Synergistic Effect of Oxygen Vacancies and Ni Species on Tuning Selectivity of Ni/ZrO2 Catalyst for Hydrogenation of Maleic Anhydride into Succinic Anhydride and γ-Butyrolacetone

 ,
,
Abstract
:
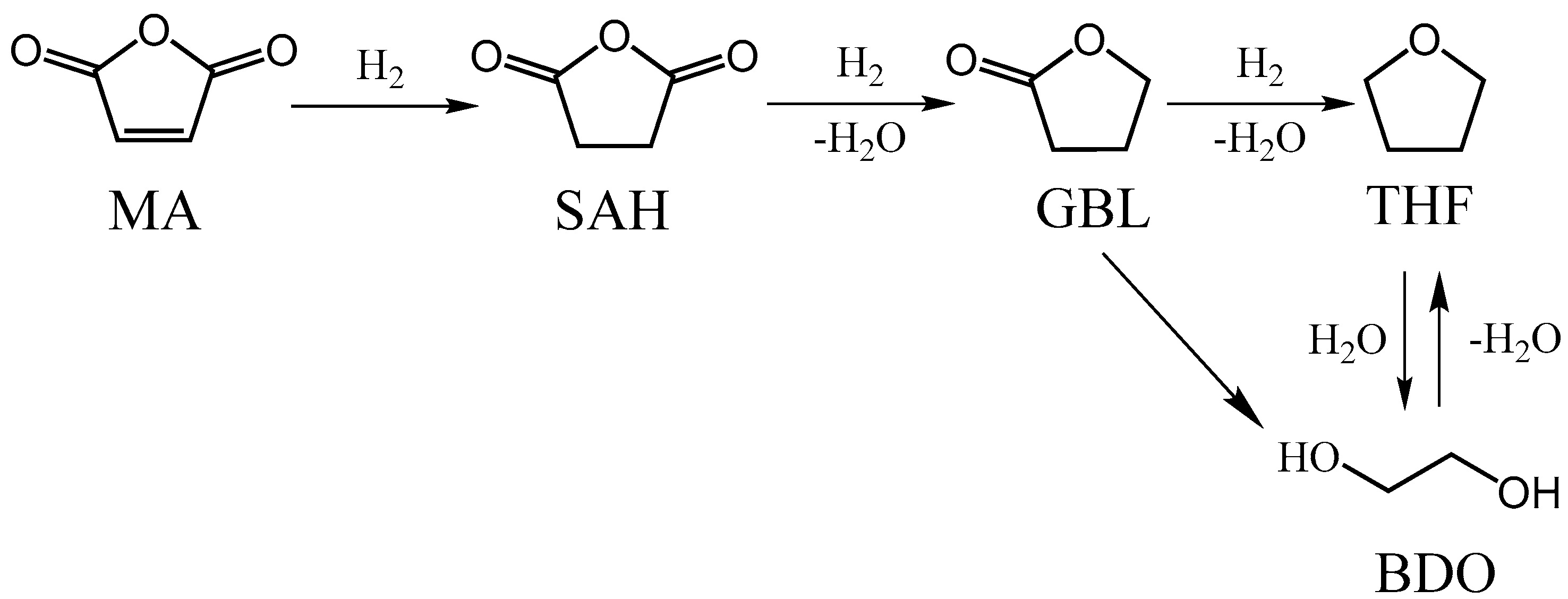
1. Introduction
2. Experimental Section
2.1. ZrO2 Preparation
2.2. Ni/ZrO2 Preparation
2.3. Structure Characterizations
2.4. Catalytic Activity Tests
3. Results
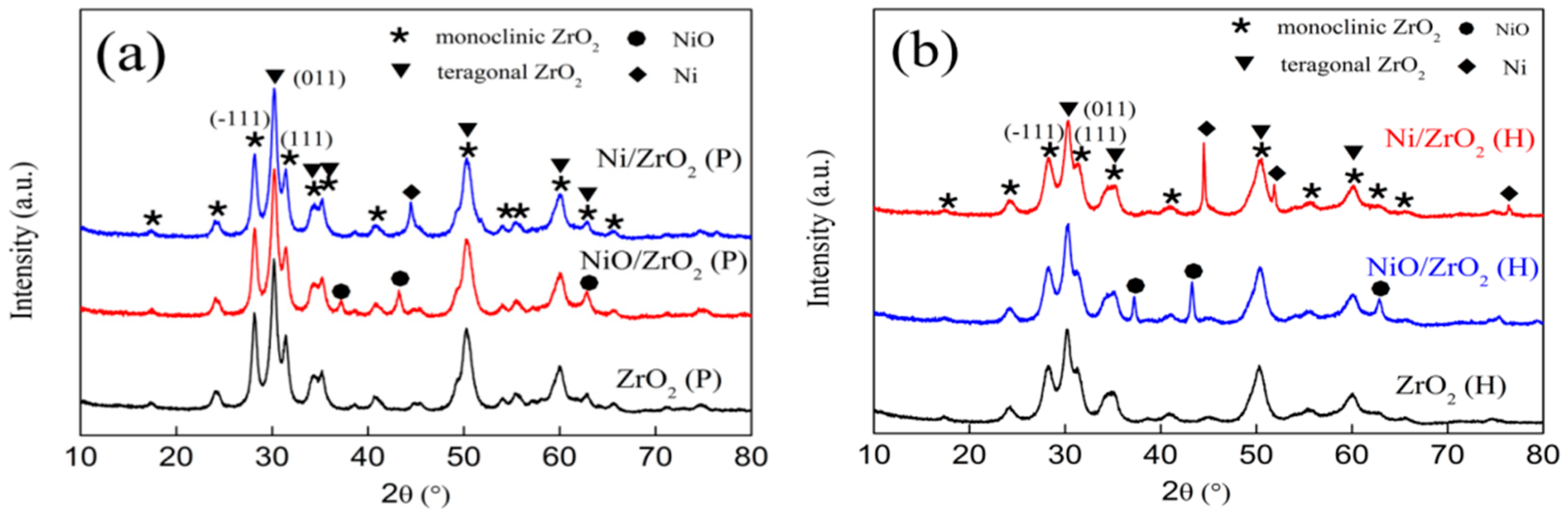
3.1. X-Ray Powder Diffraction (XRD) Patterns
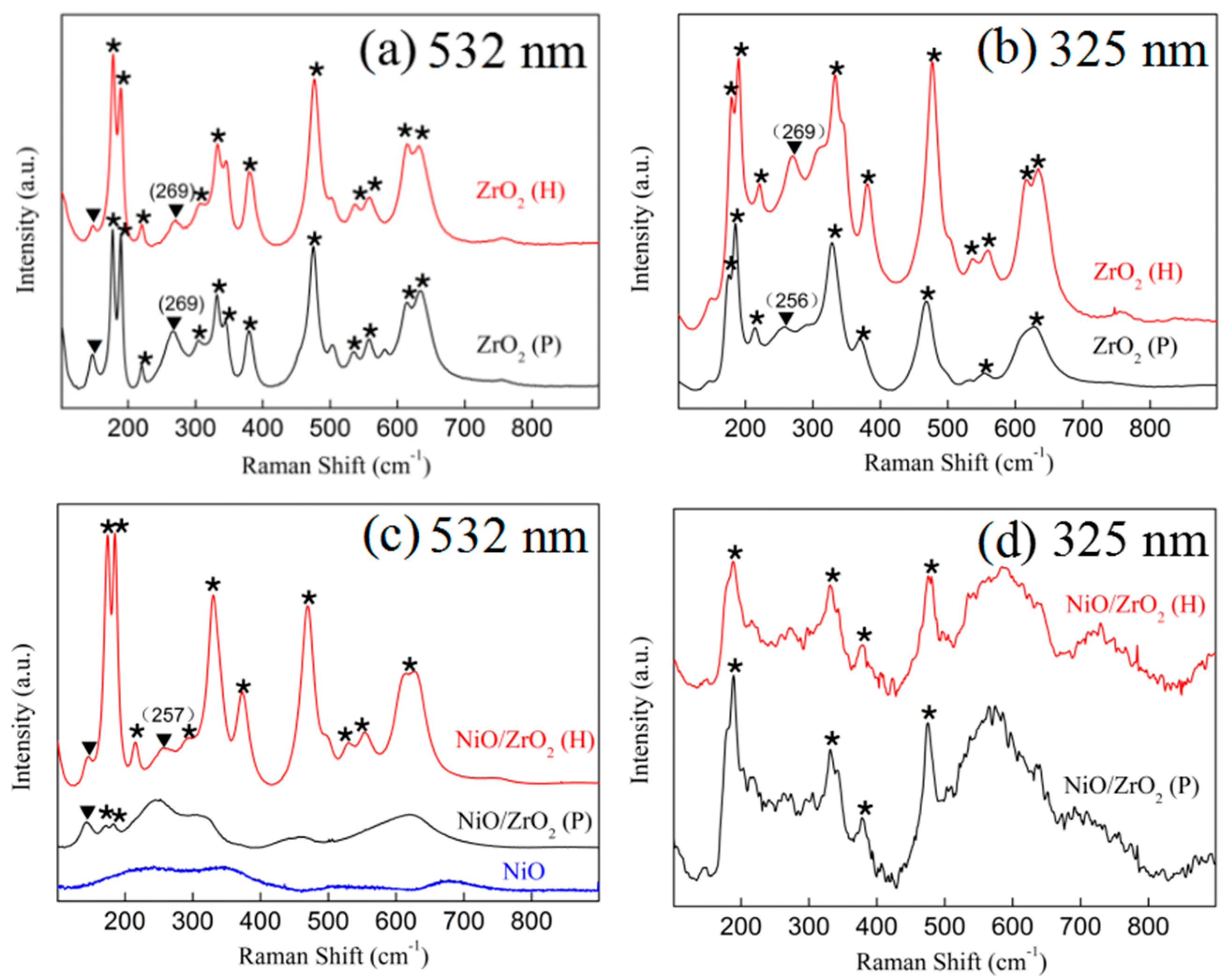
3.2. Raman Spectra
3.3. High-Resolution Transmission Electron Microscopy (HRTEM) Images
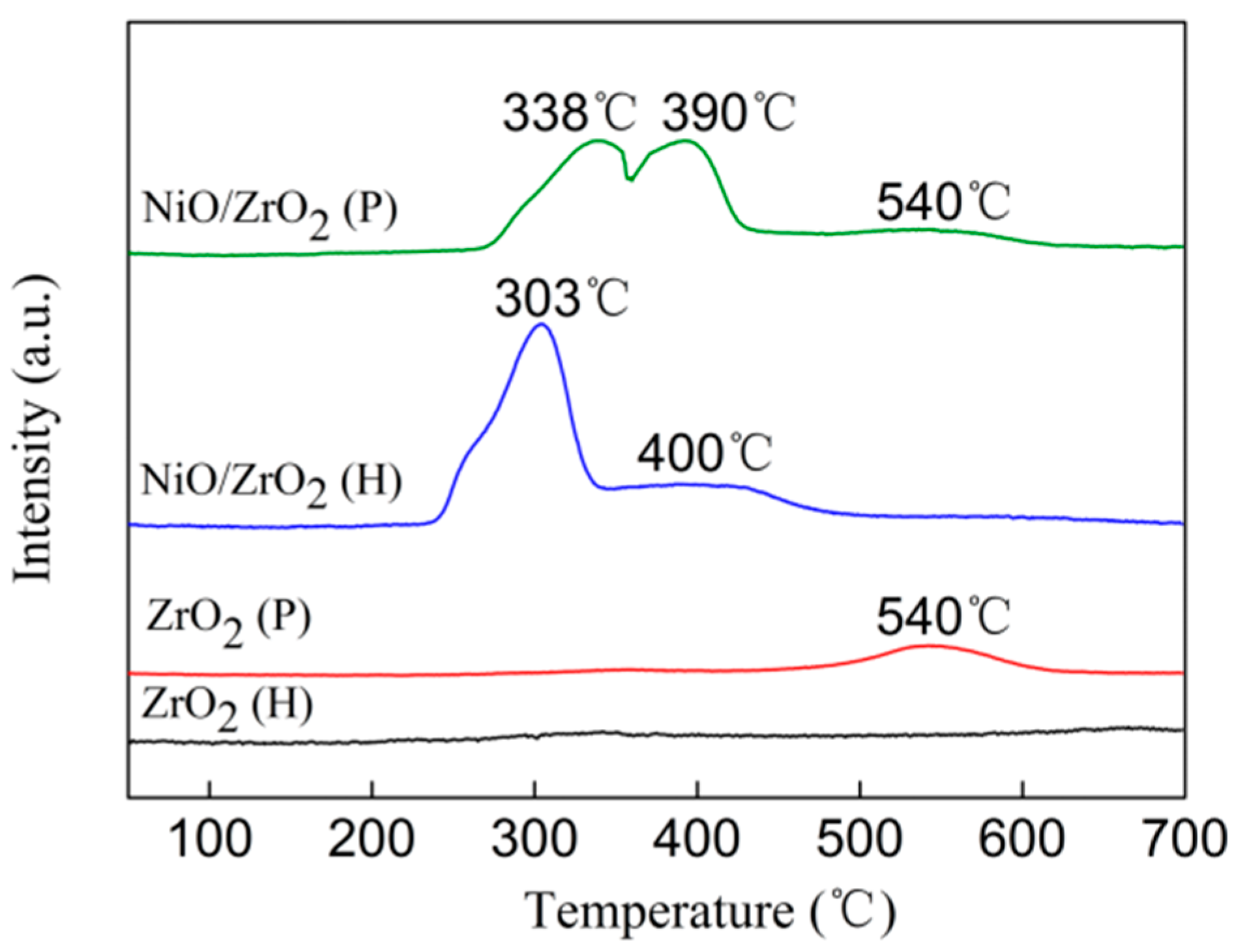
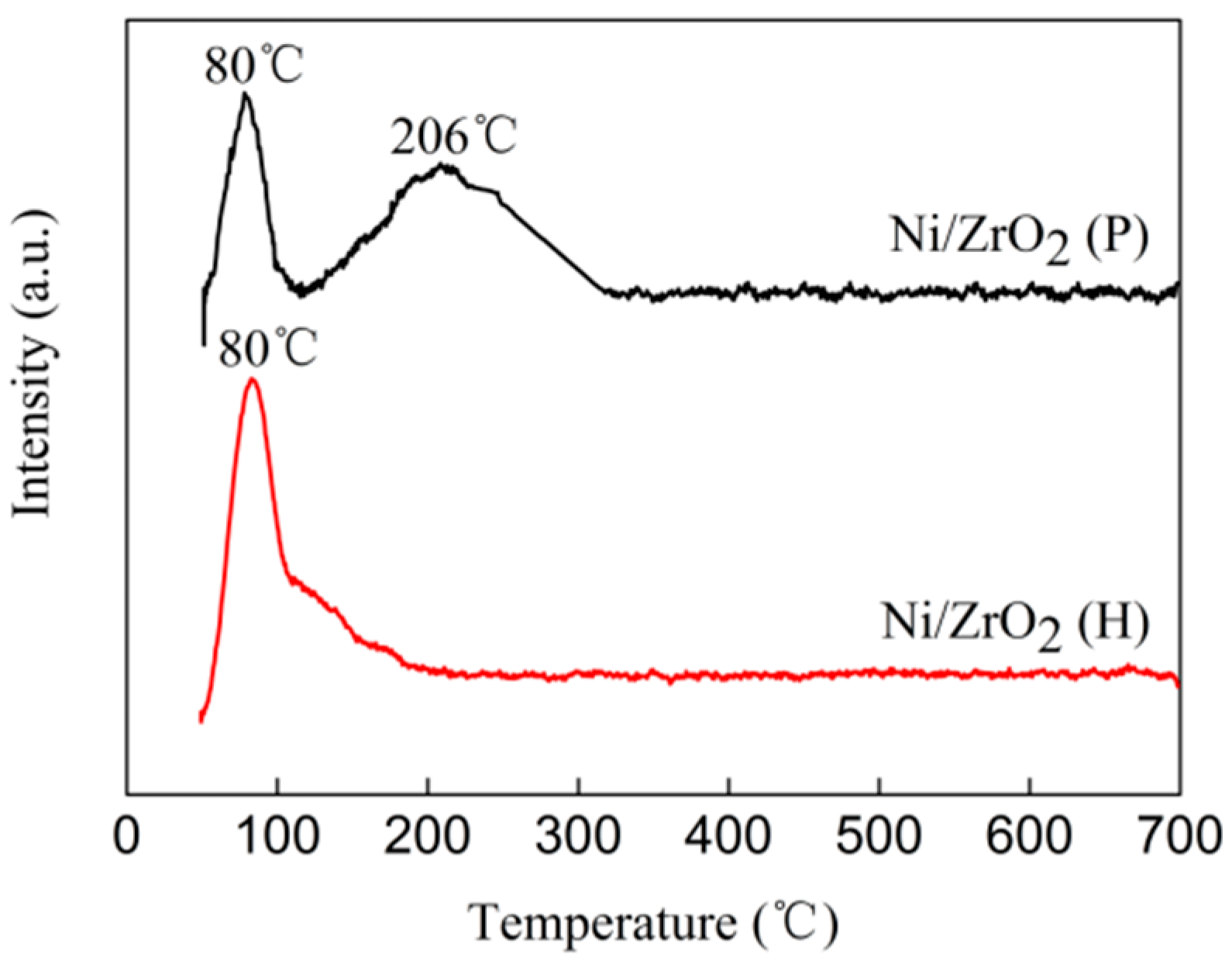
3.4. H2 Temperature-Programmed Reduction (H2-TPR)
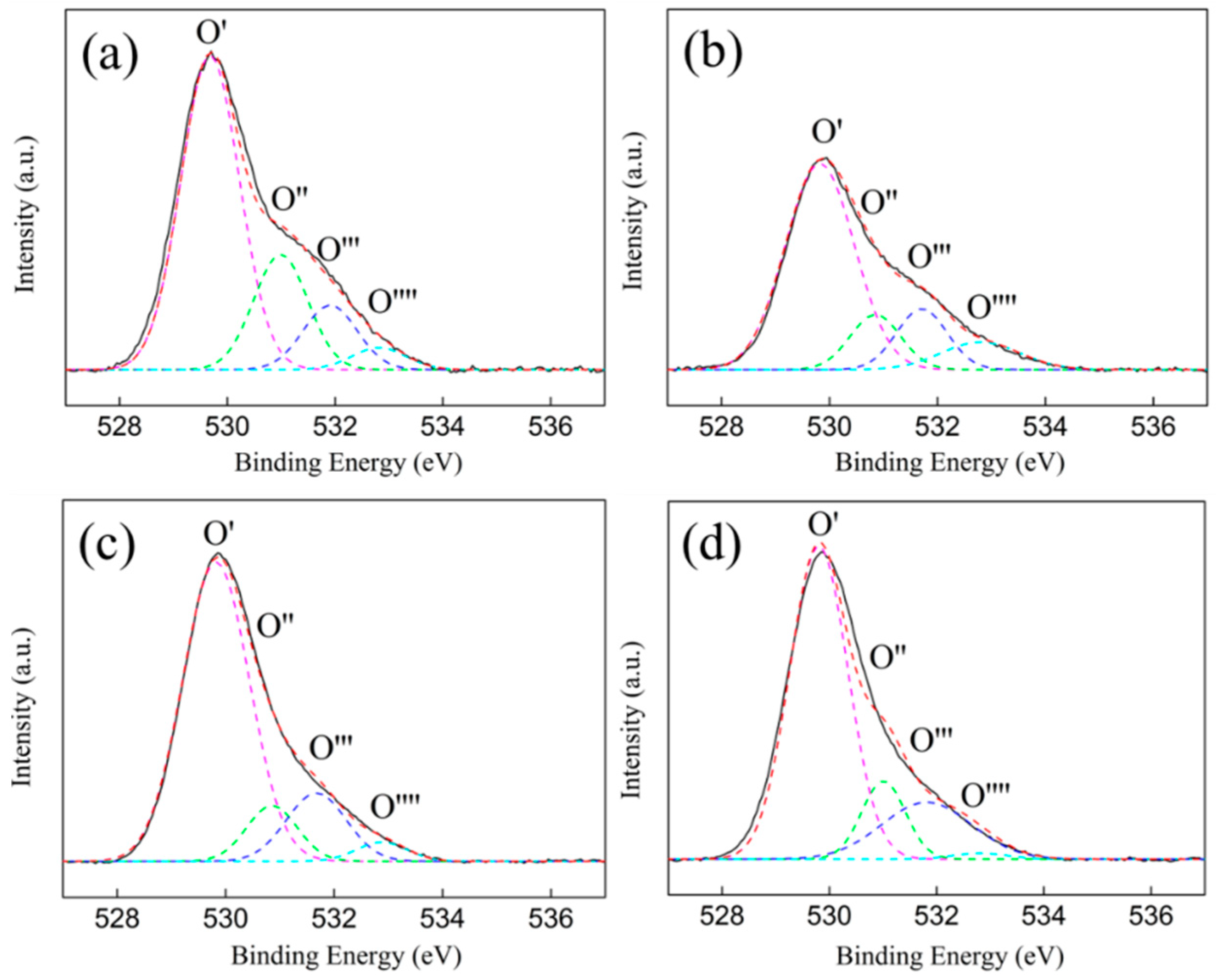
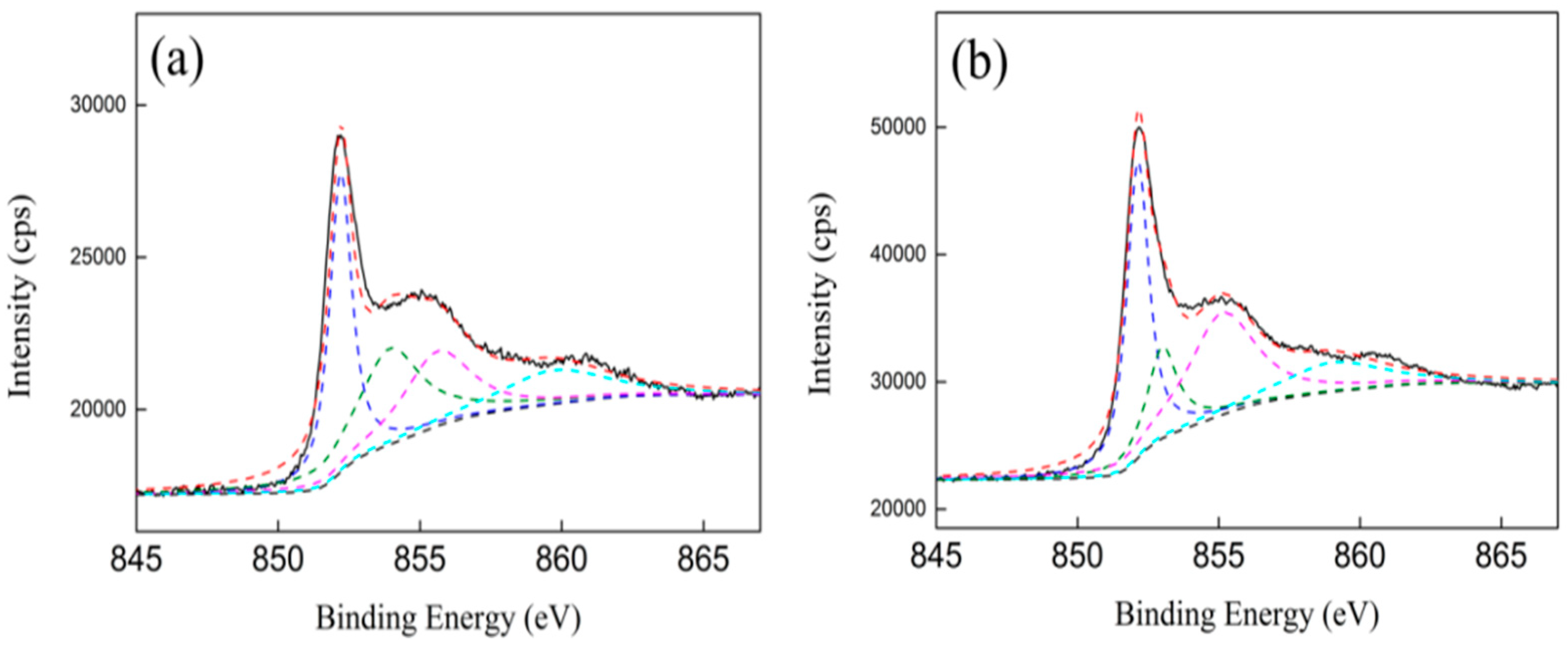
3.5. XPS Characterization
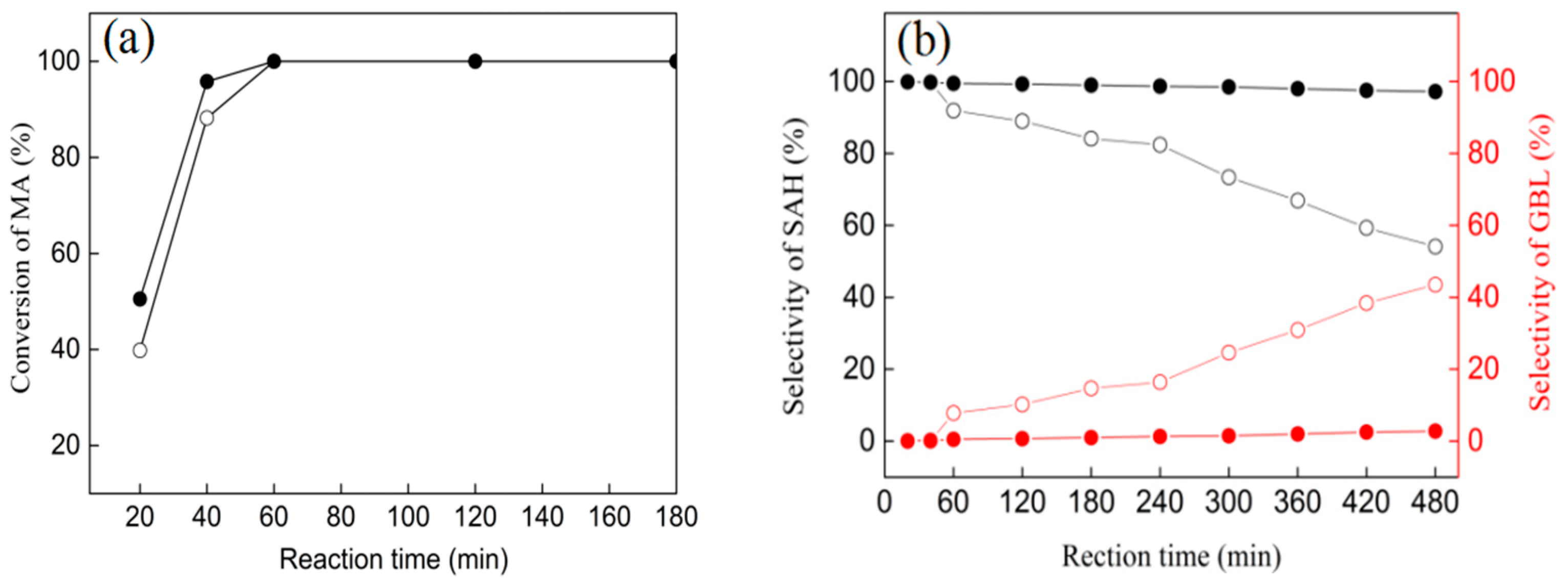
3.6. Catalytic Performances of Ni/ZrO2 (P) and Ni/ZrO2 (H) Catalysts
3.7. H2 Temperature-Programmed Desorption (H2-TPD)
3.8. In-Situ FT-IR Spectra
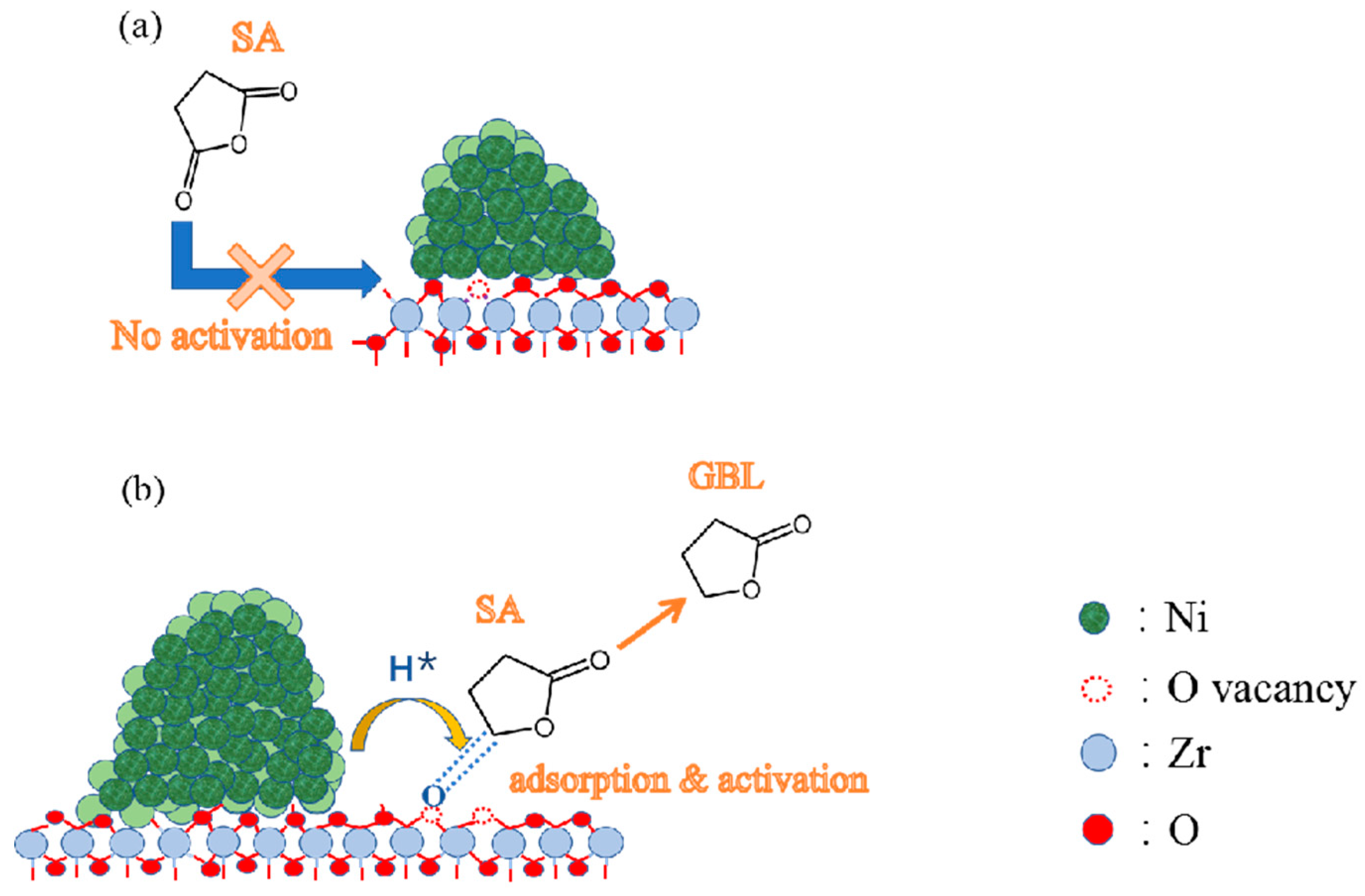
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Trivedi, B.C.; Culbertson, B.M. Maleic Anhydride, 1st ed.; Plenum Press: New York, NY, USA, 2013; pp. 132–133. [Google Scholar]
- Li, J.; Tian, W.P.; Shi, L. Hydrogenation of Maleic Anhydride to Succinic Anhydride over Ni/HY-Al2O3. Ind. Eng. Chem. Res 2010, 49, 11837–11840. [Google Scholar] [CrossRef]
- Delbecq, F.; Sautet, P. A Density Functional Study of Adsorption Structures of Unsaturated Aldehydes on Pt (111): A Key Factor for Hydrogenation Selectivity. J. Catal. 2002, 211, 398–406. [Google Scholar] [CrossRef]
- Lopez, A.; Bitzer, T.; Heller, T.; Richardson, N.V. Adsorption of maleic anhydride on Si (100)-2×1. Surf. Sci. 2001, 477, 219–226. [Google Scholar] [CrossRef]
- Jung, S.M.; Godard, E.; Jung, S.Y.; Park, K.C.; Choi, J.U. Liquid-phase hydrogenation of maleic anhydride over Pd/SiO2: Effect of tin on catalytic activity and deactivation. J. Mol. Catal. A Chem. 2003, 198, 297–302. [Google Scholar] [CrossRef]
