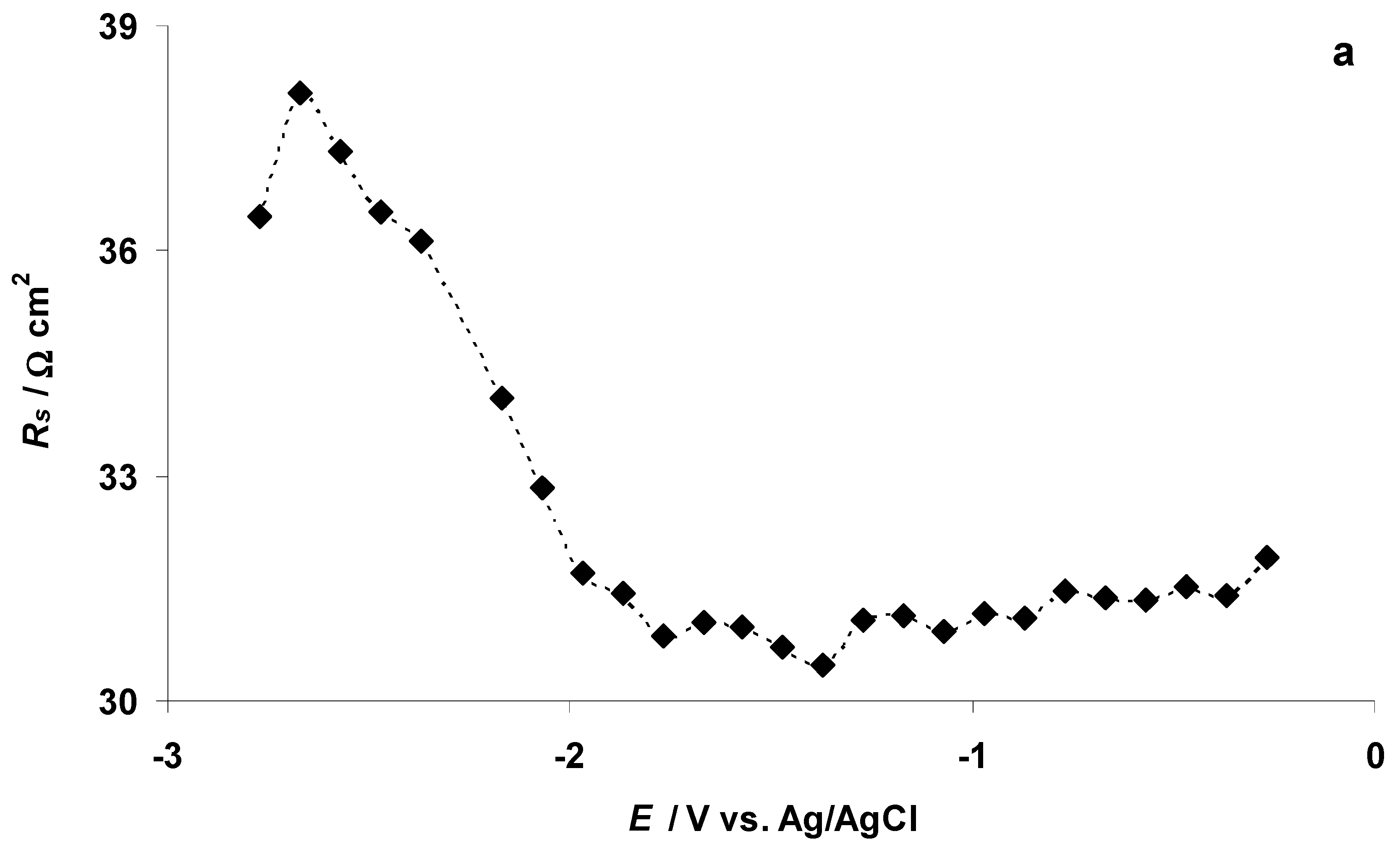
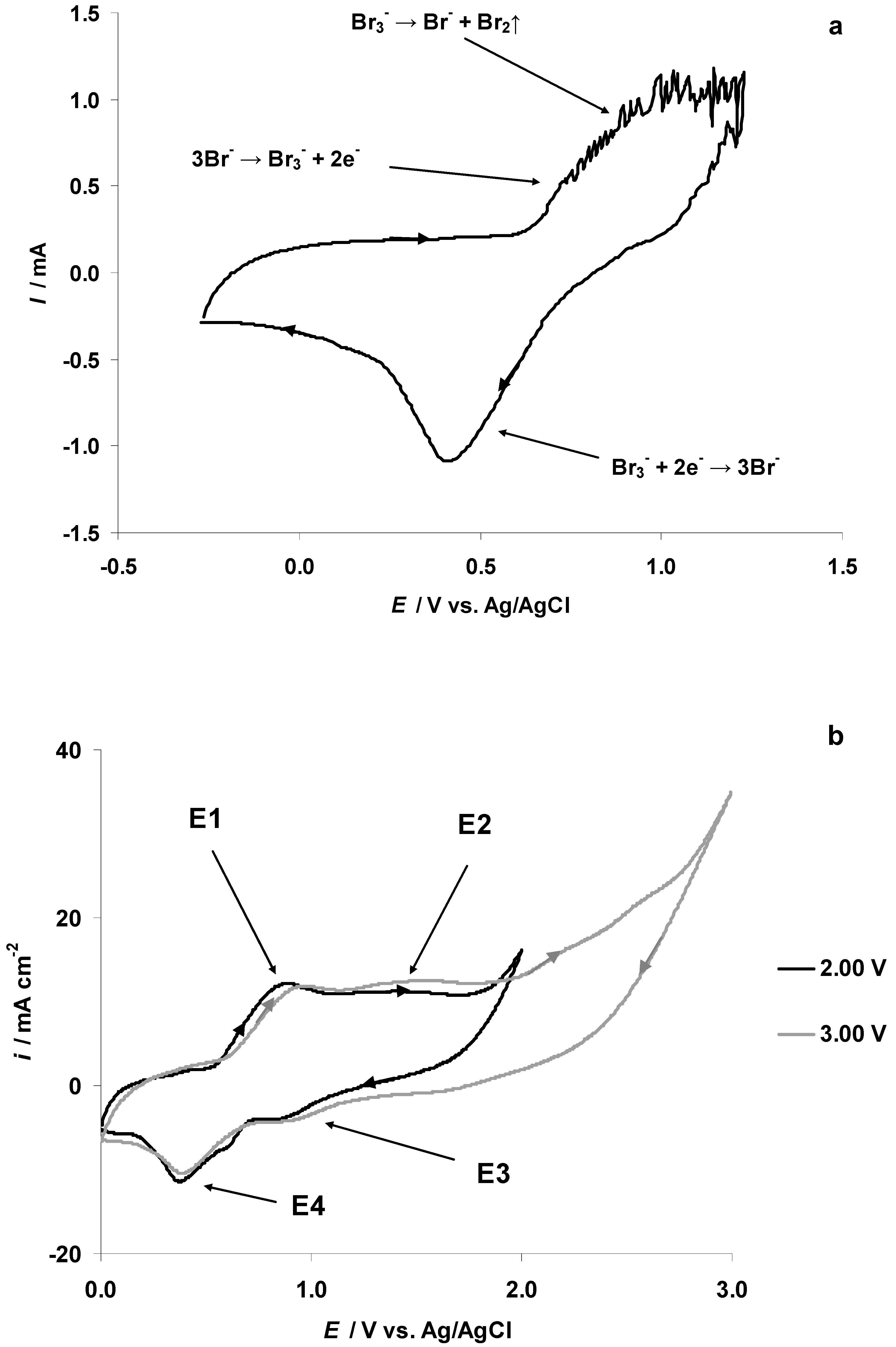
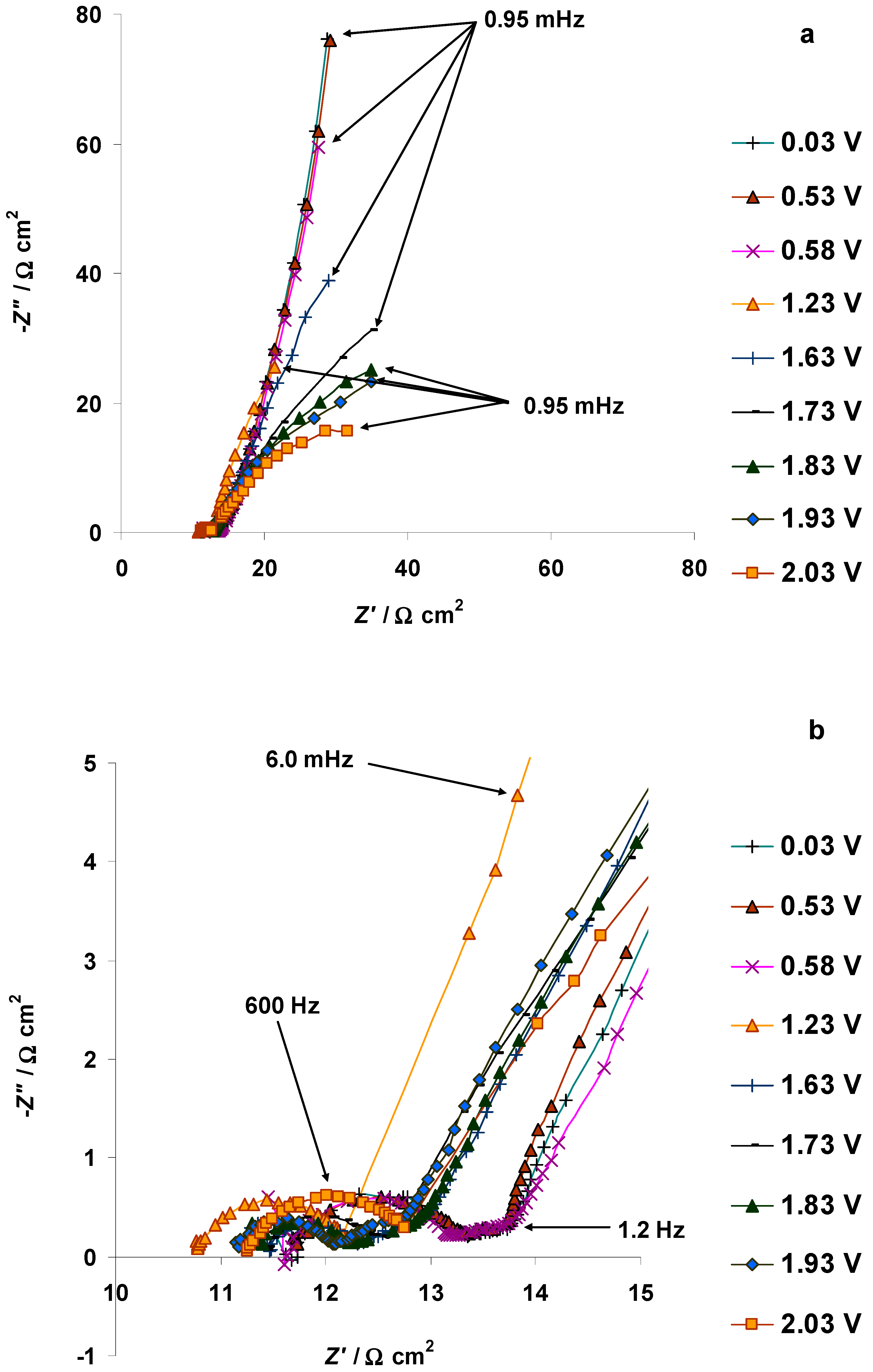
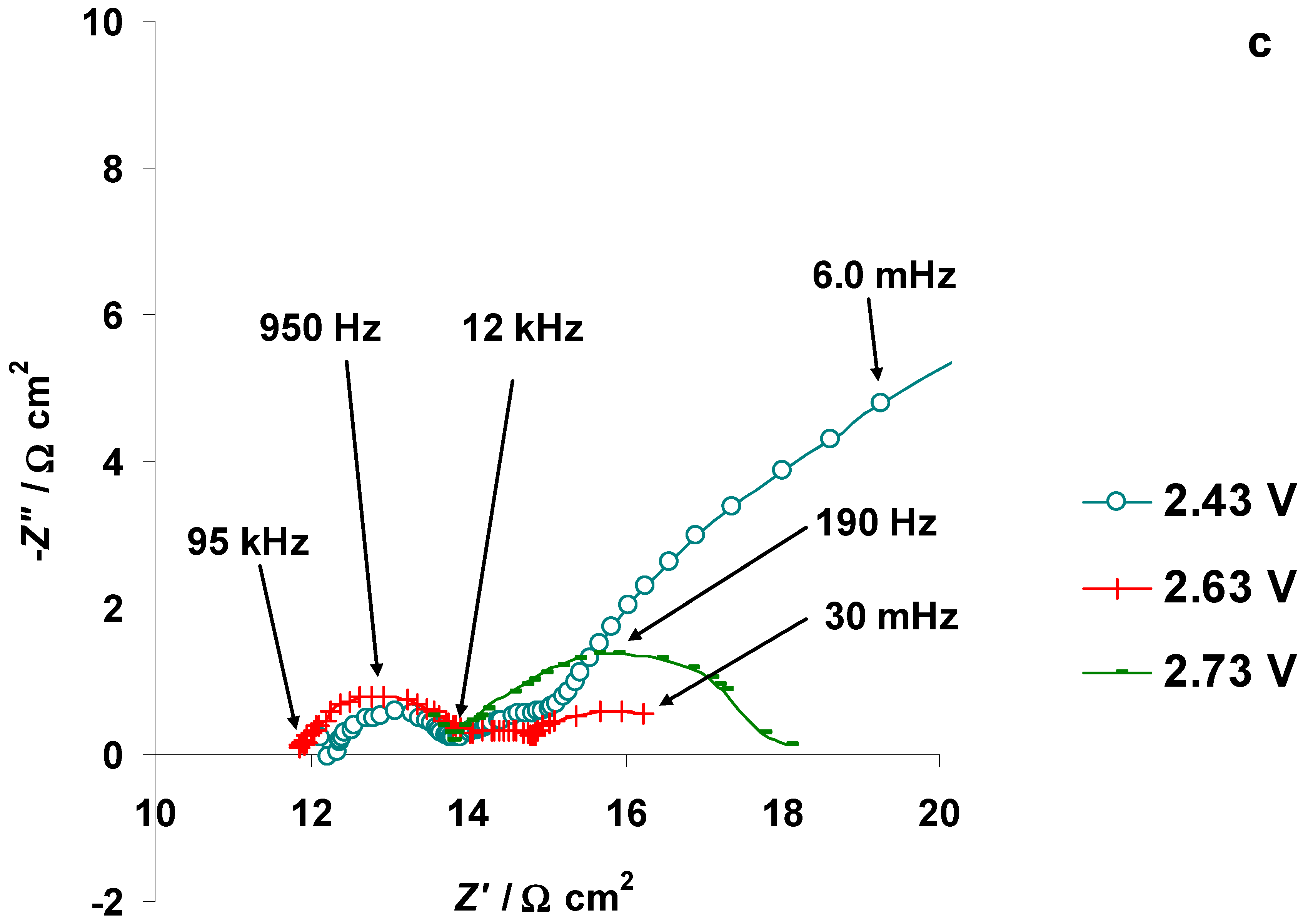
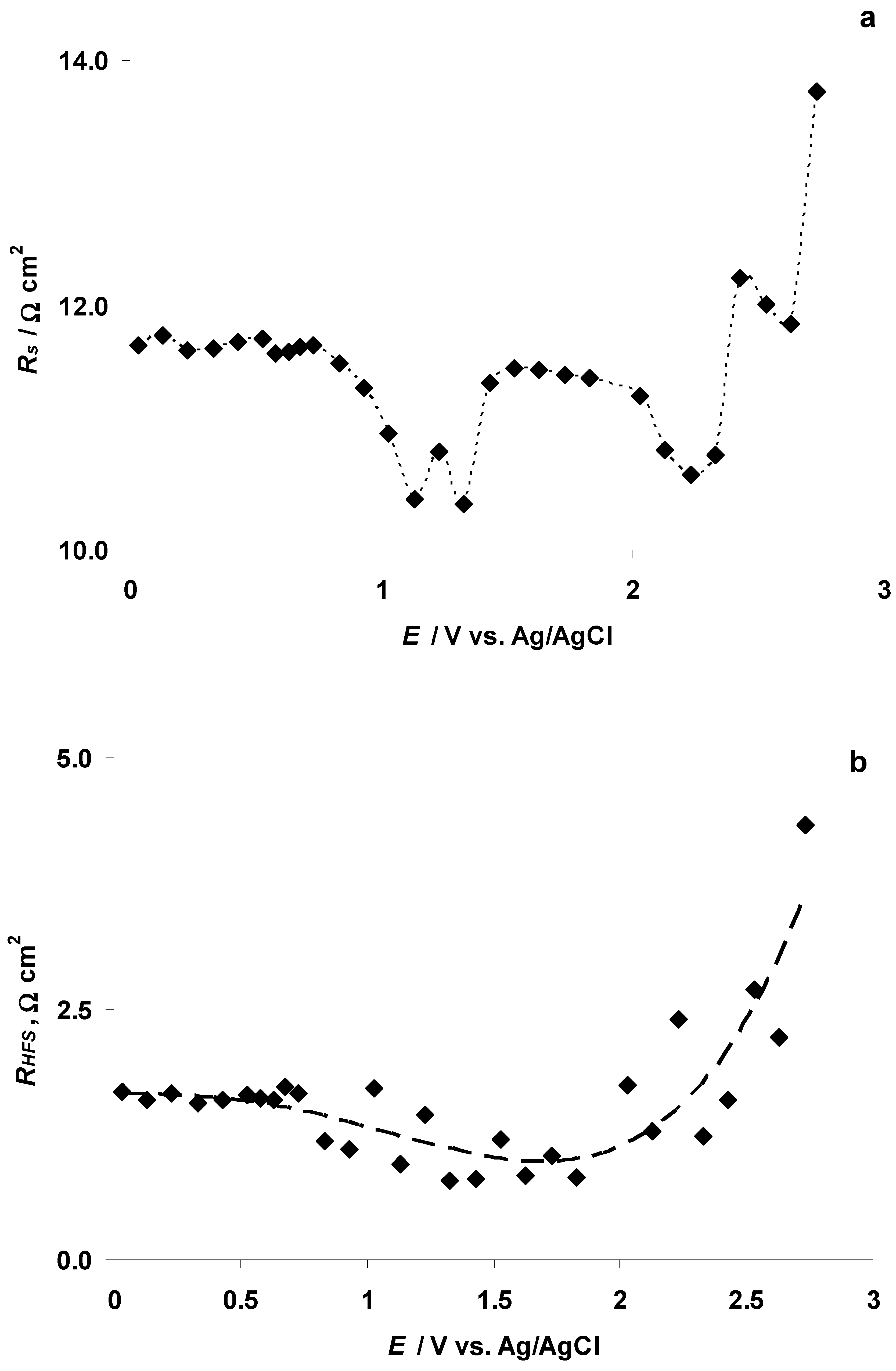
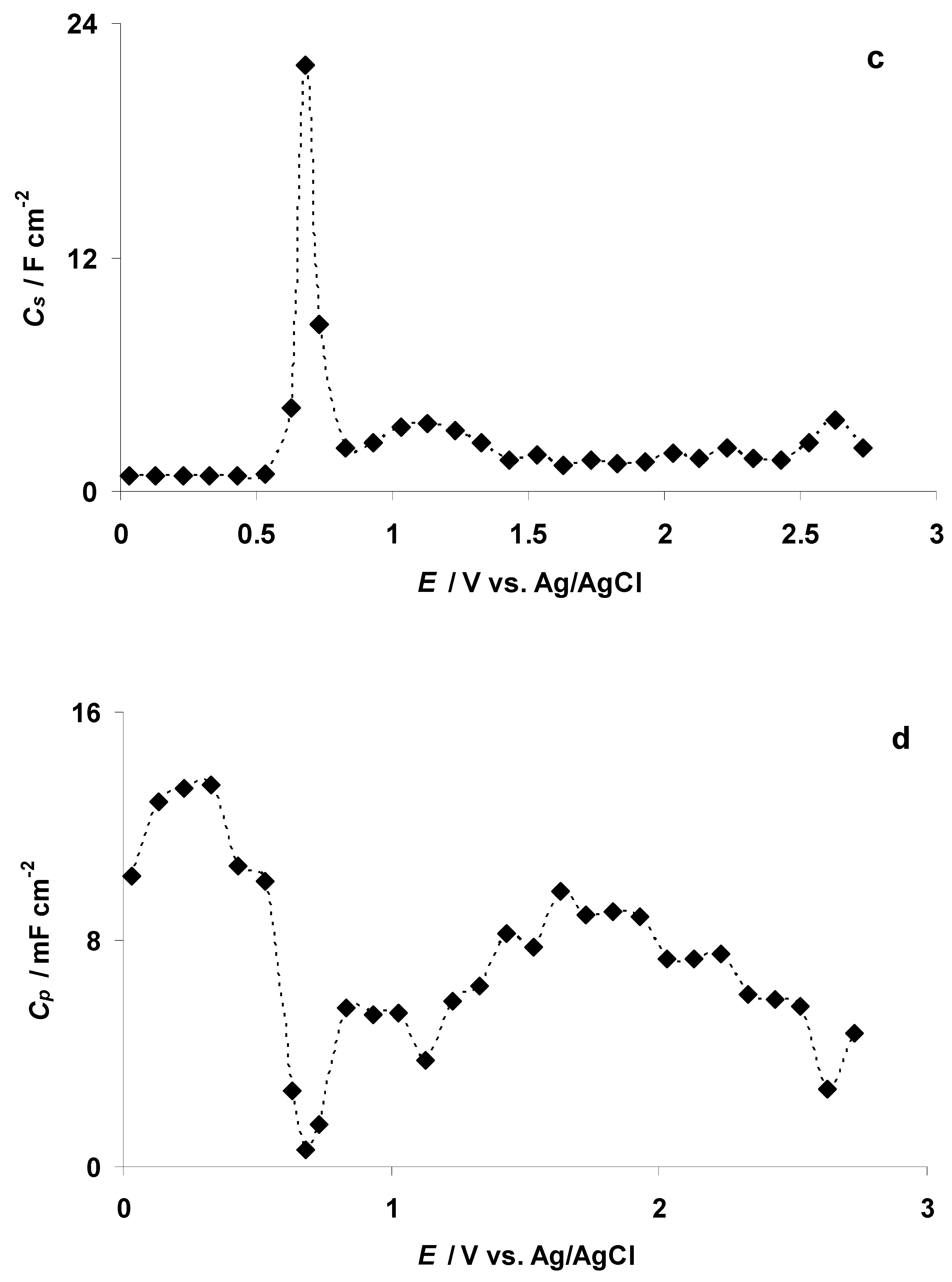
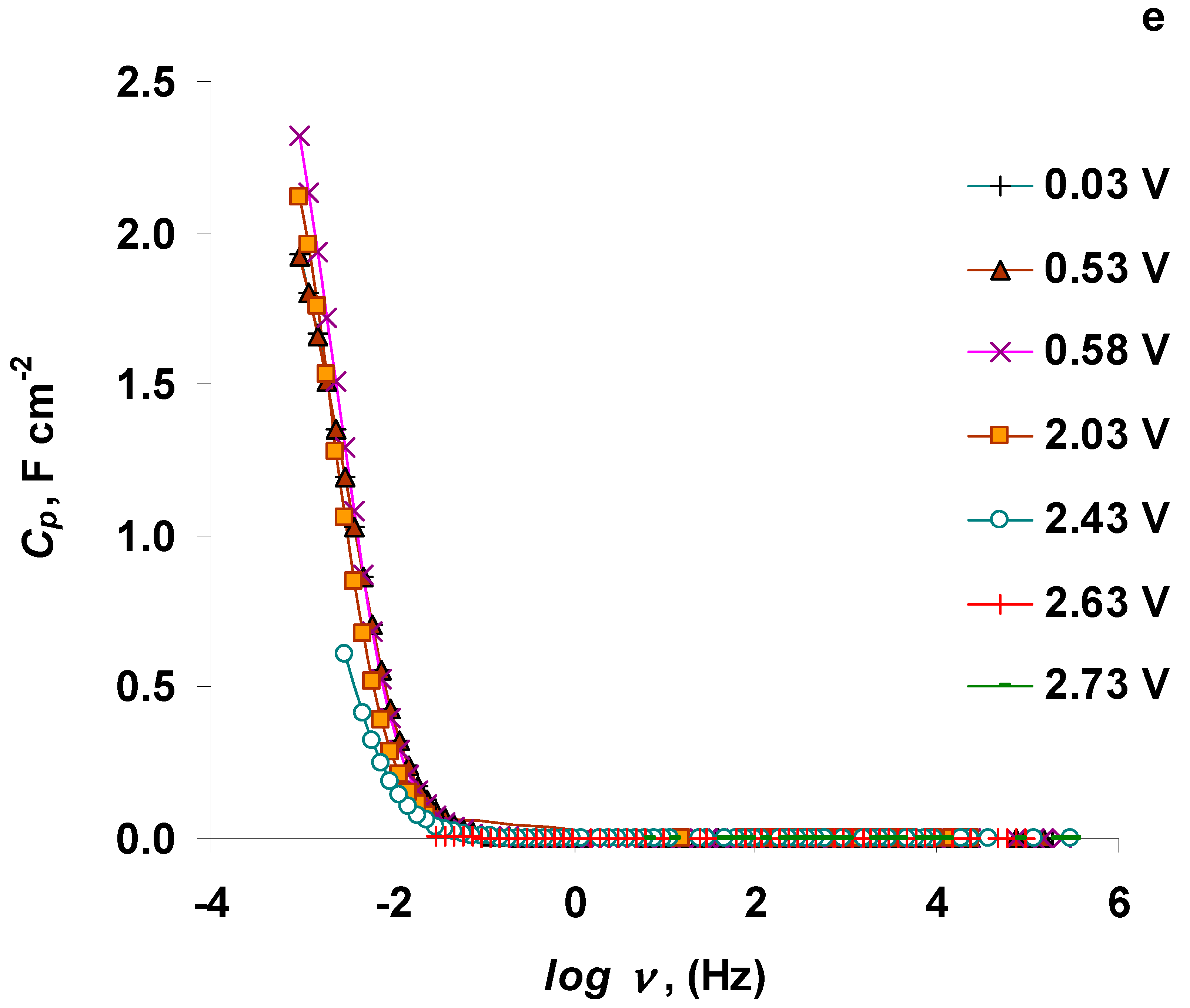
3.4. The Electrochemical Measurements Data Collected at the Positively Polarized C(Mo2C) Electrode
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, G.; Zhang, L.; Zhang, J. A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 2012, 41, 797–828. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhao, X.S. Carbon-based materials as supercapacitor electrodes. Chem. Soc. Rev. 2009, 38, 2520–2531. [Google Scholar] [CrossRef] [PubMed]
- Beguin, F.; Presser, V.; Balducci, A.; Frackowiak, E. Carbons and electrolytes for Advanced Supercapacitors. Adv. Mater. 2014, 26, 2219–2251. [Google Scholar] [CrossRef] [PubMed]
- Theerthagiri, J.; Karuppasamy, K.; Durai, G.; Rana, A.H.S.; Arunachalam, P.; Sangeetha, K.; Kuppusami, P.; Kim, H.-S. Recent Advances in Metal Chalcogenides (MX.; X = S, Se) Nanostructures for Electrochemical Supercapacitor Applications: A Brief Review. Nanomaterials 2018, 8, 256. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Lee, Y.H. Carbon-based electrochemical capacitors. Chem. Sus. Chem. 2012, 5, 480–499. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Burke, A.F. Electrochemical capacitors: Challenges and opportunities for real-world applications. Interface 2008, 17, 53–57. [Google Scholar]
