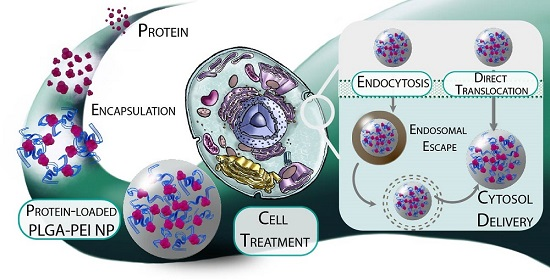
Nanocarriers for Protein Delivery to the Cytosol: Assessing the Endosomal Escape of Poly(Lactide-co-Glycolide)-Poly(Ethylene Imine) Nanoparticles



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
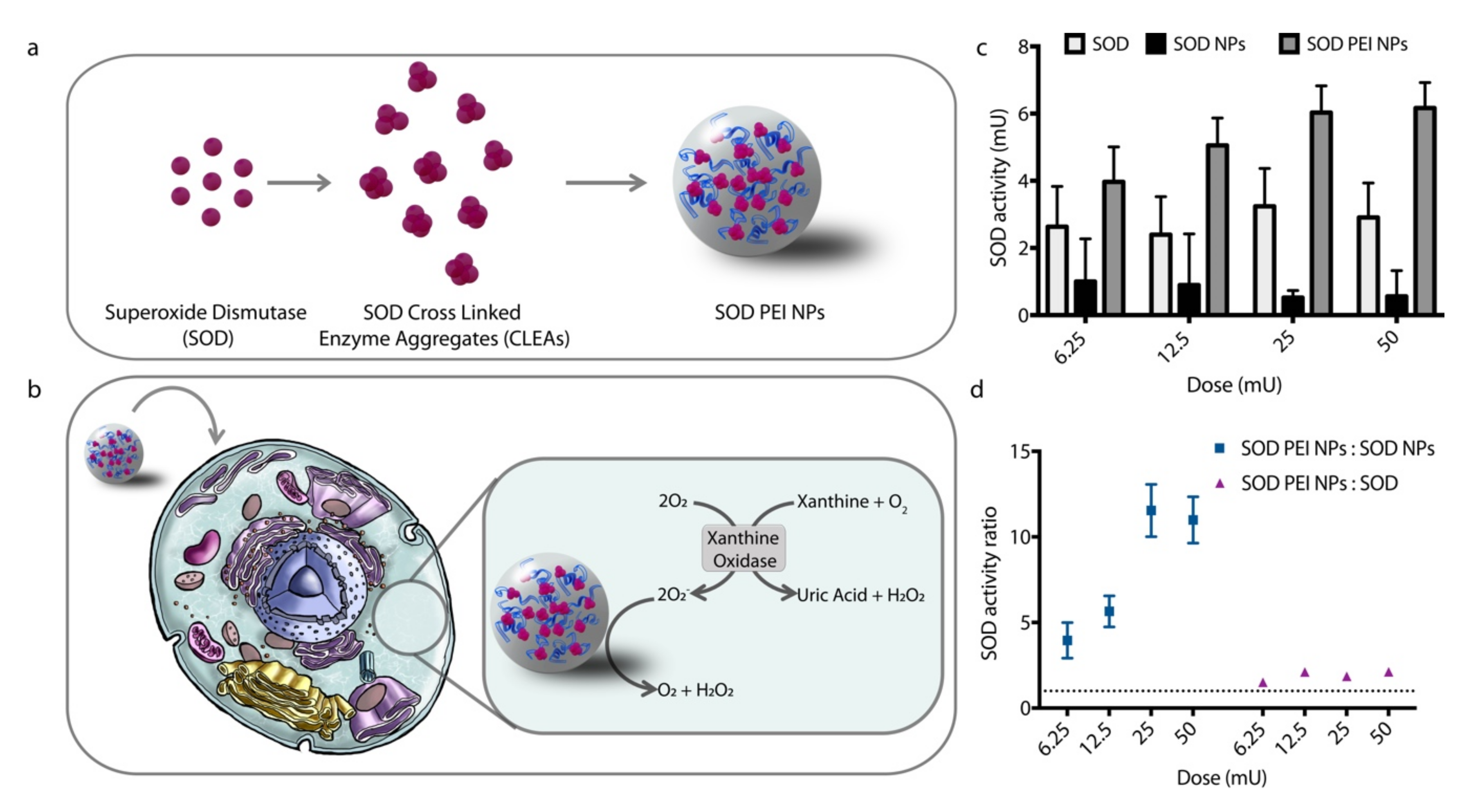