- Pallassana, V.; Neurock, M. First-Principles Periodic Density Functional Study of the Hydrogenation of Maleic Anhydride to Succinic Anhydride over Palladium (111). J. Phys. Chem. B 2000, 104, 9449–9459. [Google Scholar] [CrossRef]
- Ponec, V. On the role of promoters in hydrogenations on metals; α-β unsaturated aldehydes and ketones. Appl. Catal. A Gen. 1997, 149, 27–48. [Google Scholar] [CrossRef]
- Yu, Y.; Zhan, W.C.; Guo, Y.; Lu, G.Z.; Adjimi, S.; Guo, Y.L. Gas-phase hydrogenation of maleic anhydride to γ-butyrolactone over Cu-CeO2-Al2O3 catalyst at atmospheric pressure: Effects of the residual sodium and water in the catalyst precursor. J. Mol. Catal. A Chem. 2014, 395, 392–397. [Google Scholar] [CrossRef]
- Meyer, C.I.; Marchi, A.J.; Monzon, A.; Garetto, T.F. Deactivation and regeneration of Cu/SiO2 catalyst in the hydrogenation of maleic anhydride. Kinetic modelling. Appl. Catal. A Gen. 2009, 367, 122–129. [Google Scholar] [CrossRef]
- Feng, Y.H.; Yin, H.B.; Wang, A.L.; Xie, T.; Jiang, T.S. Selective hydrogenation of maleic anhydride to succinic anhydride catalyzed by metallic nickel catalysts. Appl. Catal. A Gen. 2012, 425-426, 205–212. [Google Scholar] [CrossRef]
- Li, J.; Ren, Y.H.; Yue, B.; He, H.Y. Ni/Al2O3 catalysts derived from spinel NiAl2O4 for low-temperature hydrogenation of maleic anhydride to succinic anhydride. Chin. J. Catal. 2017, 38, 1166–1173. [Google Scholar] [CrossRef]
- Li, J.; Tian, W.P.; Shi, L. Highly Active and Selective Nickel-Platinum Catalyst for the Low Temperature Hydrogenation of Maleic Anhydride to Succinic Anhydride and Synthesis of Succinic Acid at 40 °C. Catal. Lett. 2011, 141, 565–571. [Google Scholar] [CrossRef]
- Guo, S.F.; Shi, L. Synthesis of succinic anhydride from maleic anhydride on Ni/diatomite catalysts. Catal. Today 2013, 212, 137–141. [Google Scholar] [CrossRef]
- Meyer, C.I.; Regenhardt, S.A.; Bertone, M.E.; Marchi, A.J.; Garetto, T.F. Gas-Phase Maleic Anhydride Hydrogenation Over Ni/SiO2-Al2O3 Catalysts: Effect of Metal Loading. Catal. Lett. 2003, 143, 1067–1073. [Google Scholar] [CrossRef]
- Regenhardt, S.A.; Meyer, C.I.; Garetto, T.F.; Marchi, A.J. Selective gas phase hydrogenation of maleic anhydride over Ni-supported catalysts: Effect of support on the catalytic performance. Appl. Catal. A Gen. 2012, 449, 81–87. [Google Scholar] [CrossRef]
