Aptamer–Target–Gold Nanoparticle Conjugates for the Quantification of Fumonisin B1

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis and Characterization of Gold Nanoparticles (AuNP)
2.3. Functionalization of Gold Nanoparticles (AuNP) with Aptamers
2.4. Assays with Aptamer 40 nt
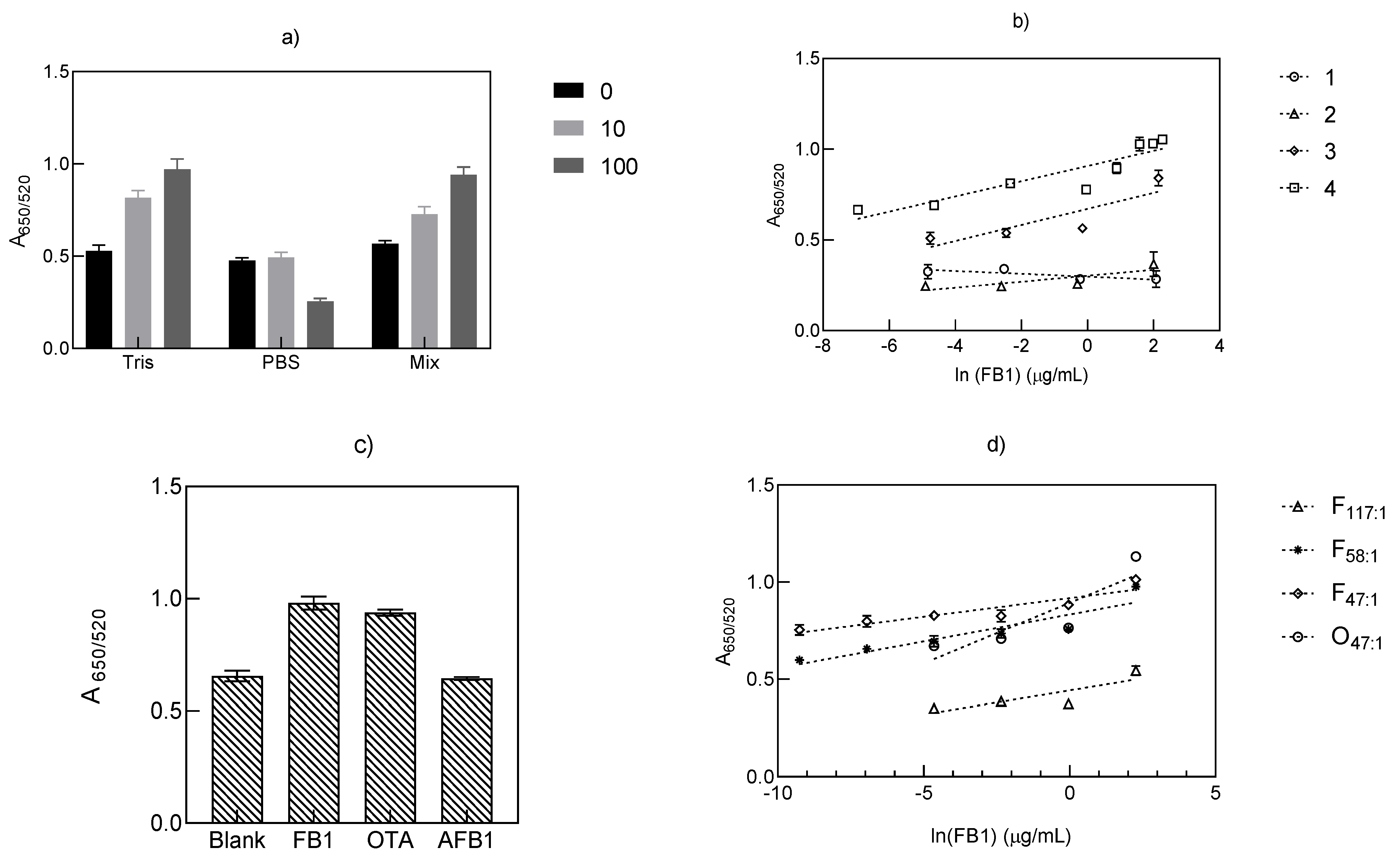
2.4.1. Effect of Tris-HCl, PBS, and Its Combination on the Binding Effect of Aptamer 40 nt
2.4.2. Effect of Different Buffers on the Performance of Aptamer 40 nt
2.4.3. Reduction in the Aptamer: AuNP Molar Ratio
2.4.4. Specificity of Aptamer 40 nt
2.5. Assays with Aptamer 96 nt
2.5.1. AuNP Functionalization with Aptamer 96 nt
2.5.2. Specificity of Aptamer 96 nt
2.5.3. Asymmetric Flow Field-Flow Fractionation (AF4)
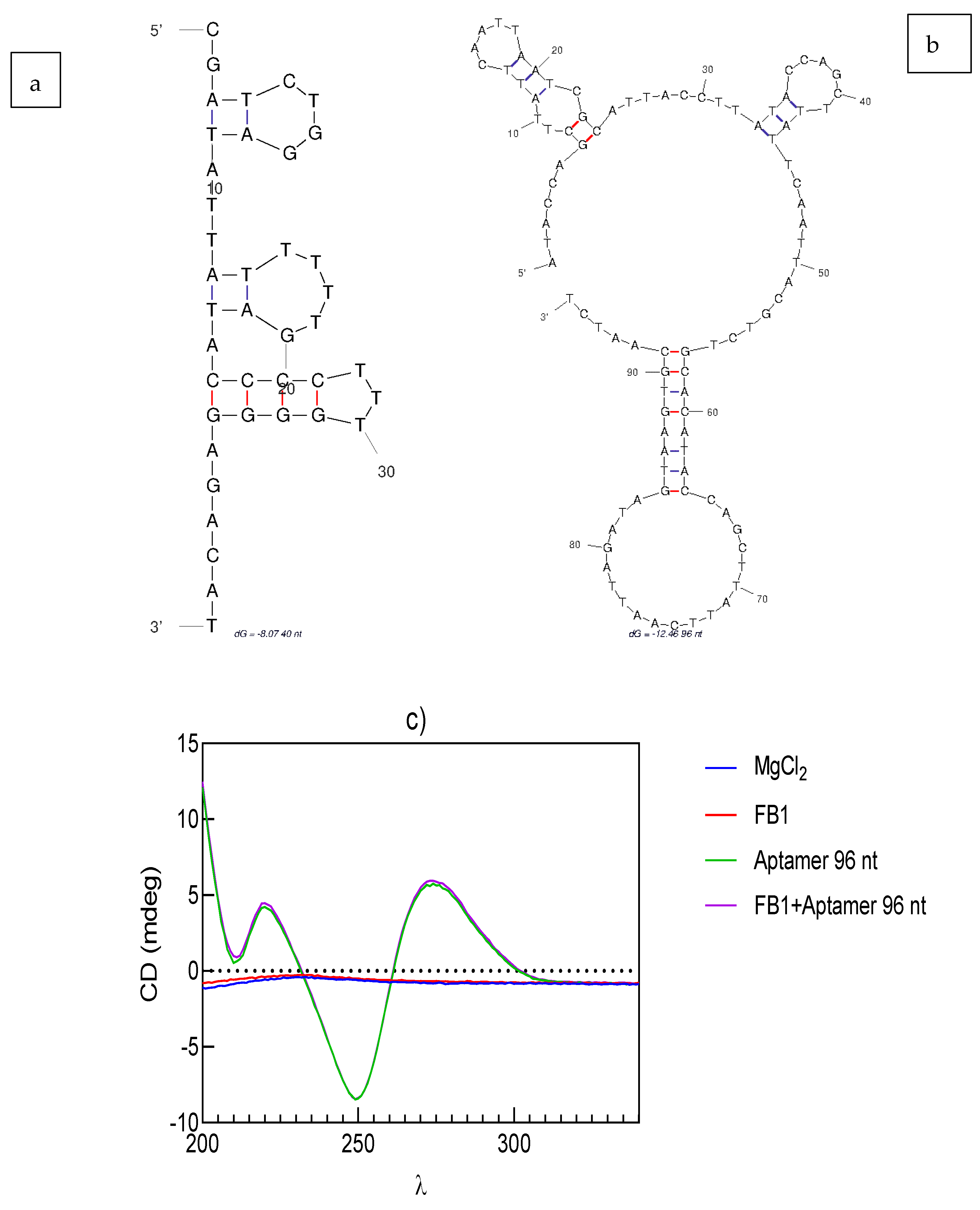
2.5.4. Prediction of the Aptamer Folded Structure
2.5.5. Circular Dichroism Spectroscopy
3. Results and Discussion
3.1. Effect of Buffer Incubation on the Quantification of FB1 with Aptamer 40 nt
3.2. Quantification of FB1 with Aptamer 96 nt
3.3. Asymmetric Flow Field-Flow Fractionation (AF4) of the Aptamer 96 nt–FB1–AuNP Conjugates
3.4. Interaction of the Conjugate Elements (Aptamer 96 nt–FB1–AuNPs)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wangia, R.N.; Githanga, D.P.; Xue, K.S.; Tang, L.; Anzala, O.A.; Wang, J.S. Validation of urinary sphingolipid metabolites as biomarker of effect for fumonisins exposure in Kenyan children. Biomarkers 2019, 24, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Wall-Martínez, H.A.; Ramírez-Martínez, A.; Wesolek, N.; Brabet, C.; Durand, N.; Rodríguez-Jimenes, G.C.; García-Alvarado, M.A.; Salgado-Cervantes, M.A.; Robles-Olvera, V.J.; Roudot, A.C. Risk assessment of exposure to mycotoxins (aflatoxins and fumonisins) through corn tortilla intake in Veracruz City (Mexico). Food Addit. Contam. A 2019, 36, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Liu, H.; Yang, J.; Wang, C.; Wang, Y.; Yang, Y.; Chen, X. Free and hidden fumonisins in raw maize and maize-based products from China. Food Addit. Contam. B 2019, 12, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Martins, C.; Vidal, A.; De Boevre, M.; De Saeger, S.; Nunes, C.; Torres, D.; Goios, A.; Lopes, C.; Assunção, R.; Alvito, P. Exposure assessment of Portuguese population to multiple mycotoxins: The human biomonitoring approach. Int. J. Hyg. Environ. Health 2019, 222, 913–925. [Google Scholar] [CrossRef]
