The Scavenging Effect of Myoglobin from Meat Extracts toward Peroxynitrite Studied with a Flow Injection System Based on Electrochemical Reduction over a Screen-Printed Carbon Electrode Modified with Cobalt Phthalocyanine: Quantification and Kinetics †




Abstract
:1. Introduction

2. Materials and Methods
2.1. Peroxynitrite Synthesis
2.2. Electrode Chemical Modification
2.3. Meat Extracts and Myoglobin Solutions
2.4. Electrochemistry
2.5. Determination of Apparent Rate Constants and Half-Lives
2.6. Surface Characterization
3. Results and Discussion
3.1. Batch Determination of Peroxynitrite Using SPCE/CoPc Electrodes
3.1.1. Characterization of the Deposited CoPc Films on the SPCE
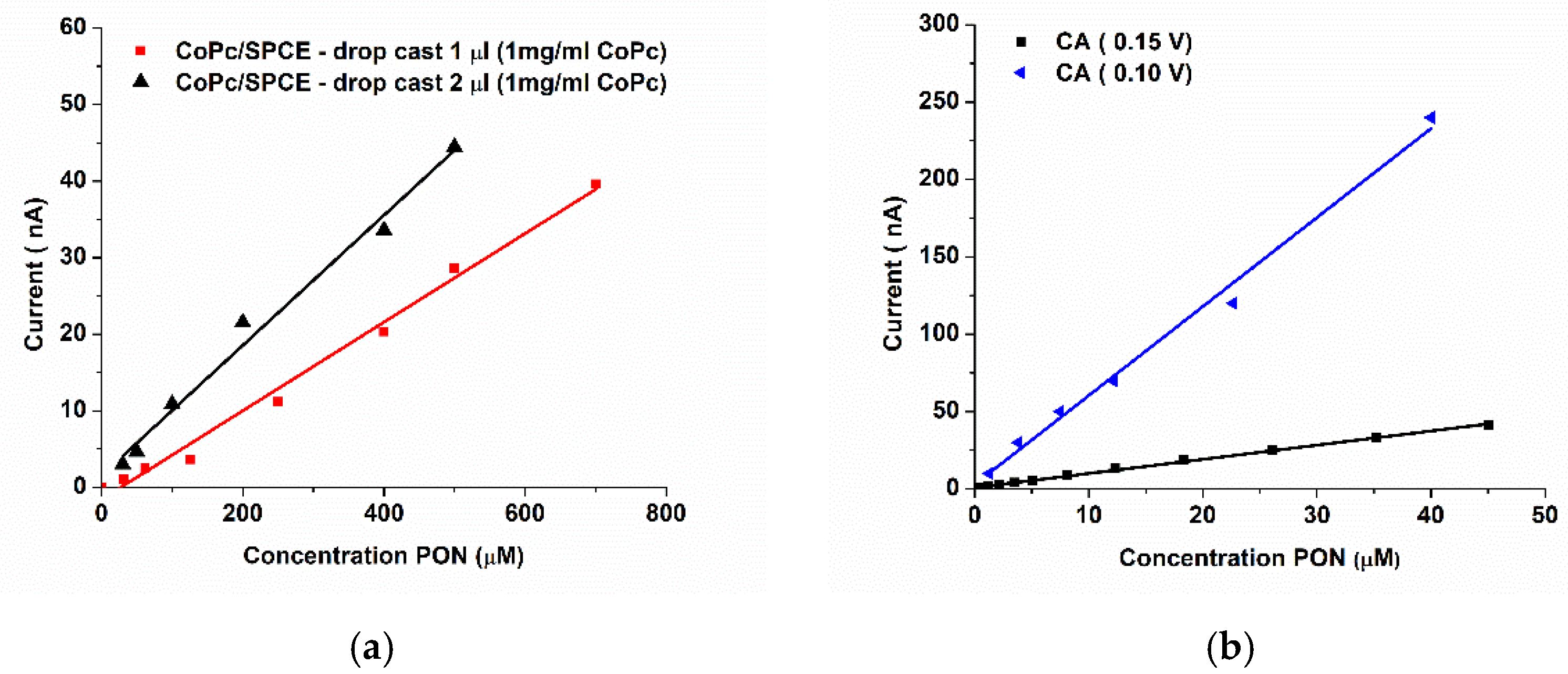
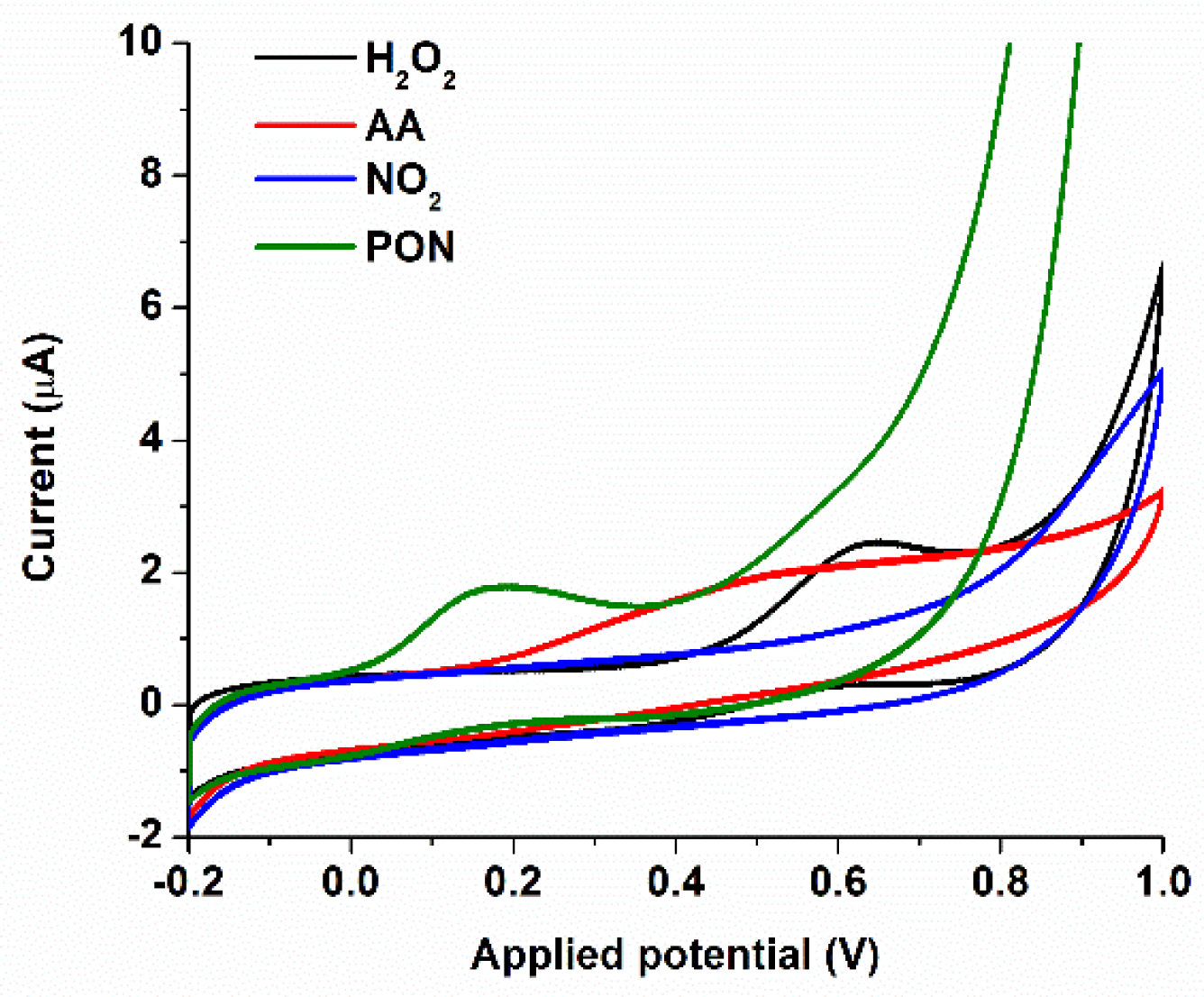
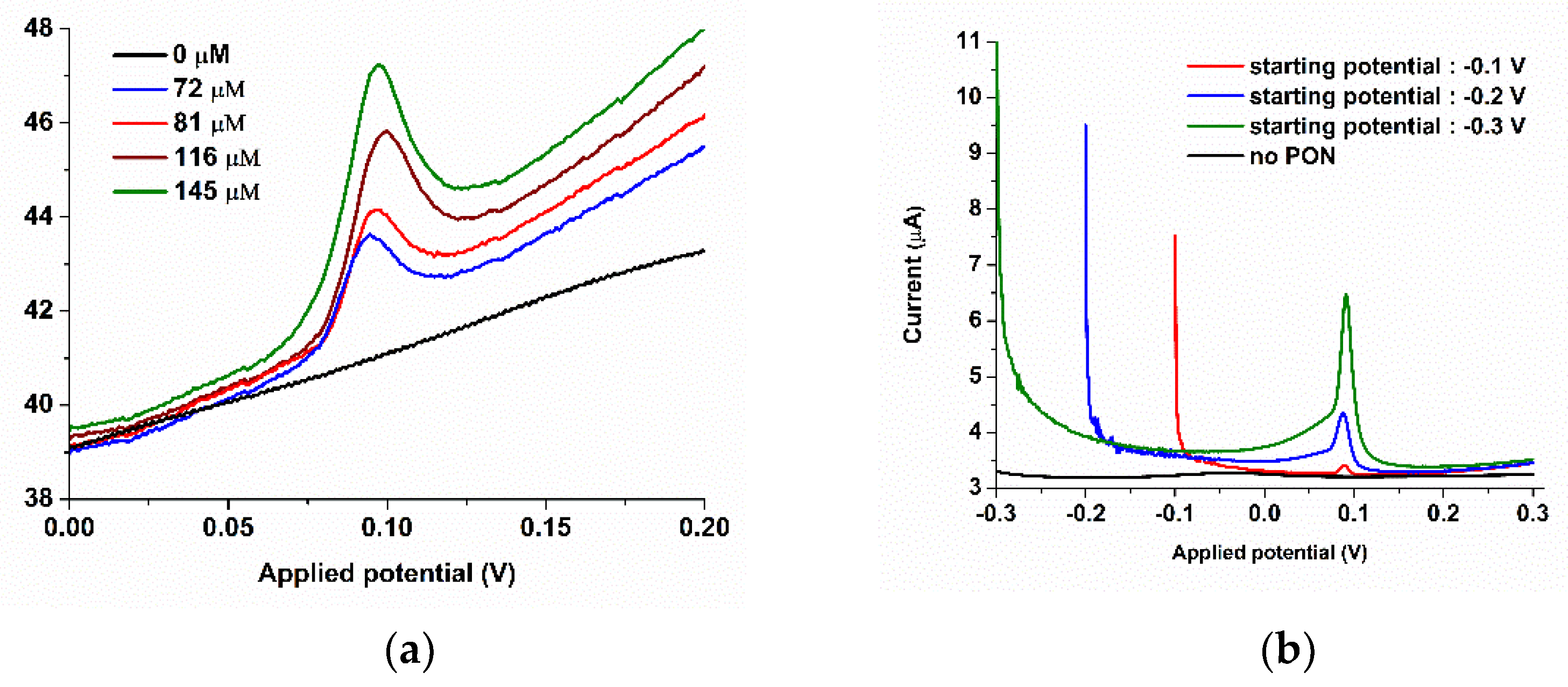
3.1.2. Batch Optimization of the CoPc-Modified Electrodes for PON Detection
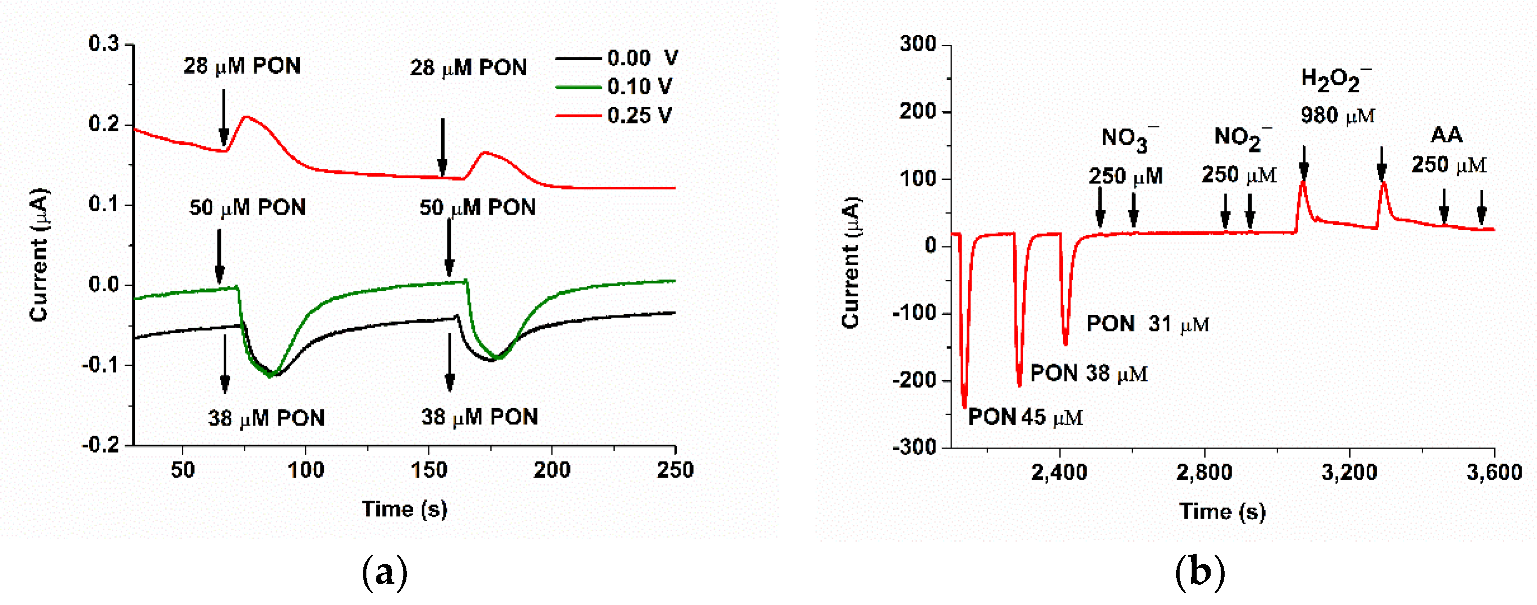
3.2. FIA Optimization of the SPCE/CoPc Electrodes for PON Detection
3.3. UV-Vis and Determination of Synthesized PON for Kinetic Studies
3.4. UV-Vis Determination of Different Forms of Myoglobin
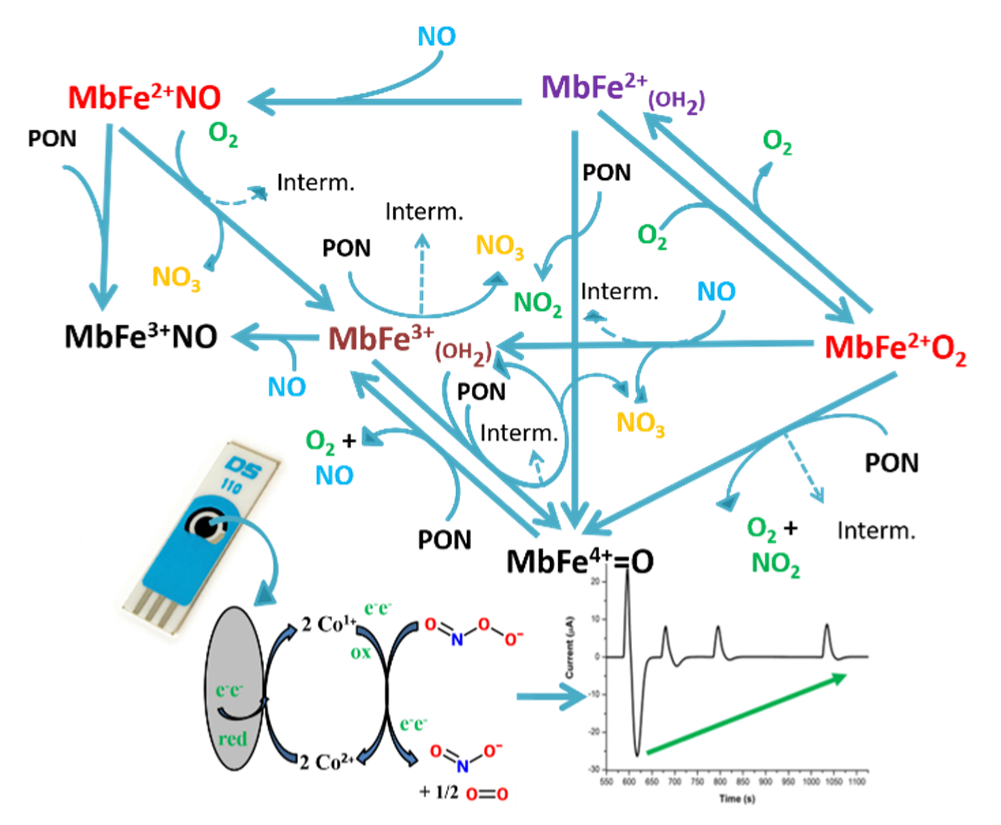
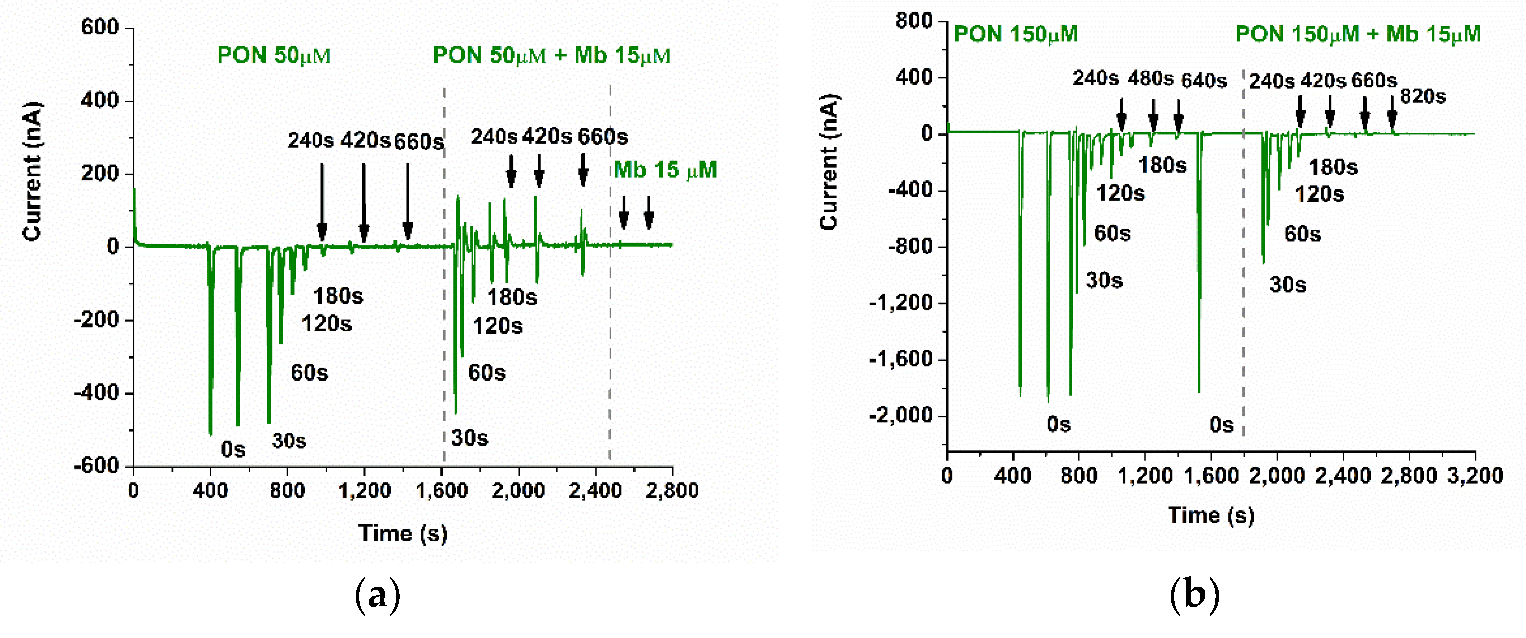
3.5. Studying the Reaction of Myoglobin with Peroxynitrite with FIA-EC
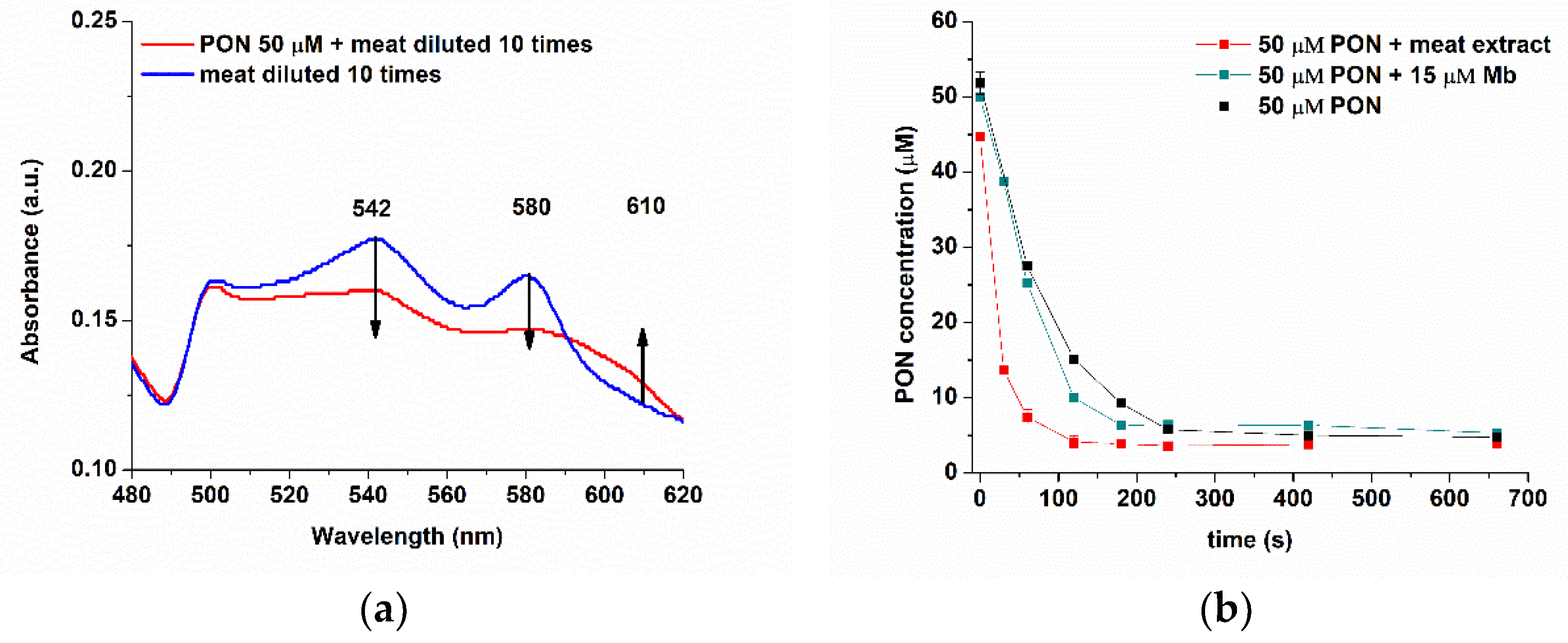
3.6. Studying the Reaction between Myoglobin from Meat Extracts and Peroxynitrite Using FIA-EC
3.6.1. Estimation of the Apparent Rate Decay Orders of PON in the Absence and Presence of Myoglobin
3.6.2. Determination of Apparent Rate Constants and Half-Lives for the Decay of PON
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hosu, I.S.; Constantinescu-Aruxandei, D.; Oancea, F.; Doni, M. Studying the Reaction of Peroxynitrite with Myoglobin for Meat Extract Samples Using Cobalt Phthalocyanine-Modified Screen-Printed Carbon Electrodes and a Flow Injection Analysis System. Proceedings 2020, 60, 46. [Google Scholar] [CrossRef]
- Exner, M.; Herold, S. Kinetic and mechanistic studies of the peroxynitrite-mediated oxidation of oxymyoglobin and oxyhemoglobin. Chem. Res. Toxicol. 2000, 13, 287–293. [Google Scholar] [CrossRef]