- Cai, J.X.; Zhu, J.X.; Zuo, L.; Fu, Y.C.; Shen, J.Y. Effect of surface acidity/basicity on the selective hydrogenation of maleic anhydride to succinic anhydride over supported nickel catalysts. Catal. Commun. 2018, 110, 93–96. [Google Scholar] [CrossRef]
- Huo, W.T.; Zhang, C.L.; Yuan, H.J.; Jia, M.J.; Ning, C.L.; Tang, Y.; Zhang, Y.; Luo, J.H.; Wang, Z.L.; Zhang, W.X. Vapor-phase selective hydrogenation of maleic anhydride to succinic anhydride over Ni/TiO2 catalysts. J. Ind. Eng. Chem. 2014, 20, 4140–4145. [Google Scholar] [CrossRef]
- Torresa, C.C.; Alderetea, J.B.; Mellab, C.; Pawelec, B. Maleic anhydride hydrogenation to succinic anhydride over mesoporous Ni/TiO2 catalysts: Effects of Ni loading and temperature. J. Mol. Catal. A Chem. 2016, 423, 441–448. [Google Scholar] [CrossRef]
- Hu, T.J.; Yin, H.B.; Zhang, R.C.; Wu, H.X.; Jiang, T.S.; Wada, Y.J. Gas phase hydrogenation of maleic anhydride to γ-butyrolactone by Cu-Zn-Ti catalysts. Catal. Commun. 2007, 8, 193–199. [Google Scholar] [CrossRef]
- Yu, Y.; Guo, Y.L.; Zhan, W.C.; Guo, Y.; Wang, Y.Q.; Wang, Y.S.; Zhang, Z.G.; Lu, G.Z. Gas-phase hydrogenation of maleic anhydride to γ-butyrolactone at atmospheric pressure over Cu-CeO2-Al2O3 catalyst. J. Mol. Catal. A Chem. 2011, 337, 77–81. [Google Scholar] [CrossRef]
- Liao, X.; Zhang, Y.; Hill, M.; Xia, X.; Zhao, Y.X.; Jiang, Z. Highly efficient Ni/CeO2 catalyst for the liquid phase hydrogenation of maleic anhydride. Appl. Catal. A Gen. 2014, 488, 256–264. [Google Scholar] [CrossRef]
- Lin, Y.P.; Zhu, Y.F.; Pan, X.L.; Bao, X.H. Modulating the methanation activity of Ni by the crystal phase of TiO2. Catal. Sci. Technol. 2017, 7, 2813–2818. [Google Scholar] [CrossRef]
- Resende, K.A.; Braga, A.H.; Noronha, F.B.; Hori, C.E. Hydrodeoxygenation of phenol over Ni/Ce1−xNbxO2 catalysts. Appl. Catal. B Environ. 2019, 245, 100–113. [Google Scholar] [CrossRef]
- Wang, Y.; Arandiyan, H.; Scott, J.; Dai, H.X.; Amal, R. Hierarchically Porous Network-Like Ni/Co3O4: Noble Metal-Free Catalysts for Carbon Dioxide Methanation. Adv. Sustain. Syst. 2018, 2, 1700119. [Google Scholar] [CrossRef]