- Abbas, H.; Vesonder, R.F.; Boyette, C.D.; Hoagland, R.E.; Krick, T. Production of fumonisins by Fusarium moniliforme cultures isolated from jimsonweed in Mississippi. J. Phytopathol. 1992, 136, 199–203. [Google Scholar] [CrossRef]
- Abbas, H.K.; Riley, R.T. The presence and phytotoxicity of fumonisins and AAL-toxin in Alternaria alternata. Toxicon 1996, 34, 503. [Google Scholar] [CrossRef]
- Mansson, M.; Klejnstrup, M.L.; Phipps, R.K.; Nielsen, K.F.; Frisvad, J.C.; Gotfredsen, C.H.; Larsen, T.O. Isolation and NMR characterization of fumonisin B2 and a new fumonisin B6 from Aspergillus niger. J. Agric. Food Chem. 2010, 58, 949–953. [Google Scholar] [CrossRef]
- Mogensen, J.M.; Møller, K.A.; Von Freiesleben, P.; Labuda, R.; Varga, E.; Sulyok, M.; Kubátová, A.; Thrane, U.; Andersen, B.; Nielsen, K.F. Production of fumonisins B 2 and B 4 in Tolypocladium species. J. Ind. Microbiol. Biotechnol. 2011, 38, 1329–1335. [Google Scholar] [CrossRef]
- Rheeder, J.P.; Marasas, W.F.; Vismer, H.F. Production of fumonisin analogs by Fusarium species. Appl. Environ. Microbiol. 2002, 68, 2101–2105. [Google Scholar] [CrossRef] [Green Version]
- Ostry, V.; Malir, F.; Toman, J.; Grosse, Y. Mycotoxins as human carcinogens—the IARC Monographs classification. Mycotoxin Res. 2017, 33, 65–73. [Google Scholar] [CrossRef]
- Liu, X.; Fan, L.; Yin, S.; Chen, H.; Hu, H. Molecular mechanisms of fumonisin B1-induced toxicities and its applications in the mechanism-based interventions. Toxicon 2019, 167, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Q.; Jiang, Y.; Fan, Y.; Ma, Y.; Lei, H.; Su, J. Fumonisin B1 induces oxidative stress and breaks barrier functions in pig iliac endothelium cells. Toxins 2019, 11, 387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.; Kim, G.; Moon, J. Performance improvement of the one-dot lateral flow immunoassay for aflatoxin B1 by using a smartphone-based reading system. Sensors 2013, 13, 5109–5116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.U.; Zourob, M.; Tamiya, E. Food Biosensors; Royal Society of Chemistry: Cambridge, UK, 2016; pp. 2–9. [Google Scholar]