2.2. Cross-linked Aggregates (CLAs) and cross-linked enzyme aggregates (CLEAs) Synthesis
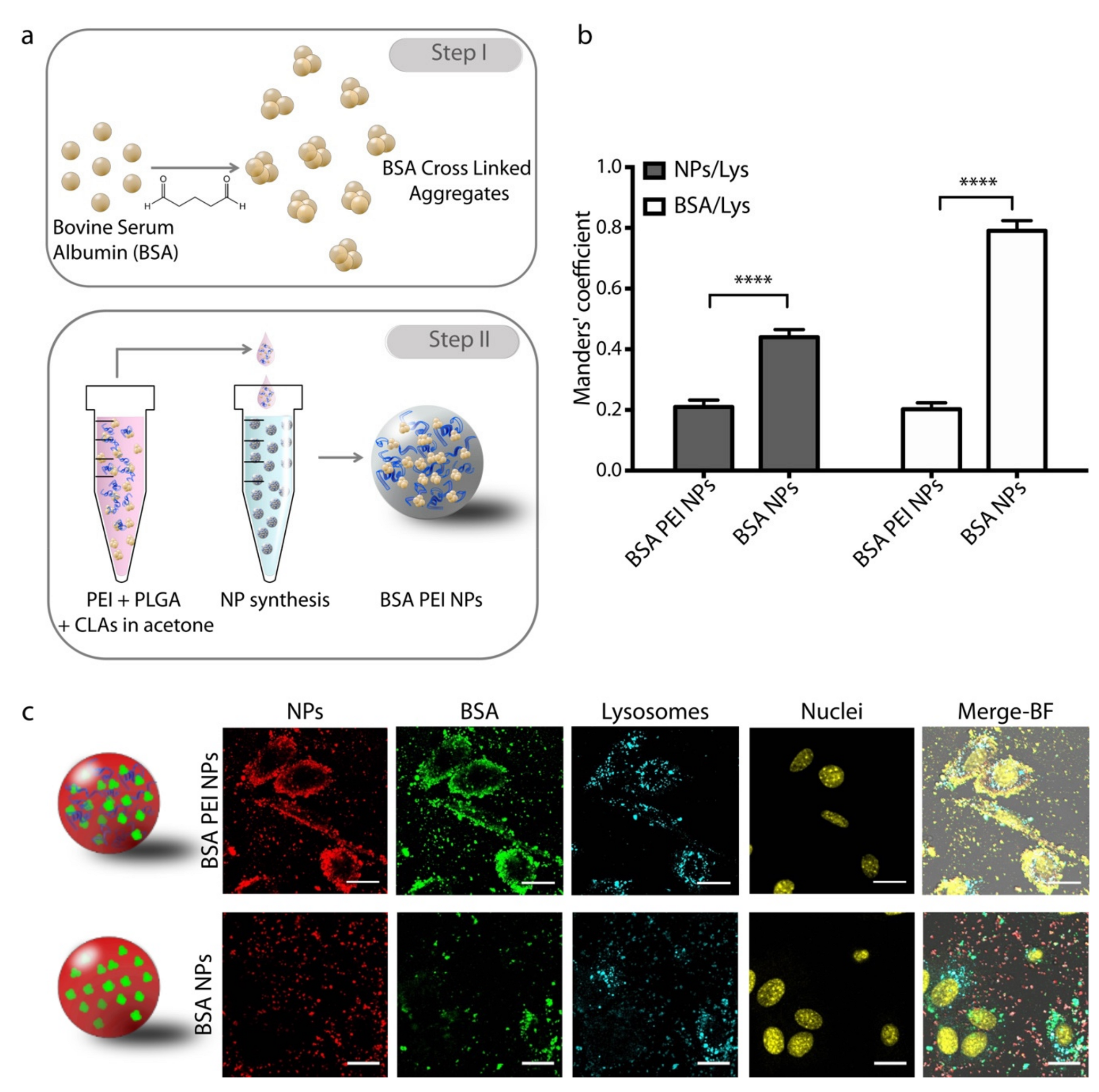
2.3. Nanoparticle (NP) Synthesis
2.4. NP Characterization
2.4.1. Dynamic Light Scattering and Zeta Potential
2.4.2. Protein Quantitation
2.5. Cell Culture
2.6. Endosomal Escape of NPs