- Vasilescu, A.; Vezeanu, A.; Liu, Y.; Hosu, I.S.; Worden, R.M.; Peteu, S.F. Meat Freshness: Peroxynitrite’s Oxidative Role, Its moltural Scavengers, and New Measuring Tools. In Instrumental Methods for the Analysis and Identification of Bioactive Molecules; Jayprakasha, G.K., Patil, B.S., Pellati, F., Eds.; American Chemical Society: Washington, DC, USA, 2014; pp. 303–332. [Google Scholar]
- Brannan, R.G.; Decker, E.A. Peroxynitrite-induced oxidation of lipids: Implications for muscle foods. J. Agric. Food Chem. 2001, 49, 3074–3079. [Google Scholar] [CrossRef] [PubMed]
- Jeong, J.Y.; Kim, G.D.; Yang, H.S.; Joo, S.T. Pigments and Color of Muscle Foods; CRC Press: Boca Raton, FL, USA, 2014; pp. 44–61. [Google Scholar]
- Møller, J.K.S.; Skibsted, L.H. Myoglobins: The link between discoloration and lipid oxidation in muscle and meat. Quim. Nova 2006, 29, 1270–1278. [Google Scholar] [CrossRef]
- Szunerits, S.; Peteu, S.; Boukherroub, R. CHAPTER 4 Peroxynitrite-Sensitive Electrochemically Active Matrices. In Peroxynitrite Detection in Biological Media: Challenges and Advances; The Royal Society of Chemistry: Cambridge, UK, 2016; pp. 63–77. [Google Scholar]
- Li, M.; Gong, X.; Li, H.-W.; Han, H.; Shuang, S.; Song, S.; Dong, C. A fast detection of peroxynitrite in living cells. Anal. Chim. Acta 2020, 1106, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Wada, M.; Kira, M.; Kido, H.; Ikeda, R.; Kuroda, N.; Nishigaki, T.; Nakashima, K. Semi-micro flow injection analysis method for evaluation of quenching effect of health foods or food additive antioxidants on peroxynitrite. Luminescence 2011, 26, 191–195. [Google Scholar] [CrossRef] [PubMed]
- Ruzicka, J.; Ruzicka, J. Flow injection analysis—A survey of its potential as solution handling and data gathering technique in chemical research and industry. Z. Anal. Chem. 1988, 329, 653–655. [Google Scholar] [CrossRef]
- Hosu, I.S.; Wang, Q.; Vasilescu, A.; Peteu, S.F.; Raditoiu, V.; Railian, S.; Zaitsev, V.; Turcheniuk, K.; Wang, Q.; Li, M.; et al. Cobalt phthalocyanine tetracarboxylic acid modified reduced graphene oxide: A sensitive matrix for the electrocatalytic detection of peroxynitrite and hydrogen peroxide. RSC Adv. 2015, 5, 1474–1484. [Google Scholar] [CrossRef]
- Cortes, J.S.; Granados, S.G.; Ordaz, A.A.; Jimenez, J.A.L.; Griveau, S.; Bedioui, F. Electropolymerized manganese tetraaminophthalocyanine thin films onto platinum ultramicroelectrode for the electrochemical detection of peroxynitrite in solution. Electroanalysis 2007, 19, 61–64. [Google Scholar] [CrossRef]