- McKeague, M.; Bradley, C.R.; Girolamo, A.D.; Visconti, A.; Miller, J.D.; DeRosa, M.C. Screening and initial binding assessment of fumonisin B1 aptamers. Int. J. Mol. Sci. 2010, 11, 4864–4881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Huang, Y.; Duan, N.; Wu, S.; Xia, Y.; Ma, X.; Zhu, C.; Jiang, Y.; Ding, Z.; Wang, Z. Selection and characterization of single stranded DNA aptamers recognizing fumonisin B 1. Microchim. Acta 2014, 181, 1317–1324. [Google Scholar] [CrossRef]
- Pamies, R.; Hernández, J.G.C.; Fernández Espín, V.; Collado-González, M.; Díaz, F.G.B.; García de la Torre, J. Aggregation behaviour of gold nanoparticles in saline aqueous media. J. Nanopart. Res. 2014, 16, 2376. [Google Scholar] [CrossRef]
- Li, H.; Rothberg, L. Colorimetric detection of DNA sequences based on electrostatic interactions with unmodified gold nanoparticles. Proc. Natl. Acad. Sci. USA 2004, 101, 14036–14039. [Google Scholar] [CrossRef] [Green Version]
- Pandey, P.C.; Pandey, G. Synthesis of gold nanoparticles resistant to pH and salt for biomedical applications; functional activity of organic amine. J. Mater. Res. 2016, 31, 3313. [Google Scholar] [CrossRef]
- Amendola, V.; Pilot, R.; Frasconi, M.; Maragò, O.M.; Iatì, M.A. Surface plasmon resonance in gold nanoparticles: A review. J. Phys. Condens. Matter 2017, 29, 203002. [Google Scholar] [CrossRef]
- Yue, S.; Jie, X.; Wei, L.; Bin, C.; Dou Dou, W.; Yi, Y.; QingXia, L.; JianLin, L.; TieSong, Z. Simultaneous detection of ochratoxin A and fumonisin B1 in cereal samples using an aptamer–photonic crystal encoded suspension array. Anal. Chem. 2014, 86, 11797–11802. [Google Scholar] [CrossRef]
- Hagendorfer, H.; Kaegi, R.; Traber, J.; Mertens, S.F.; Scherrers, R.; Ludwig, C.; Ulrich, A. Application of an asymmetric flow field flow fractionation multi-detector approach for metallic engineered nanoparticle characterization–prospects and limitations demonstrated on Au nanoparticles. Anal. Chim. Acta 2011, 706, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Mudalige, T.K.; Qu, H.; Van Haute, D.; Ansar, S.M.; Linder, S.W. Capillary electrophoresis and asymmetric flow field-flow fractionation for size-based separation of engineered metallic nanoparticles: A critical comparative review. TrAC Trends Anal. Chem. 2018, 106, 202–212. [Google Scholar] [CrossRef]
- Schachermeyer, S.; Ashby, J.; Zhong, W. Aptamer–protein binding detected by asymmetric flow field flow fractionation. J. Chromatogr. A 2013, 1295, 107–113. [Google Scholar] [CrossRef] [PubMed]
- Derbyshire, N.; White, S.J.; Bunka, D.H.; Song, L.; Stead, S.; Tarbin, J.; Sharman, M.; Zhou, D.; Stockley, P.G. Toggled RNA aptamers against aminoglycosides allowing facile detection of antibiotics using gold nanoparticle assays. Anal. Chem. 2012, 84, 6595–6602. [Google Scholar] [CrossRef] [PubMed]
- Quesada-González, D.; Stefani, C.; González, I.; De la Escosura-Muñiz, A.; Domingo, N.; Mutjé, P.; Merkoçi, A. Signal enhancement on gold nanoparticle-based lateral flow tests using cellulose nanofibers. Biosens. Bioelectron. 2019, 141, 111407. [Google Scholar] [CrossRef]
- Yang, Y.; Li, W.; Shen, P.; Liu, R.; Li, Y.; Xu, J.; Zheng, Q.; Zhang, Y.; Li, J.; Zheng, T. Aptamer fluorescence signal recovery screening for multiplex mycotoxins in cereal samples based on photonic crystal microsphere suspension array. Sens. Actuators B Chem. 2017, 248, 351–358. [Google Scholar] [CrossRef]
- Tian, H.; Sofer, Z.; Pumera, M.; Bonanni, A. Investigation on the ability of heteroatom-doped graphene for biorecognition. Nanoscale 2017, 9, 3530–3536. [Google Scholar] [CrossRef]
- Mirón-Mérida, V.A.; Gong, Y.Y.; Goycoolea, F.M. Aptamer-based detection of fumonisin B1: A systematic comparison with conventional and other novel methods. ChemRxiv 2020. (Preprint). [Google Scholar] [CrossRef]
- Frost, N.R.; McKeague, M.; Falcioni, D.; DeRosa, M.C. An in solution assay for interrogation of affinity and rational minimer design for small molecule-binding aptamers. Analyst 2015, 140, 6643–6651. [Google Scholar] [CrossRef]
- Cheng, Z.X.; Bonanni, A. All-in-one: Electroactive nanocarbon as simultaneous platform and label for single-step biosensing. Chem. Eur. J. 2018, 24, 6380–6385. [Google Scholar] [CrossRef]