- Miller, J.R.; Simon, P. Electrochemical capacitors for energy management. Science 2008, 321, 651–652. [Google Scholar] [CrossRef] [PubMed]
- Kötz, R.; Carlen, M. Principles and applications of electrochemical capacitors. Electrochim. Acta 2000, 45, 2483–2498. [Google Scholar] [CrossRef]
- Simon, P.; Gogotsi, Y. Materials for electrochemical capacitors. Nature Mater. 2008, 7, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Volfkovich, Y.M.; Serdyuk, T.M. Electrochemical capacitors. Russ. J. Electrochem. 2002, 38, 935–959. [Google Scholar] [CrossRef]
- Pell, W.G.; Conway, B.E.; Adams, W.A.; de Oliveira, J. Electrochemical efficiency in multiple discharge/recharge cycling of supercapacitors in hybrid EV applications. J. Power Sources 1999, 80, 134–141. [Google Scholar] [CrossRef]
- Jänes, A.; Eskusson, J.; Kanarbik, R.; Saar, A.; Lust, E. Surface analysis of supercapacitor electrodes after long-lasting constant current tests in organic electrolyte. J. Electrochem. Soc. 2012, 159, A1141–A1147. [Google Scholar] [CrossRef]
- Romann, T.; Oll, O.; Pikma, P.; Tamme, H.; Lust, E. Surface chemistry of carbon electrodes in 1-ethyl-3-methylimidazolium tetrafluoroborate ionic liquid—An in situ infrared study. Electrochim. Acta 2014, 125, 183–190. [Google Scholar] [CrossRef]
- Kurig, H.; Vestli, M.; Tõnurist, K.; Jänes, A.; Lust, E. Influence of room temperature ionic liquid anion chemical composition and electrical charge delocalization on the supercapacitor properties. J. Electrochem. Soc. 2012, 159, A944–A951. [Google Scholar] [CrossRef]