- Oprea, R.; Peteu, S.F.; Subramanian, P.; Qi, W.; Pichonat, E.; Happy, H.; Bayachou, M.; Boukherroub, R.; Szunerits, S. Peroxynitrite activity of hemin-functionalized reduced graphene oxide. Analyst 2013, 138, 4345–4352. [Google Scholar] [CrossRef] [Green Version]
- Koh, W.C.A.; Son, J.I.; Choe, E.S.; Shim, Y.-B. Electrochemical Detection of Peroxynitrite Using a Biosensor Based on a Conducting Polymer−Manganese Ion Complex. Anal. Chem. 2010, 82, 10075–10082. [Google Scholar] [CrossRef]
- Mason, R.; Jacob, R.; Corbalan, J.; Szczesny, D.; Matysiak, K.; Malinski, T. The favorable kinetics and balance of nebivolol-stimulated nitric oxide and peroxynitrite release in human endothelial cells. BMC Pharmacol. Toxicol. 2013, 14, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosu, I.S.; Constantinescu-Aruxandei, D.; Jecu, M.-L.; Oancea, F.; Badea Doni, M. Peroxynitrite Sensor Based on a Screen Printed Carbon Electrode Modified with a Poly(2,6-dihydroxynaphthalene) Film. Sensors 2016, 16, 1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boni, A.C.; Wong, A.; Dutra, R.A.F.; Sotomayor, M.D.T. Cobalt phthalocyanine as a biomimetic catalyst in the amperometric quantification of dipyrone using FIA. Talanta 2011, 85, 2067–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angnes, L.; Azevedo, C.M.N.; Araki, K.; Toma, H.E. Electrochemical detection of NADH and dopamine in flow analysis based on tetraruthenated porphyrin modified electrodes. Anal. Chim. Acta. 1996, 329, 91–95. [Google Scholar] [CrossRef]
- Channon, R.B.; Joseph, M.B.; Bitziou, E.; Bristow, A.W.T.; Ray, A.D.; Macpherson, J.V. Electrochemical Flow Injection Analysis of Hydrazine in an Excess of an Active Pharmaceutical Ingredient: Achieving Pharmaceutical Detection Limits Electrochemically. Anal. Chem. 2015, 87, 10064–10071. [Google Scholar] [CrossRef]
- Santos, A.M.; Silva, T.A.; Vicentini, F.C.; Fatibello-Filho, O. Flow injection analysis system with electrochemical detection for the simultaneous determination of nanomolar levels of acetaminophen and codeine. Arab. J. Chem. 2020, 13, 335–345. [Google Scholar] [CrossRef]