- Arandiyan, H.; Chang, H.Z.; Liu, C.X.; Peng, Y.; Li, J.H. Dextrose-aided hydrothermal preparation with large surface area on 1D single-crystalline perovskite La0.5Sr0.5CoO3 nanowires without template: Highly catalytic activity for methane combustion. J. Mol. Catal. A Chem. 2013, 378, 299–306. [Google Scholar] [CrossRef]
- Arandiyan, H.; Scott, J.; Wang, Y.; Dai, H.X.; Sun, H.Y.; Amal, R. Meso-Molding Three-Dimensional Macroporous Perovskites: A New Approach to Generate High-Performance Nanohybrid Catalysts. ACS Appl. Mater. Interfaces 2016, 8, 2457–2463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Y.F.; Kong, X.; Cao, D.B.; Cui, J.L.; Zhu, Y.L.; Li, Y.W. The Rise of Calcination Temperature Enhances the Performance of Cu Catalysts: Contributions of Support. ACS Catal. 2014, 4, 3675–3681. [Google Scholar] [CrossRef]
- De Souza, P.M.; Rabelo-Neto, R.C.; Borges, L.E.P.; Jacobs, G.; Davis, B.H.; Graham, U.M.; Resasco, D.E.; Noronha, F.B. Effect of Zirconia Morphology on Hydrodeoxygenation of Phenol over Pd/ZrO2. ACS Catal. 2015, 5, 7385–7398. [Google Scholar] [CrossRef]
- Samson, K.; Ś liwa, M.; Socha, R.P.; Gora-Marek, K.; Mucha, D.; Rutkowska-Zbik, D.; Paul, J-F.; Ruggiero-Mikołajczyk, M.; Grabowski, R.; Słoczynsk, J. Influence of ZrO2 Structure and Copper Electronic State on Activity of Cu/ZrO2 Catalysts in Methanol Synthesis from CO2. ACS Catal. 2014, 4, 3730–3741. [Google Scholar] [CrossRef]
- Rhodes, M.D.; Bell, A.T. The effects of zirconia morphology on methanol synthesis from CO and H2 over Cu/ZrO2 catalysts Part I. Steady-state studies. J. Catal. 2005, 233, 198–209. [Google Scholar] [CrossRef]
- Zhang, X.P.; Zhang, Q.D.; Tsubaki, N.; Tan, Y.S.; Han, Y.Z. Influence of Zirconia Phase on the Performance of Ni/ZrO2 for Carbon Dioxide Reforming of Methane; ACS Symposium Series; American Chemical Society: Washington, DC, USA, 2015; Volume 1194, pp. 135–153. [Google Scholar]
- Jung, K.T.; Bell, A.T. The effects of synthesis and pretreatment conditions on the bulk structure and surface properties of zirconia. J. Mol. Catal. A Chem. 2000, 163, 27–42. [Google Scholar] [CrossRef]