- Jänes, A.; Thomberg, T.; Kurig, H.; Lust, E. Nanoscale fine-tuning of porosity of carbide-derived carbon prepared from molybdenum carbide. Carbon 2009, 47, 23–29. [Google Scholar] [CrossRef]
- Jänes, A.; Permann, L.; Arulepp, M.; Lust, E. Electrochemical characteristics of nanoporous carbide-derived carbon materials in non-aqueous electrolyte solutions. Electrochem. Commun. 2004, 6, 313–318. [Google Scholar] [CrossRef]
- Thomberg, T.; Jänes, A.; Lust, E. Energy and power performance of electrochemical double-layer capacitors based on molybdenum carbide derived carbon. Electrochim. Acta 2010, 55, 3138–3143. [Google Scholar] [CrossRef]
- Jänes, A.; Thomberg, T.; Tõnurist, K.; Kurig, H.; Laheäär, A.; Lust, E. Micro- and mesoporous carbide-derived carbon materials and polymer membranes for supercapacitors. ECS Transact. 2008, 16, 57–67. [Google Scholar] [CrossRef]
- Von Czarnecki, P.; Ahrens, M.; Iliev, B.; Schubert, T.J.S. Ionic liquid based electrolytes for electrical storage. ECS Transact. 2017, 77, 79–87. [Google Scholar] [CrossRef]
- Chandra, A. Supercapacitors: An alternative technology for energy storage. Proc. Natl. Acad. Sci. India Sect. A Phys. Sci. 2012, 82, 79–90. [Google Scholar] [CrossRef]
- Jänes, A.; Eskusson, J.; Mattisen, L.; Lust, E. Electrochemical behaviour of hybrid devices based on Na2SO4 and Rb2SO4 neutral aqueous electrolytes and carbon electrodes within wide cell potential region. J. Solid State Electrochem. 2015, 19, 769–783. [Google Scholar] [CrossRef]