- Herold, S.; Exner, M.; Boccini, F. The mechanism of the peroxynitrite-mediated oxidation of myoglobin in the absence and presence of carbon dioxide. Chem. Res. Toxicol. 2003, 16, 390–402. [Google Scholar] [CrossRef]
- Ascenzi, P.; Marinis, E.D.; Masi, A.d.; Ciaccio, C.; Coletta, M. Peroxynitrite scavenging by ferryl sperm whale myoglobin and human hemoglobin. Biochem. Biophys. Res. Commun. 2009, 390, 27–31. [Google Scholar] [CrossRef] [Green Version]
- Connolly, B.J.; Brannan, R.G.; Decker, E.A. Potential of peroxynitrite to alter the color of myoglobin in muscle foods. J. Agric. Food Chem. 2002, 50, 5220–5223. [Google Scholar] [CrossRef] [PubMed]
- Herold, S.; Shivashankar, K. Metmyoglobin and methemoglobin catalyze the isomerization of peroxynitrite to nitrate. Biochemistry 2003, 42, 14036–14046. [Google Scholar] [CrossRef] [PubMed]
- Kalinga, S. Peroxynitrite-Induced Modifications of Myoglobin: A Kinetic and Mechanistic Study. Ph.D. Thesis, ETH Zurich, Zürich, Switzerland, 2005. [Google Scholar]
- Connolly, B.J.; Decker, E.A. Peroxynitrite induced discoloration of muscle foods. Meat Sci. 2004, 66, 499–505. [Google Scholar] [CrossRef]
- Robinson, K.M.; Beckman, J.S. Synthesis of Peroxynitrite from Nitrite and Hydrogen Peroxide. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 2005; Volume 396, pp. 207–214. [Google Scholar] [CrossRef]
- Badea, M.; Amine, A.; Benzine, M.; Curulli, A.; Moscone, D.; Lupu, A.; Volpe, G.; Palleschi, G. Rapid and Selective Electrochemical Determination of Nitrite in Cured Meat in the Presence of Ascorbic Acid. Microchim. Acta 2004, 147, 51–58. [Google Scholar] [CrossRef] [Green Version]
- Herold, S. Kinetic and spectroscopic characterization of an intermediate peroxynitrite complex in the nitrogen monoxide induced oxidation of oxyhemoglobin. FEBS Lett. 1998, 439, 85–88. [Google Scholar] [CrossRef] [Green Version]
- Molina, C.; Kissner, R.; Koppenol, W.H. Decomposition kinetics of peroxynitrite: Influence of pH and buffer. Dalton Trans. 2013, 42, 9898–9905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kissner, R.; Koppenol, W.H. Product Distribution of Peroxynitrite Decay as a Function of pH, Temperature, and Concentration. J. Am. Chem. Soc. 2002, 124, 234–239. [Google Scholar] [CrossRef]
- Levine, I. Physical Chemistry; McGraw-Hill Education: New York, NY, USA, 2009. [Google Scholar]