- Li, W.Z.; Huang, H.; Li, H.J.; Zhang, W.; Liu, H.C. Facile Synthesis of Pure Monoclinic and Tetragonal Zirconia Nanoparticles and Their Phase Effects on the Behavior of Supported Molybdena Catalysts for Methanol-Selective Oxidation. Langmuir 2008, 24, 8358–8366. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.H.; Lu, F.; Liut, D.W.; Alzyabt, B. Phonons and Phase Transitions in Zirconia. J. Raman Spectrosc. 1990, 21, 577–584. [Google Scholar] [CrossRef]
- Bish, D.L.; Howard, S.A. Quantitative phase analysis using the Rietveld method. J. Appl. Crystallogr. 1988, 21, 86–91. [Google Scholar] [CrossRef] [Green Version]
- Shukla, S.; Seal, S. Mechanisms of room temperature metastable tetragonal phase stabilisation in zirconia. Int. Mater. Rev. 2005, 50, 45–64. [Google Scholar] [CrossRef]
- Yashima, M.; Ohtake, K.; Kakihana, M.; Arashis, H.; Yoshimura, M. Determination of tetragonal-cubic phase boundary of Zr1−xRxO2−x/2 (R = Nd, Sm, Y, Er and Yb) by Raman scattering. J. Phys. Chem. Solids 1996, 57, 17–24. [Google Scholar] [CrossRef]
- Li, C.; Li, M.J. UV Raman spectroscopic study on the phase transformation of ZrO2, Y2O3-ZrO2 and SO42−/ZrO2. J. Raman Spectrosc. 2002, 33, 301–308. [Google Scholar] [CrossRef]
- Shi, L.; Tin, K.C.; Wong, N.B. Thermal stability of zirconia membranes. J. Mater. Sci. 1999, 34, 3367–3374. [Google Scholar] [CrossRef]
- Lopez, E.F.; Escribano, V.S.; Panizza, M.; Carnascialic, M.M.; Busca, G. Vibrational and electronic spectroscopic properties of zirconia Powders. J. Mater. Chem. 2001, 11, 1891–1897. [Google Scholar] [CrossRef]
- Thackeray, D.P.C. The Raman spectrum of zirconium dioxide. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 1974, 30, 549–550. [Google Scholar] [CrossRef]
- Yashima, M.; Ohtake, K.; Arashi, H.; Kakihana, M.; Yoshimura, M. Determination of cubic-tetragonal phase boundary in Zr1-XYXO2-X/2 solid solutions by Raman spectroscopy. J. Appl. Phys. 1993, 74, 7603–7605. [Google Scholar] [CrossRef]
- Chen, S.Q.; Li, L.P.; Hu, W.B.; Huang, X.S.; Li, Q.; Xu, Y.S.; Zuo, Y.Y.; Li, G.S. Anchoring High-Concentration Oxygen Vacancies at Interfaces of CeO2-x/Cu toward Enhanced Activity for Preferential CO Oxidation. ACS Appl. Mater. Interfaces 2015, 7, 22999–23007. [Google Scholar] [CrossRef] [PubMed]