2.6.1. Intracellular Localization of NPs
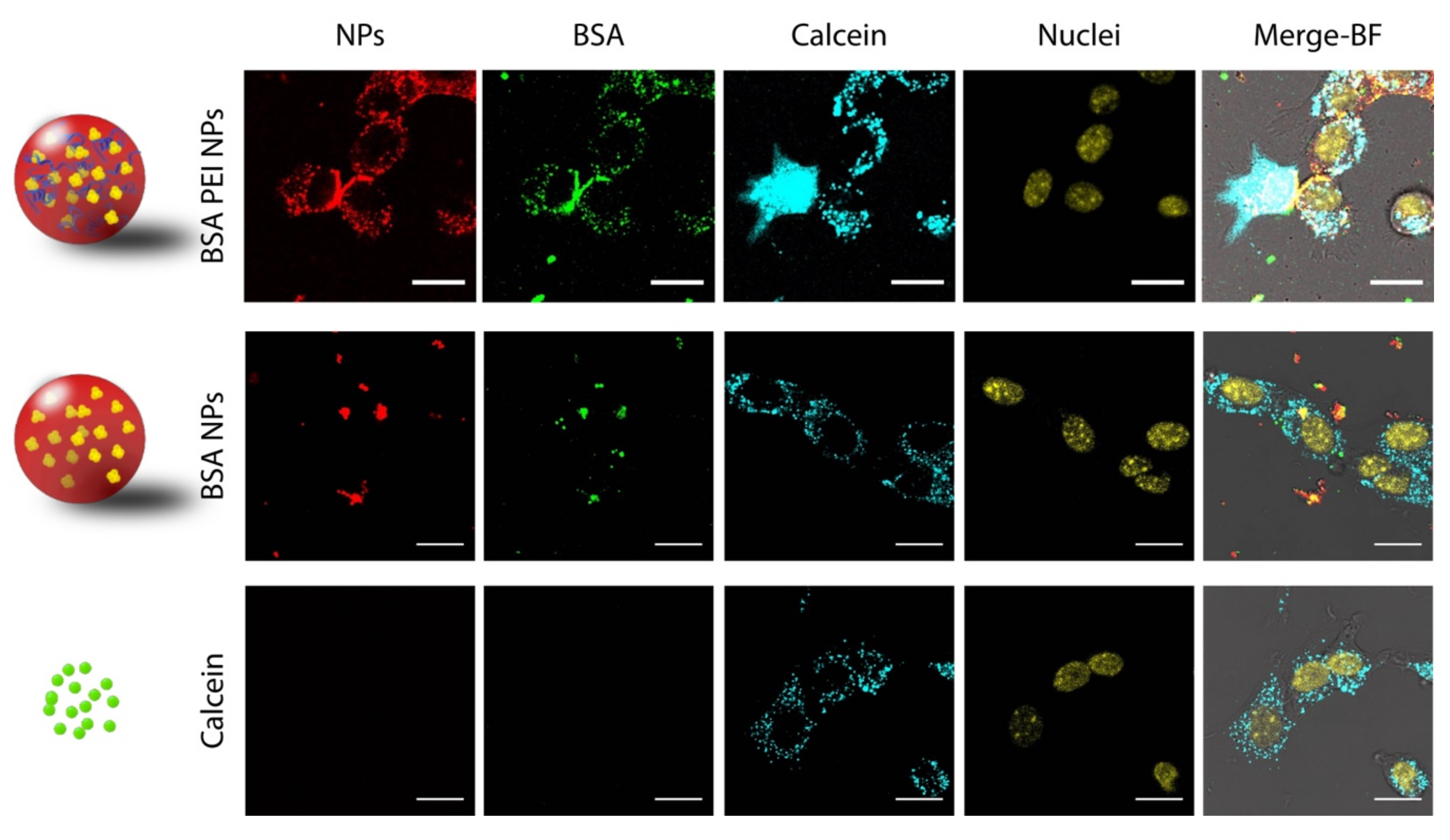
2.6.2. Calcein Leakage Assay
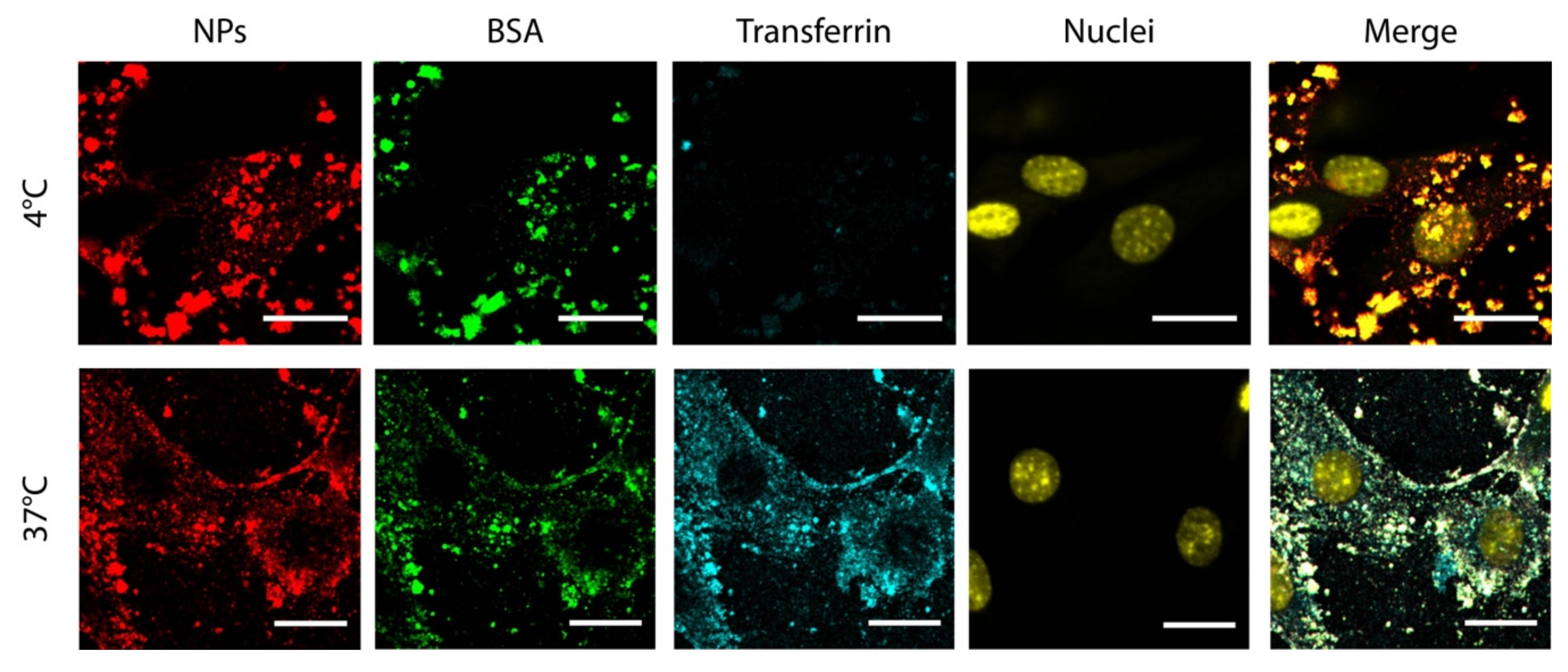
2.6.3. Endocytosis Inhibition
2.6.4. Confocal Imaging
2.7. SOD Enzymatic Activity
- (a)
- Assay buffer (10 mM sodium carbonate, 20 mM EDTA).