- Ciriaco, F.; De Leo, V.; Catucci, L.; Pascale, M.; Logrieco, A.F.; DeRosa, M.C.; De Girolamo, A. An in-silico pipeline for rapid screening of DNA aptamers against mycotoxins: The case-study of Fumonisin B1, Aflatoxin B1 and Ochratoxin A. Polymers 2020, 12, 2983. [Google Scholar] [CrossRef]
- Baglieri, A.; Reyneri, A.; Gennari, M.; Nègre, M. Organically modified clays as binders of fumonisins in feedstocks. J. Environ. Sci. Health Part B 2013, 48, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Liu, N.; Yang, L.; Wang, J.; Song, S.; Nie, D.; Yang, X.; Hou, J.; Wu, A. Cross-linked chitosan polymers as generic adsorbents for simultaneous adsorption of multiple mycotoxins. Food Control 2015, 57, 362–369. [Google Scholar] [CrossRef]
- Chávez, J.L.; Hagen, J.A.; Kelley-Loughnane, N. Fast and selective plasmonic serotonin detection with aptamer-gold nanoparticle conjugates. Sensors 2017, 17, 681. [Google Scholar] [CrossRef] [Green Version]
- Riley, K.R.; El Hadri, H.; Tan, J.; Hackley, V.A.; MacCrehan, W.A. High separation efficiency of gold nanomaterials of different aspect ratio and size using capillary transient isotachophoresis. J. Chromatogr. A 2019, 1598, 216–222. [Google Scholar] [CrossRef] [PubMed]
- González-Espinosa, Y.; Sabagh, B.; Moldenhauer, E.; Clarke, P.; Goycoolea, F.M. Characterisation of chitosan molecular weight distribution by multi-detection asymmetric flow-field flow fractionation (AF4) and SEC. Int. J. Biol. Macromol. 2019, 136, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Cho, T.J.; Hackley, V.A. Fractionation and characterization of gold nanoparticles in aqueous solution: Asymmetric-flow field flow fractionation with MALS, DLS, and UV–Vis detection. Anal. Bioanal. Chem. 2010, 398, 2003–2018. [Google Scholar] [CrossRef]
- Schmidt, B.; Loeschner, K.; Hadrup, N.; Mortensen, A.; Sloth, J.J.; Bender Koch, C.; Larsen, E.H. Quantitative characterization of gold nanoparticles by field-flow fractionation coupled online with light scattering detection and inductively coupled plasma mass spectrometry. Anal. Chem. 2011, 83, 2461–2468. [Google Scholar] [CrossRef] [Green Version]