- Puigdollers, A.R.; Illas, F.; Pacchioni, G. Structure and Properties of Zirconia Nanoparticles from Density Functional Theory Calculations. J. Phys. Chem. C 2016, 120, 4392–4402. [Google Scholar] [CrossRef]
- Tosoni, S.; Chen, H.Y.T.; Pacchioni, G. A DFT study of Ni clusters deposition on titania and zirconia (101) surfaces. Surf. Sci. 2016, 646, 230–238. [Google Scholar] [CrossRef]
- Yang, F.F.; Liu, D.; Zhao, Y.T.; Wang, H.; Han, J.Y.; Ge, Q.F.; Zhu, X.L. Size Dependence of Vapor Phase Hydrodeoxygenation of m-Cresol on Ni/SiO2 Catalysts. ACS Catal. 2018, 8, 1672–1682. [Google Scholar] [CrossRef]
- Rahman, M.A.; Rout, S.; Thomas, J.P.; McGillivray, D.; Leung, K.T. Defect-Rich Dopant-Free ZrO2 Nanostructures with Superior Dilute Ferromagnetic Semiconductor Properties. J. Am. Chem. Soc. 2016, 138, 11896–11906. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.J.; Li, M.; Hua, Q.; Zhang, L.J.; Ma, Y.S.; Ye, B.J.; Huang, W.X. Shape-dependent interplay between oxygen vacancies and Ag-CeO2 interaction in Ag/CeO2 catalysts and their influence on the catalytic activity. J. Catal. 2012, 293, 195–204. [Google Scholar] [CrossRef]
- Puigdollers, A.R.; Schlexer, P.; Tosoni, S.; Pacchioni, G. Increasing Oxide Reducibility: The Role of Metal/Oxide Interfaces in the Formation of Oxygen Vacancies. ACS Catal. 2017, 7, 6493–6513. [Google Scholar] [CrossRef]
- Hengne, A.M.; Samal, A.K.; Enakonda, L.R.; Harb, M.; Gevers, L.E.; Anjum, D.H.; Hedhili, M.N.; Saih, Y.; Huang, K.W.; Marie Basset, J. Ni-Sn-Supported ZrO2 Catalysts Modified by Indium for Selective CO2 Hydrogenation to Methanol. ACS Omega 2018, 3, 3688–3701. [Google Scholar] [CrossRef]
- Martensson, A.S.; Nyberg, C.; Andersson, S. Adsorption of hydrogen on a stepped nickel surface. Surf. Sci 1988, 205, 12–24. [Google Scholar] [CrossRef]