- (b)
- 740 μg/mL Nitro Blue Tetrazolium chloride (NBT), 300 μg/mL xanthine in assay buffer (for NP assay) or DMEM (for in-cell assay).
- (c)
- 28 μg/mL Xanthine Oxidase from milk (XOD) in assay buffer (for NP assay) or DMEM (for in-cell assay).
3. Results and Discussion
3.1. NP synthesis and Characterization
3.2. Intracellular Localization of NPs
3.3. Calcein Leakage Assay
3.4. Internalization Kinetics and Mechanism Study
3.5. Delivery of a Cytosolic Enzyme
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Leader, B.; Baca, Q.J.; Golan, D.E. Protein therapeutics: A summary and pharmacological classification. Nat. Rev. Drug Discov. 2008, 7, 23–99. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.-H.; Liu, C.-H.; Liao, S.-H.; Lin, Y.-Y.; Tang, H.-W.; Liu, S.-Y.; Lai, I.-R.; Wu, K.C.-W. Cosynthesis of Cargo-Loaded Hydroxyapatite/Alginate Core–Shell Nanoparticles (HAP@Alg) as pH-Responsive Nanovehicles by a Pre-gel Method. ACS Appl. Mater. Interfaces 2012, 4, 6720–6727. [Google Scholar] [CrossRef]
- Wang, L.; Liu, C.-H.; Nemoto, Y.; Fukata, N.; Wu, K.C.-W.; Yamauchi, Y. Rapid synthesis of biocompatible gold nanoflowers with tailored surface textures with the assistance of amino acid molecules. RSC Adv. 2012, 2, 4608–4611. [Google Scholar] [CrossRef]
- Liao, S.-H.; Liu, C.-H.; Bastakoti, B.P.; Suzuki, N.; Chang, Y.; Yamauchi, Y.; Lin, F.-H.; Wu, K.C.-W. Functionalized magnetic iron oxide/alginate core-shell nanoparticles for targeting hyperthermia. Int. J. Nanomed. 2015, 10, 3315–3328. [Google Scholar] [Green Version]
- Liao, Y.-T.; Liu, C.-H.; Yu, J.; Wu, K.C.-W. Liver cancer cells: targeting and prolonged-release drug carriers consisting of mesoporous silica nanoparticles and alginate microspheres. Int. J. Nanomed. 2014, 9, 2767–2778. [Google Scholar] [Green Version]
- Shieh, F.-K.; Wang, S.-C.; Yen, C.-I.; Wu, C.-C.; Dutta, S.; Chou, L.-Y.; Morabito, J.V.; Hu, P.; Hsu, M.-H.; Wu, K.C.-W.; et al. Imparting Functionality to Biocatalysts via Embedding Enzymes into Nanoporous Materials by a de Novo Approach: Size-Selective Sheltering of Catalase in Metal–Organic Framework Microcrystals. J. Am. Chem. Soc. 2015, 137, 4276–4279. [Google Scholar] [CrossRef]
- Yu, M.; Wu, J.; Shi, J.; Farokhzad, O.C. Nanotechnology for protein delivery: Overview and perspectives. J. Control. Release 2016, 240, 24–37. [Google Scholar] [CrossRef]
- Lian, H.-Y.; Hu, M.; Liu, C.-H.; Yamauchi, Y.; Wu, K.C.-W. Highly biocompatible, hollow coordination polymer nanoparticles as cisplatin carriers for efficient intracellular drug delivery. Chem. Commun. 2012, 48, 5151–5153. [Google Scholar] [CrossRef]
- Bastakoti, B.P.; Hsu, Y.-C.; Liao, S.-H.; Wu, K.C.-W.; Inoue, M.; Yusa, S.; Nakashima, K.; Yamauchi, Y. Inorganic–Organic Hybrid Nanoparticles with Biocompatible Calcium Phosphate Thin Shells for Fluorescence Enhancement. Chem. An Asian J. 2013, 8, 1301–1305. [Google Scholar] [CrossRef]
- Rosalia, R.A.; Cruz, L.J.; van Duikeren, S.; Tromp, A.T.; Silva, A.L.; Jiskoot, W.; de Gruijl, T.; Löwik, C.; Oostendorp, J.; van der Burg, S.H.; et al. CD40-targeted dendritic cell delivery of PLGA-nanoparticle vaccines induce potent anti-tumor responses. Biomaterials 2015, 40, 88–97. [Google Scholar] [CrossRef]
- Kocbek, P.; Obermajer, N.; Cegnar, M.; Kos, J.; Kristl, J. Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J. Control. Release 2007, 120, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Morita, T.; Sakamura, Y.; Horikiri, Y.; Suzuki, T.; Yoshino, H. Protein encapsulation into biodegradable microspheres by a novel S/O/W emulsion method using poly (ethylene glycol) as a protein micronization adjuvant. J. Control. Release 2000, 69, 435–444. [Google Scholar] [CrossRef]
- Bilati, U.; Allémann, E.; Doelker, E. Development of a nanoprecipitation method intended for the entrapment of hydrophilic drugs into nanoparticles. Eur. J. Pharm. Sci. 2005, 24, 67–75. [Google Scholar] [CrossRef]