- Gao, X.; Zu, L.; Cai, X.; Li, C.; Lian, H.; Liu, Y.; Wang, X.; Cui, X. High Performance of Supercapacitor from PEDOT:PSS Electrode and Redox Iodide Ion Electrolyte. Nanomaterials 2018, 8, 335. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.; Lian, K. Proton-conducting polymer electrolytes and their applications in solid supercapacitors: A review. RSC Adv. 2014, 4, 33091–33113. [Google Scholar] [CrossRef]
- Ehsani, A.; Heidari, A.A.; Shiri, H.M. Electrochemical pseudocapacitors based on ternary nanocomposite of conductive polymer/graphene/metal oxide: An introduction and review to it in recent studies. Chem. Rec. 2018, 18, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Thangavel, R.; Kannan, A.G.; Ponraj, R.; Thangavel, V.; Kim, D.-W.; Lee, Y.-S. High-energy green supercapacitor driven by ionic liquid electrolytes as an ultra-high stable next-generation energy storage device. J. Power Sources 2018, 383, 102–109. [Google Scholar] [CrossRef]
- Gonzales, A.; Goikolea, E.; Barrena, J.A.; Mysyk, R. Review on supercapacitors: Technologies and materials. Renew. Sustain. Energy Rev. 2016, 58, 1189–1206. [Google Scholar] [CrossRef]
- Meng, Q.; Cai, K.; Chen, Y.; Chen, L. Research progress on conducting polymer based supercapacitor electrode materials. Nano Energy 2017, 36, 268–285. [Google Scholar] [CrossRef]