- Cui, J.L.; Tan, J.J.; Zhu, Y.L.; Cheng, F.Q. Aqueous Hydrogenation of Levulinic Acid to 1,4-Pentanediol over Mo-Modified Ru/Activated Carbon Catalyst. ChemSusChem 2018, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Bertero, N.M.; Trasarti, A.F.; Apesteguía, C.R.; Marchi, A.J. Solvent effect in the liquid-phase hydrogenation of acetophenone over Ni/SiO2: A comprehensive study of the phenomenon. Appl. Catal. A Gen. 2011, 394, 228–238. [Google Scholar] [CrossRef]
- Hu, Q.; Yang, L.; Fan, G.L.; Li, F. Hydrogenation of biomass-derived compounds containing a carbonyl group over a copper-based nanocatalyst: Insight into the origin and influence of surface oxygen vacancies. J. Catal. 2016, 340, 184–195. [Google Scholar] [CrossRef]
- Han, J.S.; Kim, Y.H.; Jang, H.S.; Hwang, S.Y.; Jegal, J.; Kim, J.W.; Lee, Y.S. Heterogeneous zirconia-supported ruthenium catalyst for highly selective hydrogenation of 5- hydroxymethyl-2-furaldehyde to 2,5-bis(hydroxymethyl)furans in various n-alcohol solvents. RSC Adv. 2016, 6, 93394–93397. [Google Scholar] [CrossRef]
- Manyar, H.G.; Paun, C.; Pilus, R.; Rooney, D.W.; Thompson, J.M.; Hardacre, C. Highly selective and efficient hydrogenation of carboxylic acids to alcohols using titania supported Pt catalysts. Chem. Commun. 2010, 46, 6279–6281. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Catalysts | Metal Content [a] [wt %] | SBET [m2g−1] | Pore Diameter [nm] | Tetragonal Phase [b] [%] | Tetragonal Phase [c] [%] | Tetragonal Phase [d] [%] | Crystalline Size of ZrO2 [e] [nm] | Crystalline Size of NiO [f] [nm] | Crystalline Size of Ni [g] [nm] |
|---|---|---|---|---|---|---|---|---|---|
| ZrO2 (P) | - | 98 | 7.1 | 48 | distorted | 23 | 16 | - | - |
| NiO/ZrO2 (P) | - | 73 | 5.8 | 47 | - | disordered | 17 | 16 | - |
| Ni/ZrO2 (P) | 9.42 | 53 | 3.5 | 46 | - | - | 17 | - | 18 |
| ZrO2 (H) | - | 71 | 3.8 | 39 | 35 | 11 | 10 | - | - |
| NiO/ZrO2 (H) | - | 58 | 3.7 | 41 | - | distorted | 10 | 26 | - |
| Ni/ZrO2 (H) | 9.39 | 25 | 3.6 | 41 | - | - | 10 | - | 40 |
| Samples | Binding Energy (eV) | I (%) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| BE1 | BE2 | BE3 | BE4 | I1 (%) | I2 (%) | I3 (%) | I4 (%) | I2 + I3 (%) | |
| ZrO2 (P) | 529.7 | 531.0 | 531.9 | 532.8 | 62.9 | 21.2 | 11.9 | 4.0 | 33.1 |
| ZrO2 (H) | 529.8 | 530.8 | 531.7 | 532.8 | 71.0 | 11.6 | 12.9 | 4.5 | 24.5 |