- Gaspar, M.M.; Blanco, D.; Cruz, M.E.M.; Alonso, M.J. Formulation of l-asparaginase-loaded poly(lactide-co-glycolide) nanoparticles: influence of polymer properties on enzyme loading, activity and in vitro release. J. Control. Release 1998, 52, 53–62. [Google Scholar] [CrossRef]
- Morales-Cruz, M.; Flores-Fernández, G.M.; Morales-Cruz, M.; Orellano, E.A.; Rodriguez-Martinez, J.A.; Ruiz, M.; Griebenow, K. Two-step nanoprecipitation for the production of protein-loaded PLGA nanospheres. Results Pharma Sci. 2012, 2, 79–85. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galliani, M.; Santi, M.; del Grosso, A.; Cecchettini, A.; Santorelli, F.M.; Hofmann, S.L.; Lu, J.Y.; Angella, L.; Cecchini, M.; Signore, G. Cross Linked Enzyme Aggregates as Versatile Tool for Enzyme Delivery: Application to Polymeric Nanoparticles. Bioconjugate Chem. 2018, 29, 2225–2231. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; van Rantwijk, F.; Sheldon, R.A. Cross-Linked Enzyme Aggregates: A Simple and Effective Method for the Immobilization of Penicillin Acylase. Org. Lett. 2000, 2, 1361–1364. [Google Scholar] [CrossRef]
- Schoevaart, R.; Wolbers, M.W.; Golubovic, M.; Ottens, M.; Kieboom, A.P.G.; van Rantwijk, F.; van der Wielen, L.A.M.; Sheldon, R.A. Preparation, optimization, and structures of cross-linked enzyme aggregates (CLEAs). Biotechnol. Bioeng. 2004, 87, 754–762. [Google Scholar] [CrossRef]
- Lai, S.K.; Hida, K.; Man, S.T.; Chen, C.; Machamer, C.; Schroer, T.A.; Hanes, J. Privileged delivery of polymer nanoparticles to the perinuclear region of live cells via a non-clathrin, non-degradative pathway. Biomaterials 2007, 28, 2876–2884. [Google Scholar] [CrossRef]
- Trif, M.; Florian, P.E.; Roseanu, A.; Moisei, M.; Craciunescu, O.; Astete, C.E.; Sabliov, C.M. Cytotoxicity and intracellular fate of PLGA and chitosan-coated PLGA nanoparticles in Madin–Darby bovine kidney (MDBK) and human colorectal adenocarcinoma (Colo 205) cells. J. Biomed. Mater. Res. 2015, 103, 3599–3611. [Google Scholar] [CrossRef]
- Behzadi, S.; Serpooshan, V.; Tao, W.; Hamaly, M.A.; Alkawareek, M.Y.; Dreaden, E.C.; Brown, D.; Alkilany, A.M.; Farokhzad, O.C.; Mahmoudi, M. Cellular uptake of nanoparticles: Journey inside the cell. Chem. Soc. Rev. 2017, 46, 4218–4244. [Google Scholar] [CrossRef]
- Wattiaux, R.; Laurent, N.; Wattiaux-De Coninck, S.; Jadot, M. Endosomes, lysosomes: Their implication in gene transfer. Adv. Drug Deliv. Rev. 2000, 41, 201–208. [Google Scholar] [CrossRef]
- Selby, L.I.; Cortez-Jugo, C.M.; Such, G.K.; Johnston, A.P.R. Nanoescapology: progress toward understanding the endosomal escape of polymeric nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1452. [Google Scholar] [CrossRef]
- Salomone, F.; Cardarelli, F.; Signore, G.; Boccardi, C.; Beltram, F. In Vitro Efficient Transfection by CM18-Tat11 Hybrid Peptide: A New Tool for Gene-Delivery Applications. PLoS ONE 2013, 8, e70108. [Google Scholar] [CrossRef]
- Salomone, F.; Cardarelli, F.; Di Luca, M.; Boccardi, C.; Nifosì, R.; Bardi, G.; di Bari, L.; Serresi, M.; Beltram, F. A novel chimeric cell-penetrating peptide with membrane-disruptive properties for efficient endosomal escape. J. Control. Release 2012, 163, 293–303. [Google Scholar] [CrossRef]
- Dong, K.; Wang, Z.; Zhang, Y.; Ren, J.; Qu, X. Metal–Organic Framework-Based Nanoplatform for Intracellular Environment-Responsive Endo/Lysosomal Escape and Enhanced Cancer Therapy. ACS Appl. Mater. Interfaces 2018, 10, 31998–32005. [Google Scholar] [CrossRef]
- Perche, F.; Yi, Y.; Hespel, L.; Mi, P.; Dirisala, A.; Cabral, H.; Miyata, K.; Kataoka, K. Hydroxychloroquine-conjugated gold nanoparticles for improved siRNA activity. Biomaterials 2016, 90, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Thomas, M.; Klibanov, A.M.; Langer, R. Exploring polyethylenimine-mediated DNA transfection and the proton sponge hypothesis. J. Gene Med. 2005, 7, 657–663. [Google Scholar] [CrossRef] [PubMed]