- Samokhvalov, A.V.; Safenkova, I.V.; Eremin, S.A.; Zherdev, A.V.; Dzantiev, B.B. Use of anchor protein modules in fluorescence polarisation aptamer assay for ochratoxin A determination. Anal. Chim. Acta 2017, 962, 80–87. [Google Scholar] [CrossRef]
- Baaske, P.; Wienken, C.J.; Reineck, P.; Duhr, S.; Braun, D. Optical thermophoresis for quantifying the buffer dependence of aptamer binding. Angew. Chem. Int. Ed. 2010, 49, 2238–2241. [Google Scholar] [CrossRef]
- Wu, S.; Duan, N.; Li, X.; Tan, G.; Ma, X.; Xia, Y.; Wang, Z.; Wang, H. Homogenous detection of fumonisin B1 with a molecular beacon based on fluorescence resonance energy transfer between NaYF4: Yb, Ho upconversion nanoparticles and gold nanoparticles. Talanta 2013, 116, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Chen, R.H.; Lee, C.H.; Chang, Y.; Chen, C.S.; Chen, W.Y. Studies of the binding mechanism between aptamers and thrombin by circular dichroism, surface plasmon resonance and isothermal titration calorimetry. Colloids Surf. B 2011, 88, 552–558. [Google Scholar] [CrossRef] [PubMed]
- Jing, M.; Bowser, M.T. Methods for measuring aptamer-protein equilibria: A review. Anal. Chim. Acta 2011, 686, 9–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assay | Binding Buffer | Incubation with AuNP Time/Temperature (min/°C) | Equation A650/A520 = | r2 | Range of Tested Concentrations (µg/mL) | Limit of Detection(LOD) (µg/mL) |
|---|---|---|---|---|---|---|
| 1 | MgCl2 0.19 mM | 120/37 | −0.008 ln [X] + 0.2977 | 0.6542 | 0.008–8.0 | 4.16 |
| 2 | NaCl 0.06M + MgCl2 0.1mM | 120/37 | 0.0164 ln [X] + 0.3025 | 0.6795 | 0.0074–7.4 | 0.35 |
| 3 | PBS (eq. 12 mM NaCl) + Tris-HCl buffer 17 mM | 120/37 | 0.0442 ln [X] + 0.6709 | 0.737 | 0.0086–8.6 | 0.11 |
| 4 | Tris HCl buffer 14.06 mM | 105/20 | 0.0419 ln [X] + 0.9072 | 0.8311 | 0.0096–9.66 | 0.03 |
| Assay 1 | Equation 2 A650/520 = | r2 | Range of Tested Concentrations (µg/mL) | LOD (µg/mL) |
|---|---|---|---|---|
| F117:1 | 0.0248 ln[X] + 0.4434 | 0.695 | 0.0096–9.66 | 0.54 |
| F58:1 | 0.0276 ln [X] + 0.8332 | 0.833 | 0.00096–9.66 | 0.001 |
| F47:1 | 0.0192 ln [X] + 0.9169 | 0.84 | 0.000096–9.66 | 0.0007 |