- Zhang, W.; Feng, P.; Chen, J.; Sun, Z.; Zhao, B. Electrically conductive hydrogels for flexible energy storage systems. Prog. Polym. Sci. 2019, 88, 220–240. [Google Scholar] [CrossRef]
- Shi, Y.; Zhang, J.; Pan, L.; Shi, Y.; Yu, G. Energy gels: A bio-inspired material platform for advanced energy applications. Nano Today 2016, 11, 738–762. [Google Scholar] [CrossRef]
- Väärtnõu, M.; Lust, E. Impedance characteristics of iodide ions adsorption on Bi single crystal planes in ethanol. J. Electroanal. Chem. 2004, 565, 211–218. [Google Scholar] [CrossRef]
- Kim, K.-S.; Shin, B.-K.; Lee, H. Physical and electrochemical properties of 1-butyl-3-metylimidazolium bromide, 1-butyl-3-methylimidazolium iodide, and 1-butyl-3-metylimidazolium tetrafluoroborate. Korean J. Chem. Eng. 2004, 21, 1010. [Google Scholar] [CrossRef]
- Siinor, L.; Siimenson, C.; Lust, K.; Lust, E. Mixture of 1-ethyl-3-methylimidazolium tetrafluoroborate and 1-ethyl-3-methylimidazolium iodide: A new potential high capacitance electrolyte for EDLCs. Electrochem. Commun. 2013, 35, 5–7. [Google Scholar] [CrossRef]
- Siinor, L.; Poom, J.; Siimenson, C.; Lust, K.; Lust, E. Electrochemical characteristics pyrolytic graphite|mixture of 1-ethyl-3-methylimidazolium tetrafluoroborate and 1-ethyl-3-methylimidazolium iodide interface. J. Electroanal. Chem. 2014, 719, 133–137. [Google Scholar] [CrossRef]
- Siimenson, C.; Siinor, L.; Lust, K.; Lust, E. The electrochemical characteristics of the mixture of 1-ethyl-3-methylimidazolium tetrafluoroborate and 1-ethyl-3-methylimidazolium iodide. J. Electroanal. Chem. 2014, 730, 59–64. [Google Scholar] [CrossRef]