- Sonawane, N.D.; Szoka, F.C.; Verkman, A.S. Chloride Accumulation and Swelling in Endosomes Enhances DNA Transfer by Polyamine-DNA Polyplexes. J. Biol. Chem. 2003, 278, 44826–44831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, I.; Thibault, M.; de Crescenzo, G.; Buschmann, M.D.; Lavertu, M. Ionization Behavior of Chitosan and Chitosan-DNA Polyplexes Indicate That Chitosan Has a Similar Capability to Induce a Proton-Sponge Effect as PEI. Biomacromolecules 2013, 14, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Boussif, O.; Lezoualc’h, F.; Zanta, M.A.; Mergny, M.D.; Scherman, D.; Demeneix, B.; Behr, J.P. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. PNAS 1995, 92, 7297–7301. [Google Scholar] [CrossRef] [PubMed]
- Chertok, B.; David, A.E.; Yang, V.C. Polyethyleneimine-modified iron oxide nanoparticles for brain tumor drug delivery using magnetic targeting and intra-carotid administration. Biomaterials 2010, 31, 6317–6324. [Google Scholar] [CrossRef] [Green Version]
- Starcher, B. A Ninhydrin-Based Assay to Quantitate the Total Protein Content of Tissue Samples. Anal. Biochem. 2001, 292, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sabliov, C. PLA/PLGA nanoparticles for delivery of drugs across the blood-brain barrier. Nanotechnol. Rev. 2013, 2, 241–257. [Google Scholar] [CrossRef]
- Patel, T.; Zhou, J.; Piepmeier, J.M.; Saltzman, W.M. Polymeric Nanoparticles for Drug Delivery to the Central Nervous System. Adv. Drug Deliv. Rev. 2012, 64, 701–705. [Google Scholar] [CrossRef]
- Liu, M.; Li, H.; Luo, G.; Liu, Q.; Wang, Y. Pharmacokinetics and biodistribution of surface modification polymeric nanoparticles. Arch. Pharm. Res. 2008, 31, 547–554. [Google Scholar] [CrossRef]
- Lian, H.-Y.; Liang, Y.-H.; Yamauchi, Y.; Wu, K.C.-W. A Hierarchical Study on Load/Release Kinetics of Guest Molecules into/from Mesoporous Silica Thin Films. J. Phys. Chem. C 2011, 115, 6581–6590. [Google Scholar] [CrossRef]
- Rietscher, R.; Czaplewska, J.A.; Majdanski, T.C.; Gottschaldt, M.; Schubert, U.S.; Schneider, M.; Lehr, C.M. Impact of PEG and PEG-b-PAGE modified PLGA on nanoparticle formation, protein loading and release. Int. J. Pharm. 2016, 500, 187–195. [Google Scholar] [CrossRef]
- Ali, H.; Weigmann, B.; Collnot, E.; Khan, S.A.; Windbergs, M.; Lehr, C. Budesonide Loaded PLGA Nanoparticles for Targeting the Inflamed Intestinal Mucosa—Pharmaceutical Characterization and Fluorescence Imaging. Pharm. Res. 2016, 33, 1085–1092. [Google Scholar] [CrossRef]
- Zolnik, B.S.; Burgess, D.J. Effect of acidic pH on PLGA microsphere degradation and release. J. Control. Release 2007, 122, 338–344. [Google Scholar] [CrossRef]
- Kolte, A.; Patil, S.; Lesimple, P.; Hanrahan, J.W.; Misra, A. PEGylated composite nanoparticles of PLGA and polyethylenimine for safe and efficient delivery of pDNA to lungs. Int. J. Pharm. 2017, 524, 382–396. [Google Scholar] [CrossRef] [PubMed]
- Bexiga, M.G.; Varela, J.A.; Wang, F.; Fenaroli, F.; Salvati, A.; Lynch, I.; Simpson, J.C.; Dawson, K.A. Cationic nanoparticles induce caspase 3-, 7- and 9-mediated cytotoxicity in a human astrocytoma cell line. Nanotoxicology 2011, 5, 557–567. [Google Scholar] [CrossRef]
- Fröhlich, E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int. J. Nanomed. 2012, 7, 5577–5591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.-P.; Pei, Y.-Y.; Zhang, X.-Y.; Gu, Z.-H.; Zhou, Z.-H.; Yuan, W.-F.; Zhou, J.-J.; Zhu, J.-H.; Gao, X.-J. PEGylated PLGA nanoparticles as protein carriers: synthesis, preparation and biodistribution in rats. J. Control. Release 2001, 71, 203–211. [Google Scholar] [CrossRef]