- Ischiropoulos, H.; Al-Mehdi, A.B. Peroxynitrite-mediated oxidative protein modifications. FEBS Lett. 1995, 364, 279–282. [Google Scholar] [CrossRef] [Green Version]
- Carballal, S.; Bartesaghi, S.; Radi, R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 768–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herold, S.; Shivashankar, K.; Mehl, M. Myoglobin Scavenges Peroxynitrite without Being Significantly Nitrated. Biochemistry 2002, 41, 13460–13472. [Google Scholar] [CrossRef]
- Dashteh, M.; Safaiee, M.; Baghery, S.; Zolfigol, M.A. Application of cobalt phthalocyanine as a nanostructured catalyst in the synthesis of biological henna-based compounds. Appl. Organomet. Chem. 2019, 33, e4690. [Google Scholar] [CrossRef]
- Kumar, P.; Kumar, A.; Sreedhar, B.; Sain, B.; Ray, S.S.; Jain, S.L. Cobalt Phthalocyanine Immobilized on Graphene Oxide: An Efficient Visible–Active Catalyst for the Photoreduction of Carbon Dioxide. Chem. A Eur. J. 2014, 20, 6154–6161. [Google Scholar] [CrossRef] [Green Version]
- Tackley, D.R.; Dent, G.; Ewen Smith, W. IR and Raman assignments for zinc phthalocyanine from DFT calculations. Phys. Chem. Chem. Phys. 2000, 2, 3949–3955. [Google Scholar] [CrossRef]
- Szybowicz, M.; Bała, W.; Dümecke, S.; Fabisiak, K.; Paprocki, K.; Drozdowski, M. Temperature and orientation study of cobalt phthalocyanine CoPc thin films deposited on silicon substrate as studied by micro-Raman scattering spectroscopy. Thin Solid Film. 2011, 520, 623–627. [Google Scholar] [CrossRef]
- Aroca, R.; Pieczonka, N.; Kam, A.P. Surface-enhanced Raman scattering and SERRS imaging of phthalocyanine mixed films. J. Porphyr. Phthalocyanines 2001, 5, 25–32. [Google Scholar] [CrossRef] [Green Version]
- Shih, Y.; Zen, J.M.; Kumar, A.S.; Chen, P.Y. Flow injection analysis of zinc pyrithione in hair care products on a cobalt phthalocyanine modified screen-printed carbon electrode. Talanta 2004, 62, 912–917. [Google Scholar] [CrossRef]
- Fotouhi, L.; Hashkavayi, A.B.; Heravi, M.M. Electrochemical behaviour and voltammetric determination of sulphadiazine using a multi-walled carbon nanotube composite film-glassy carbon electrode. J. Exp. Nanosci. 2013, 8, 947–956. [Google Scholar] [CrossRef] [Green Version]