| Ni/ZrO2 (P) | 529.8 | 530.8 | 531.7 | 532.8 | 71.8 | 10.1 | 14.6 | 3.5 | 24.7 |
| Ni/ZrO2 (H) | 529.8 | 531.0 | 531.9 | 532.8 | 70.5 | 13.4 | 13.0 | 3.1 | 26.4 |
| Samples | Ni0 (Ni 2P3/2) | NiO (Ni 2P3/2) | Ni2+ (Ni 2P3/2) | C(Ni0) (%) | C(NiO) (%) | C(Ni2+) (%) |
|---|---|---|---|---|---|---|
| Ni/ZrO2 (P) | 852.2 | 853.9 | 855.6 | 57.8 | 22.5 | 19.7 |
| Ni/ZrO2 (H) | 852.2 | 853.0 | 855.1 | 58.4 | 19.2 | 22.4 |
| Catalysts | Temperature (°C) | Pressure (MPa) | Conv. (%) | GBL Selec. (%) |
|---|---|---|---|---|
| Ni/ZrO2 (P) | 210 | 5 | 100 | 2.8 |
| 240 | 5 | 100 | 4.9 | |
| 210 | 7 | 100 | 3.2 | |
| Ni/ZrO2 (H) | 210 | 5 | 100 | 43.5 |
| 240 | 5 | 100 | 60.6 | |
| 210 | 7 | 100 | 46.2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhao, L.; Zhao, J.; Wu, T.; Zhao, M.; Yan, W.; Zhang, Y.; Li, H.; Wang, Y.; Xiao, T.; Zhao, Y. Synergistic Effect of Oxygen Vacancies and Ni Species on Tuning Selectivity of Ni/ZrO2 Catalyst for Hydrogenation of Maleic Anhydride into Succinic Anhydride and γ-Butyrolacetone. Nanomaterials 2019, 9, 406. https://doi.org/10.3390/nano9030406
Zhao L, Zhao J, Wu T, Zhao M, Yan W, Zhang Y, Li H, Wang Y, Xiao T, Zhao Y. Synergistic Effect of Oxygen Vacancies and Ni Species on Tuning Selectivity of Ni/ZrO2 Catalyst for Hydrogenation of Maleic Anhydride into Succinic Anhydride and γ-Butyrolacetone. Nanomaterials. 2019; 9(3):406. https://doi.org/10.3390/nano9030406
Chicago/Turabian StyleZhao, Lili, Jianghong Zhao, Tianjie Wu, Min Zhao, Wenjun Yan, Yin Zhang, Haitao Li, Yongzhao Wang, Tiancun Xiao, and Yongxiang Zhao. 2019. "Synergistic Effect of Oxygen Vacancies and Ni Species on Tuning Selectivity of Ni/ZrO2 Catalyst for Hydrogenation of Maleic Anhydride into Succinic Anhydride and γ-Butyrolacetone" Nanomaterials 9, no. 3: 406. https://doi.org/10.3390/nano9030406
APA StyleZhao, L., Zhao, J., Wu, T., Zhao, M., Yan, W., Zhang, Y., Li, H., Wang, Y., Xiao, T., & Zhao, Y. (2019). Synergistic Effect of Oxygen Vacancies and Ni Species on Tuning Selectivity of Ni/ZrO2 Catalyst for Hydrogenation of Maleic Anhydride into Succinic Anhydride and γ-Butyrolacetone. Nanomaterials, 9(3), 406. https://doi.org/10.3390/nano9030406