- Rodrigues de Azevedo, C.; von Stosch, M.; Costa, M.S.; Ramos, A.M.; Cardoso, M.M.; Danhier, F.; Préat, V.; Oliveira, R. Modeling of the burst release from PLGA micro- and nanoparticles as function of physicochemical parameters and formulation characteristics. Int. J. Pharm. 2017, 532, 229–240. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Kidston, E.M.; Beck, F.; Lloyd, J.B., II. Kinetics of Protein Uptake and Digestion by Rat Yolk Sac. J. Cell Biol. 1975, 64, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Marwaha, R.; Sharma, M. DQ-Red BSA Trafficking Assay in Cultured Cells to Assess Cargo Delivery to Lysosomes. Bio Protoc. 2017, 7, e2571. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Litwin, T.; Nagaraja, A.R.; Kwong, B.; Katz, J.; Watson, N.; Irvine, D.J. Cytosolic Delivery of Membrane-Impermeable Molecules in Dendritic Cells Using pH-Responsive Core−Shell Nanoparticles. Nano Lett. 2007, 7, 3056–3064. [Google Scholar] [CrossRef]
- Jones, R.A.; Cheung, C.Y.; Black, F.E.; Zia, J.K.; Stayton, P.S.; Hoffman, A.S.; Wilson, M.R. Poly(2-alkylacrylic acid) polymers deliver molecules to the cytosol by pH-sensitive disruption of endosomal vesicles. Biochem. J. 2003, 372, 65–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qaddoumi, M.G.; Ueda, H.; Yang, J.; Davda, J.; Labhasetwar, V.; Lee, V.H.L. The Characteristics and Mechanisms of Uptake of PLGA Nanoparticles in Rabbit Conjunctival Epithelial Cell Layers. Pharm. Res. 2004, 21, 641–648. [Google Scholar] [CrossRef]
- Cartiera, M.S.; Johnson, K.M.; Rajendran, V.; Caplan, M.J.; Saltzman, W.M. The uptake and intracellular fate of PLGA nanoparticles in epithelial cells. Biomaterials 2009, 30, 2790–2798. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Alexander-Katz, A. Cell Membranes Open “Doors” for Cationic Nanoparticles/Biomolecules: Insights into Uptake Kinetics. ACS Nano 2013, 7, 10799–10808. [Google Scholar] [CrossRef]
- Porciani, D.; Signore, G.; Marchetti, L.; Mereghetti, P.; Nifosì, R.; Beltram, F. Two Interconvertible Folds Modulate the Activity of a DNA Aptamer Against Transferrin Receptor. Mol. Ther. Nucleic Acids 2014, 3, e144. [Google Scholar] [CrossRef] [PubMed]
- Ng, P.P.; Cruz, J.S.D.; Sorour, D.N.; Stinebaugh, J.M.; Shin, S.-U.; Shin, D.S.; Morrison, S.L.; Penichet, M.L. An anti-transferrin receptor-avidin fusion protein exhibits both strong proapoptotic activity and the ability to deliver various molecules into cancer cells. PNAS 2002, 99, 10706–10711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, S.L.; Carter, L.L. ATP is required for receptor-mediated endocytosis in intact cells. J. Cell Biol. 1990, 111, 2307–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Pisa, M.; Chassaing, G.; Swiecicki, J. Translocation mechanism(s) of cell-penetrating peptides: Biophysical studies using artificial membrane bilayers. Biochemistry 2015, 54, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Arvizo, R.R.; Miranda, O.R.; Thompson, M.A.; Pabelick, C.M.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Prakash, Y.S.; Mukherjee, P. Effect of Nanoparticle Surface Charge at the Plasma Membrane and Beyond. Nano Lett. 2010, 10, 2543–2548. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Terazzi, E.; Seemann, R.; Fleury, J.B.; Baulin, V.A. Direct proof of spontaneous translocation of lipid-covered hydrophobic nanoparticles through a phospholipid bilayer. Sci. Adv. 2016, 2, e1600261. [Google Scholar] [CrossRef] [Green Version]
- Dombu, C.Y.; Kroubi, M.; Zibouche, R.; Matran, R.; Betbeder, D. Characterization of endocytosis and exocytosis of cationic nanoparticles in airway epithelium cells. Nanotechnology 2010, 21, 355102. [Google Scholar] [CrossRef]
- Cerutti, P.A. Prooxidant states and tumor promotion. Science 1985, 227, 375–381. [Google Scholar] [CrossRef]
- Corvo, M.L.; Jorge, J.C.S.; van’t Hof, R.; Cruz, M.E.M.; Crommelin, D.J.A.; Storm, G. Superoxide dismutase entrapped in long-circulating liposomes: formulation design and therapeutic activity in rat adjuvant arthritis. Biochim. Biophys. Acta 2002, 1564, 227–236. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-P.; Chen, C.-T.; Hung, Y.; Chou, C.-M.; Liu, T.-P.; Liang, M.-R.; Chen, C.-T.; Mou, C.-Y. A New Strategy for Intracellular Delivery of Enzyme Using Mesoporous Silica Nanoparticles: Superoxide Dismutase. J. Am. Chem. Soc. 2013, 135, 1516–1523. [Google Scholar] [CrossRef]