- Foster, C.W.; Pillay, J.; Metters, J.P.; Banks, C.E. Cobalt phthalocyanine modified electrodes utilised in electroanalysis: Nano-structured modified electrodes vs. bulk modified screen-printed electrodes. Sensors 2014, 14, 21905–21922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedioui, F.; Quinton, D.; Griveau, S.; Nyokong, T. Designing molecular materials and strategies for the electrochemical detection of nitric oxide, superoxide and peroxynitrite in biological systems. Phys. Chem. Chem. Phys. 2010, 12, 9976–9988. [Google Scholar] [CrossRef]
- Storkey, C.; Pattison, D.I.; Ignasiak, M.T.; Schiesser, C.H.; Davies, M.J. Kinetics of reaction of peroxynitrite with selenium- and sulfur-containing compounds: Absolute rate constants and assessment of biological significance. Free Radic. Biol. Med. 2015, 89, 1049–1056. [Google Scholar] [CrossRef]
- Møinichen, C. Microfluidic Flow Cells for Studies of Electrochemical Reactions. Master’s Thesis, Institutt for Materialteknologi, Oslo, Norway, 2012. [Google Scholar]
- Ai, F.; Chen, H.; Zhang, S.H.; Liu, S.Y.; Wei, F.; Dong, X.Y.; Cheng, J.K.; Huang, W.H. Real-time monitoring of oxidative burst from single plant protoplasts using microelectrochemical sensors modified by platinum nanoparticles. Anal. Chem. 2009, 81, 8453–8458. [Google Scholar] [CrossRef]
- Amatore, C.; Arbault, S.; Bruce, D.; de Oliveira, P.; Erard, M.; Vuillaume, M. Characterization of the electrochemical oxidation of peroxynitrite: Relevance to oxidative stress bursts measured at the single cell level. Chem.Eur. J. 2001, 7, 4171–4179. [Google Scholar] [CrossRef]
- Kissner, R.; Nauser, T.; Bugnon, P.; Lye, P.G.; Koppenol, W.H. Formation and properties of peroxynitrite as studied by laser flash photolysis, high-pressure stopped-flow technique, and pulse radiolysis. Chem. Res. Toxicol. 1998, 11, 1285–1292. [Google Scholar] [CrossRef]
- Kirsch, M.; Korth, H.-G.; Wensing, A.; Sustmann, R.; de Groot, H. Product formation and kinetic simulations in the pH range 1–14 account for a free-radical mechanism of peroxynitrite decomposition. Arch. Biochem. Biophys. 2003, 418, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Ascenzi, P.; De Simone, G.; Tundo, G.R.; Platas-Iglesias, C.; Coletta, M. Ferric nitrosylated myoglobin catalyzes peroxynitrite scavenging. J. Biol. Inorg. Chem. 2020, 25, 361–370. [Google Scholar] [CrossRef]