- Lust, E.; Siinor, L.; Kurig, H.; Romann, T.; Ivaništšev, V.; Siimenson, C.; Thomberg, T.; Kruusma, J.; Lust, K.; Pikma, P.; et al. Characteristics of Capacitors Based on Ionic Liquids: From Dielectric Polymers to Redox-Active Adsorbed Species. ECS Transact. 2016, 75, 161–170. [Google Scholar] [CrossRef]
- Oll, O.; Siimenson, C.; Lust, K.; Gorbatovski, G.; Lust, E. Specific adsorption from an ionic liquid: Impedance study of iodide ion adsorption from a pure halide ionic liquid at bismuth single crystal planes. Electrochim. Acta 2017, 247, 910–919. [Google Scholar] [CrossRef]
- Yamazaki, S.; Ito, T.; Yamagata, M.; Ishikawa, M. Non-aqueous electrochemical capacitor utilizing electrolytic redox reactions of bromide species in ionic liquid. Electrochim. Acta 2012, 86, 294–297. [Google Scholar] [CrossRef]
- Siimenson, C.; Lembinen, M.; Oll, O.; Läll, L.; Tarkanovskaja, M.; Ivaništšev, V.; Siinor, L.; Thomberg, T.; Lust, K.; Lust, E. Electrochemical Investigation of 1-Ethyl-3-methylimidazolium Bromide and Tetrafluoroborate Mixture at Bi(111) Electrode Interface. J. Electrochem. Soc. 2016, 163, H723–H730. [Google Scholar] [CrossRef]
- Gastrol, D.; Walkowiak, J.; Fic, K.; Frackowiak, E. Enhancement of the carbon electrode capacitance by brominated hydroquinones. J. Power Sources 2016, 326, 587–594. [Google Scholar] [CrossRef]