- Lee, S.; Yang, S.C.; Heffernan, M.J.; Taylor, W.R.; Murthy, N. Polyketal Microparticles: A New Delivery Vehicle for Superoxide Dismutase. Bioconjugate Chem. 2007, 18, 4–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nukolova, N.V.; Aleksashkin, A.D.; Abakumova, T.O.; Morozova, A.Y.; Gubskiy, I.L.; Kirzhanova, Е.А.; Abakumov, M.A.; Chekhonin, V.P.; Klyachko, N.L.; Kabanov, A.V. Multilayer polyion complex nanoformulations of superoxide dismutase 1 for acute spinal cord injury. J. Control. Release 2018, 270, 226–236. [Google Scholar] [CrossRef]
- Crapo, J.D.; Oury, T.; Rabouille, C.; Slot, J.W.; Chang, L.Y. Copper, zinc superoxide dismutase is primarily a cytosolic protein in human cells. Proc. Natl Acad. Sci. USA 1992, 89, 10405–10409. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, C.; Fridovich, I. Superoxide dismutase: Improved assays and an assay applicable to acrylamide gels. Anal. Biochem. 1971, 44, 276–287. [Google Scholar] [CrossRef]
- Deng, H.X.; Hentati, A.; Tainer, J.A.; Iqbal, Z.; Cayabyab, A.; Hung, W.Y.; Getzoff, E.D.; Hu, P.; Herzfeldt, B.; Roos, R.P.; et al. Amyotrophic lateral sclerosis and structural defects in Cu, Zn superoxide dismutase. Science 1993, 261, 1047–1051. [Google Scholar] [CrossRef] [PubMed]
- Namboodiri, A.M.; Peethambaran, A.; Mathew, R.; Sambhu, P.A.; Hershfield, J.; Moffett, J.R.; Madhavarao, C.N. Canavan disease and the role of N-acetylaspartate in myelin synthesis. Mol. Cell. Endocrinol. 2006, 252, 216–223. [Google Scholar] [CrossRef] [PubMed]
- Hershfield, J.R.; Madhavarao, C.N.; Moffett, J.R.; Benjamins, J.A.; Garbern, J.Y.; Namboodiri, A. Aspartoacylase is a regulated nuclear-cytoplasmic enzyme. FASEB J. 2006, 20, 2139–2141. [Google Scholar] [CrossRef]
- Santi, M.; Maccari, G.; Mereghetti, P.; Voliani, V.; Rocchiccioli, S.; Ucciferri, N.; Luin, S.; Signore, G. Rational Design of a Transferrin-Binding Peptide Sequence Tailored to Targeted Nanoparticle Internalization. Bioconjugate Chem. 2017, 28, 471–480. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulation | Hydrodynamic Diameter (nm) (SEM) | Zeta Potential (mV) (SEM) | EE% (SEM) |
|---|---|---|---|
| BSA PEI NPs | 230 (3) | 48.5 (1.0) | 45 (1) |
| BSA NPs | 264 (1) | −20.8 (0.6) | 47 (3) |
| PEI NPs | 279 (1) | 42.7 (0.2) | - |
| NPs | 284 (2) | −25.6 (0.4) | - |
| Formulation | Hydrodynamic Diameter nm (SEM) | Zeta Potential mV (SEM) | Encapsulation Efficiency (%) | Activity Yield (%) |
|---|---|---|---|---|
| SOD PEI NPs | 187 (1) | 38 (1) | 63 (11) | 29 (5) |
| SOD NPs | 321 (9) | −21 (1) | 70 (10) | 30 (4) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galliani, M.; Tremolanti, C.; Signore, G. Nanocarriers for Protein Delivery to the Cytosol: Assessing the Endosomal Escape of Poly(Lactide-co-Glycolide)-Poly(Ethylene Imine) Nanoparticles. Nanomaterials 2019, 9, 652. https://doi.org/10.3390/nano9040652
Galliani M, Tremolanti C, Signore G. Nanocarriers for Protein Delivery to the Cytosol: Assessing the Endosomal Escape of Poly(Lactide-co-Glycolide)-Poly(Ethylene Imine) Nanoparticles. Nanomaterials. 2019; 9(4):652. https://doi.org/10.3390/nano9040652
Chicago/Turabian StyleGalliani, Marianna, Chiara Tremolanti, and Giovanni Signore. 2019. "Nanocarriers for Protein Delivery to the Cytosol: Assessing the Endosomal Escape of Poly(Lactide-co-Glycolide)-Poly(Ethylene Imine) Nanoparticles" Nanomaterials 9, no. 4: 652. https://doi.org/10.3390/nano9040652
APA StyleGalliani, M., Tremolanti, C., & Signore, G. (2019). Nanocarriers for Protein Delivery to the Cytosol: Assessing the Endosomal Escape of Poly(Lactide-co-Glycolide)-Poly(Ethylene Imine) Nanoparticles. Nanomaterials, 9(4), 652. https://doi.org/10.3390/nano9040652