- Rehmann, F.-J. Mechanistic Studies of the Nitrogen Monoxide-and Nitrite-Mediated Reduction of Ferryl Myoglobin and Ferryl Hemoglobin. Ph.D. Thesis, ETH Zürich, Zürich, Switzerland, 2002. [Google Scholar]
- Tang, J.; Faustman, C.; Hoagland, T.A. Krzywicki Revisited: Equations for Spectrophotometric Determination of Myoglobin Redox Forms in Aqueous Meat Extracts. J. Food Sci. 2004, 69, C717–C720. [Google Scholar] [CrossRef]
- Carballal, S.; Cuevasanta, E.; Yadav, P.K.; Gherasim, C.; Ballou, D.P.; Alvarez, B.; Banerjee, R. Kinetics of Nitrite Reduction and Peroxynitrite Formation by Ferrous Heme in Human Cystathionine β-Synthase*. J. Biol. Chem. 2016, 291, 8004–8013. [Google Scholar] [CrossRef] [Green Version]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Biosensor | Potential (V) | Sensitivity (nA mM−1) | LOD (nM) | pH | Ref. |
|---|---|---|---|---|---|
| Microelectrode Pt/Mn-pDPB (manganese-[poly-2,5-di-(2-thienyl)-1H-pyrrole)-1-(p-benzoicacid)]) coated with PEI (polyethyleneimine) | 0.2 | 157.0 | 1.9 | 7.4 | [14] |
| Nanoelectrode carbon fibers/manganese(III)-[2]paracyclophenylporphyrin | −0.35 | 1 | 50 | - | [15] |
| Microelectrode Pt/MnTPAc (manganese tetraaminophthalocyanine) | −0.45 | 14.6 | 5000 | 10.2 | [12] |
| Electrode SPCE/2,6-dihydroxynaphthalene | 0.15 | 4.12 | 200 | 9–12 | [16] |
| Electrode SPCE/cobalt phthalocyanine | 0.1 | 10.84 | 400 | 9 | this work |
| Pseudo First-Order Decay Rates | k (s−1) | ||||
|---|---|---|---|---|---|
| Method | SPCE/CoPc | R2 | UV-Vis | R2 | |
| Samples | |||||
| PON 50 µM | 0.0084 ± 0.0011 | 0.9830 | 0.0089 ± 0.0010 | 0.9985 | |
| PON 150 µM | 0.0028 ± 0.0012 | 0.9975 | 0.0025 ± 0.0004 | 0.9877 | |
| Mb 15 µM + PON 50 µM | 0.0134 ± 0.0010 | 0.9815 | Not possible | - | |
| Mb 15 µM + PON 150 µM | 0.0086 ± 0.0020 | 0.9842 | Not possible | - | |
| Meat diluted 10 + PON 50 µM | 0.0200 ± 0.0048 | 0.9020 | Not possible | - | |
| Half-Lives and Rate Orders | t1/2 (s) | Calculated Values for Rate Order * | ||
|---|---|---|---|---|
| SPCE/CoPc | UV-Vis | SPCE/CoPc | UV-Vis | |
| PON 50 µM | 81.33 ± 8.69 | 84.73 ± 3.53 | 1.0000 ± 0.0014 | 1.0030 ± 0.0016 |
| PON 150 µM | 252.12 ± 2.97 | 230.75 ± 5.13 | 1.0001 ± 0.0001 | 1.0054 ± 0.0009 |
| Mb 15 µM + PON 50 µM | 64.83 ± 1.64 | Not possible | 1.0004 ± 0.0008 | Not possible |