- Kruusma, J.; Tõnisoo, A.; Pärna, R.; Nõmmiste, E.; Lust, E. Influence of the negative potential of molybdenum carbide derived carbon electrode on the in situ synchrotron radiation activated X-ray photoelectron spectra of 1-ethyl-3-methylimidazolium tetrafluoroborate. Electrochim. Acta 2016, 206, 419–426. [Google Scholar] [CrossRef]
- Tõnisoo, A.; Kruusma, J.; Pärna, R.; Kikas, A.; Hirsimäki, M.; Nõmmiste, E.; Lust, E. In Situ XPS Studies of Electrochemically Negatively Polarized Molybdenum Carbide Derived Carbon Double Layer Capacitor Electrode. J. Electrochem. Soc. 2013, 160, A1084–A1093. [Google Scholar] [CrossRef]
- Foelske-Schmitz, A.; Weingarth, D.; Kötz, R. Quasi in situ XPS study of electrochemical oxidation and reduction of highly oriented pyrolytic graphite in [1-ethyl-3-methylimidazolium][BF4] electrolytes. Electrochim. Acta 2011, 56, 10321–10331. [Google Scholar] [CrossRef]
- Smith, E.F.; Rutten, F.J.M.; Villar-Garcia, I.J.; Briggs, D.; Licence, P. Ionic Liquids in Vacuo: Analysis of Liquid Surfaces Using Ultra-High-Vacuum Techniques. Langmuir 2006, 22, 9386–9392. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Villar-Garcia, I.J.; Briggs, D.; Licence, P. Ionic liquids in vacuo; solution-phase X-ray photoelectron spectroscopy. Chem. Commun. 2005, 5633–5635. [Google Scholar] [CrossRef] [PubMed]
- NIST X-ray Photoelectron Spectroscopy Database, NIST Standard Reference Database 20, Version 4.1. Available online: http://srdata.nist.gov/xps/EnergyTypeValSrch.aspx (accessed on 14 February 2019).