| O47:1 | 0.0624 ln [X] + 0.8937 | 0.76 | 0.0096–9.66 | 0.06 |
| Signal | Equation | r2 | Range of Tested Concentrations (µg/mL) | LOD (µg/mL) | |
|---|---|---|---|---|---|
| A650/520 | A650/520 = −0.074 ln [X] + 0.5153 | 0.9179 | 0.01–10 | 0.003 | |
| Absolute Area | AA = 1.34 ln [X] + 8.298 | 0.9243 | 0.01–10 | 0.002 | |
| Peak Area 520 nm | Peak 1 | Area = 0.0002 ln [X] + 0.0036 | 0.9763 | 0.001–10 | 1.68 |
| Peak 2 | Area = − 0.0005 ln [X] + 0.0065 | 0.8735 | 0.001–10 | 0.0001 | |
| Peak Area 600 nm | Peak 1 | Area = 0.0017 ln [X] + 0.0334 | 0.872 | 0.001–10 | 7.83 |
| Peak 2 | Area= − 0.006 ln [X] + 0.0946 | 0.858 | 0.001–10 | 0.000000056 | |
| Peak 2 Area /Peak 1 Area | 520 nm | P1/P2 = − 0.268 ln [X] + 1.8427 | 0.7639 | 0.001–10 | 0.007 |
| 600 nm | P1/P2 = − 0.364 ln [X] + 2.9186 | 0.9637 | 0.001–10 | 0.0006 | |
| Peak 2 Area–Peak 1 Area | 600 nm | P–P2 = − 0.008 ln [X] + 0.059 | 0.9525 | 0.001–10 | 0.0000016 |
| 28o | Area = − 0.07 ln [X] + 0.4632 | 0.9128 | 0.001–10 | 0.00000016 | |
| Diameter | D = − 9.498 ln [X] + 124.61 | 0.9514 | 0.001–10 | 0.000959 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mirón-Mérida, V.A.; González-Espinosa, Y.; Collado-González, M.; Gong, Y.Y.; Guo, Y.; Goycoolea, F.M. Aptamer–Target–Gold Nanoparticle Conjugates for the Quantification of Fumonisin B1. Biosensors 2021, 11, 18. https://doi.org/10.3390/bios11010018
Mirón-Mérida VA, González-Espinosa Y, Collado-González M, Gong YY, Guo Y, Goycoolea FM. Aptamer–Target–Gold Nanoparticle Conjugates for the Quantification of Fumonisin B1. Biosensors. 2021; 11(1):18. https://doi.org/10.3390/bios11010018
Chicago/Turabian StyleMirón-Mérida, Vicente Antonio, Yadira González-Espinosa, Mar Collado-González, Yun Yun Gong, Yuan Guo, and Francisco M. Goycoolea. 2021. "Aptamer–Target–Gold Nanoparticle Conjugates for the Quantification of Fumonisin B1" Biosensors 11, no. 1: 18. https://doi.org/10.3390/bios11010018
APA StyleMirón-Mérida, V. A., González-Espinosa, Y., Collado-González, M., Gong, Y. Y., Guo, Y., & Goycoolea, F. M. (2021). Aptamer–Target–Gold Nanoparticle Conjugates for the Quantification of Fumonisin B1. Biosensors, 11(1), 18. https://doi.org/10.3390/bios11010018