| Mb 15 µM + PON 150 µM | 88.12 ± 0.03 | Not possible | 1.0001 ± 0.0001 | Not possible |
| Meat diluted 10 + PON 50 µM | 19.77 ± 0.10 | Not possible | 1.9442 ± 0.0587 | Not possible |
| Apparent First-Order Rate Constants | k (s−1) | |
|---|---|---|
| SPCE/CoPc | UV-Vis | |
| PON 50 µM | 0.00862 ± 0.0007 | 0.0080 ± 0.0018 |
| PON 150 µM | 0.00275 ± 0.0012 | 0.0030 ± 0.0004 |
| Mb 15 µM + PON 50 µM | 0.01690 ± 0.0010 | Not possible |
| Mb 15 µM + PON 150 µM | 0.00780 ± 0.0020 | Not possible |
| Apparent Second-Order Rate Constants | k (M−1 s−1) | |
|---|---|---|
| SPCE/CoPc | UV-Vis | |
| Mb 15 µM + PON 50 µM | 311.87 ± 7.9600 | Not possible |
| Meat diluted 10 + PON 50 µM | 891.76 ± 220.54 | Not possible |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hosu, I.S.; Constantinescu-Aruxandei, D.; Oancea, F.; Doni, M. The Scavenging Effect of Myoglobin from Meat Extracts toward Peroxynitrite Studied with a Flow Injection System Based on Electrochemical Reduction over a Screen-Printed Carbon Electrode Modified with Cobalt Phthalocyanine: Quantification and Kinetics. Biosensors 2021, 11, 220. https://doi.org/10.3390/bios11070220
Hosu IS, Constantinescu-Aruxandei D, Oancea F, Doni M. The Scavenging Effect of Myoglobin from Meat Extracts toward Peroxynitrite Studied with a Flow Injection System Based on Electrochemical Reduction over a Screen-Printed Carbon Electrode Modified with Cobalt Phthalocyanine: Quantification and Kinetics. Biosensors. 2021; 11(7):220. https://doi.org/10.3390/bios11070220
Chicago/Turabian StyleHosu, Ioana Silvia, Diana Constantinescu-Aruxandei, Florin Oancea, and Mihaela Doni. 2021. "The Scavenging Effect of Myoglobin from Meat Extracts toward Peroxynitrite Studied with a Flow Injection System Based on Electrochemical Reduction over a Screen-Printed Carbon Electrode Modified with Cobalt Phthalocyanine: Quantification and Kinetics" Biosensors 11, no. 7: 220. https://doi.org/10.3390/bios11070220
APA StyleHosu, I. S., Constantinescu-Aruxandei, D., Oancea, F., & Doni, M. (2021). The Scavenging Effect of Myoglobin from Meat Extracts toward Peroxynitrite Studied with a Flow Injection System Based on Electrochemical Reduction over a Screen-Printed Carbon Electrode Modified with Cobalt Phthalocyanine: Quantification and Kinetics. Biosensors, 11(7), 220. https://doi.org/10.3390/bios11070220