- Kruusma, J.; Tõnisoo, A.; Pärna, R.; Nõmmiste, E.; Vahtrus, M.; Siinor, L.; Tallo, I.; Romann, T.; Lust, E. Influence of Iodide Ions Concentration on the Stability of 1-Ethyl-3-methylimidazolium Tetrafluoroborate | Molybdenum Carbide Derived Carbon Electrode Interface. J. Electrochem. Soc. 2017, 164, A1110–A1119. [Google Scholar] [CrossRef]
- Kruusma, J.; Tõnisoo, A.; Pärna, R.; Nõmmiste, E.; Kuusik, I.; Vahtrus, M.; Tallo, I.; Romann, T.; Lust, E. The Electrochemical Behavior of 1-Ethyl-3-Methyl Imidazolium Tetracyanoborate Visualized by In Situ X-ray Photoelectron Spectroscopy at the Negatively and Positively Polarized Micro-Mesoporous Carbon Electrode. J. Electrochem. Soc. 2017, 164, A3393–A3402. [Google Scholar] [CrossRef]
- Cheek, G.T.; O’Grady, W.E.; Lawrence, S.H. Determination of Water in 1-Ethyl-3-methylimidazolium tetrafluoroborate. ECS Transact. 2007, 2, 1–5. [Google Scholar] [CrossRef]
- Hamm, U.W.; Lazarescu, V.; Kolb, D.M. Adsorption of pyrazine on Au(111) and Ag(111) electrodes an ex situ XPS study. J. Chem. Soc. Faraday Trans. 1996, 92, 3785–3790. [Google Scholar] [CrossRef]
- Zhou, W.; Kolb, D.M. Influence of an electrostatic potential at the metal/electrolyte interface on the electron binding energy of adsorbates as probed by X-ray photoelectron spectroscopy. Surf. Sci. 2004, 573, 176–182. [Google Scholar] [CrossRef]
- Allen, G.D.; Buzzeo, M.C.; Villagrán, C.; Hardacre, C.; Compton, R.G. A mechanistic study of the electro-oxidation of bromide in acetonitrile and the room temperature ionic liquid, 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)imide at platinum electrodes. J. Electroanal. Chem. 2005, 575, 311–320. [Google Scholar] [CrossRef]
- Bennett, B.; Chang, J.; Bard, A.J. Mechanism of the Br−/Br2 Redox Reaction on Platinum and Glassy Carbon Electrodes in Nitrobenzene by Cyclic Voltammetry. Electrochim. Acta 2016, 219, 1–9. [Google Scholar] [CrossRef]



























© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruusma, J.; Tõnisoo, A.; Pärna, R.; Nõmmiste, E.; Lust, E. In Situ X-ray Photoelectron Spectroscopic and Electrochemical Studies of the Bromide Anions Dissolved in 1-Ethyl-3-Methyl Imidazolium Tetrafluoroborate. Nanomaterials 2019, 9, 304. https://doi.org/10.3390/nano9020304
Kruusma J, Tõnisoo A, Pärna R, Nõmmiste E, Lust E. In Situ X-ray Photoelectron Spectroscopic and Electrochemical Studies of the Bromide Anions Dissolved in 1-Ethyl-3-Methyl Imidazolium Tetrafluoroborate. Nanomaterials. 2019; 9(2):304. https://doi.org/10.3390/nano9020304
Chicago/Turabian StyleKruusma, Jaanus, Arvo Tõnisoo, Rainer Pärna, Ergo Nõmmiste, and Enn Lust. 2019. "In Situ X-ray Photoelectron Spectroscopic and Electrochemical Studies of the Bromide Anions Dissolved in 1-Ethyl-3-Methyl Imidazolium Tetrafluoroborate" Nanomaterials 9, no. 2: 304. https://doi.org/10.3390/nano9020304
APA StyleKruusma, J., Tõnisoo, A., Pärna, R., Nõmmiste, E., & Lust, E. (2019). In Situ X-ray Photoelectron Spectroscopic and Electrochemical Studies of the Bromide Anions Dissolved in 1-Ethyl-3-Methyl Imidazolium Tetrafluoroborate. Nanomaterials, 9(2), 304. https://doi.org/10.3390/nano9020304