Microfluidic-Assisted Fabrication of Dual-Coated pH-Sensitive Mesoporous Silica Nanoparticles for Protein Delivery

 ,
,  ,
,  and
and 
Abstract
:1. Introduction
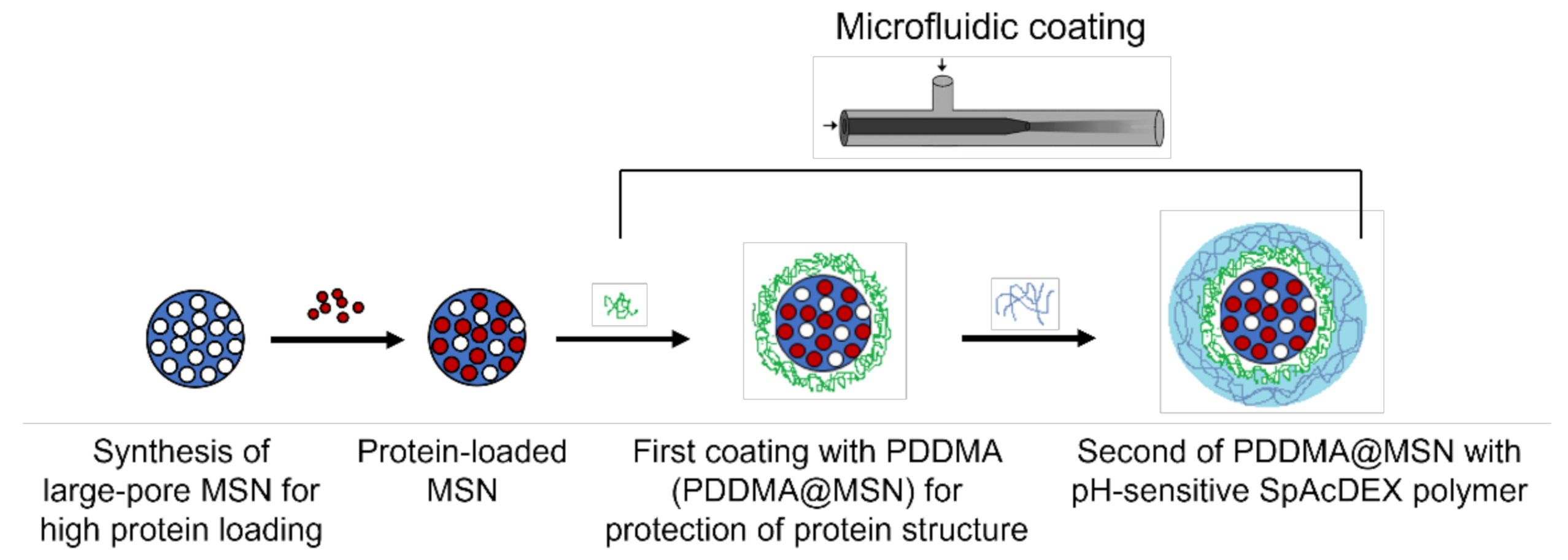
2. Results and Discussion
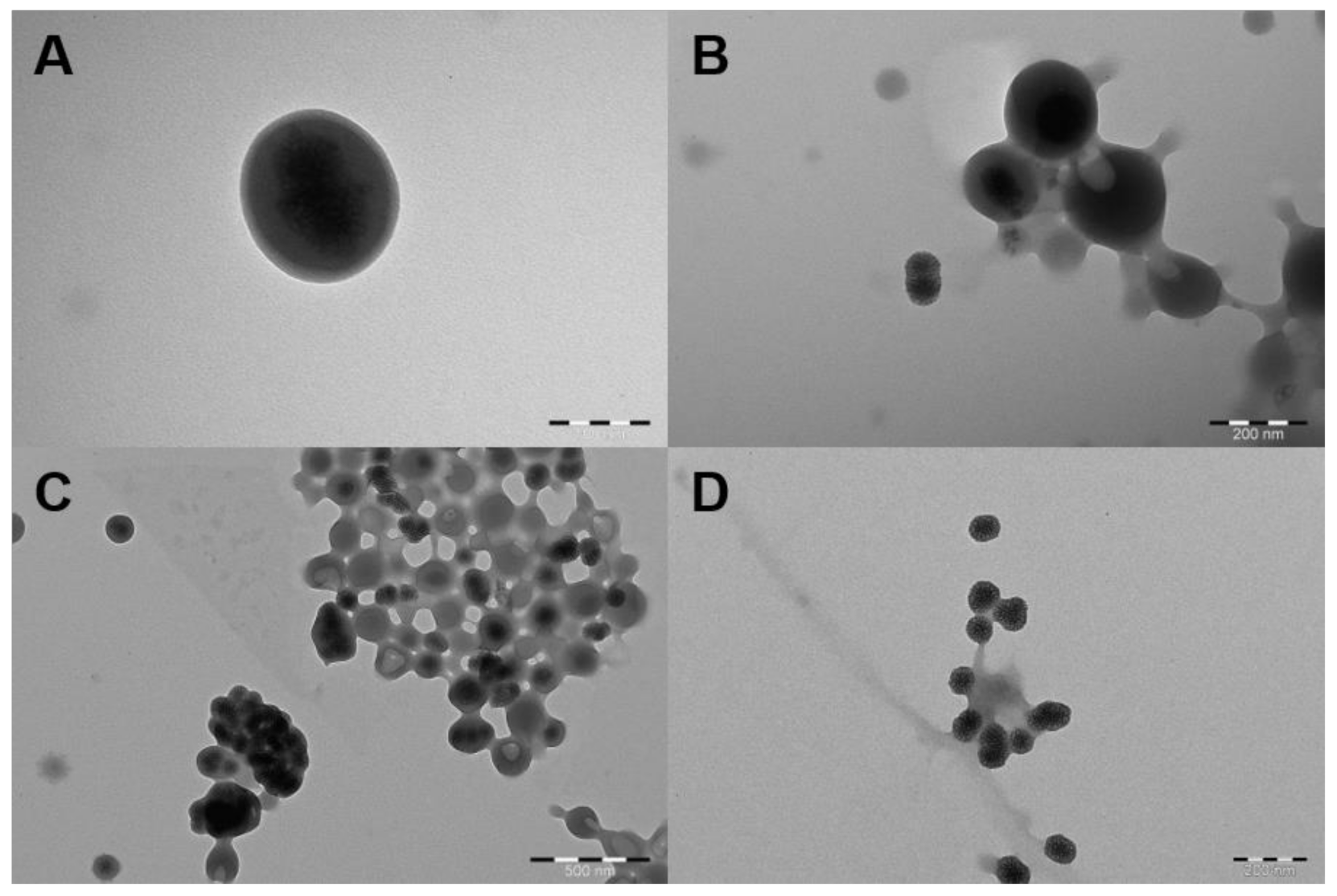
2.1. Characterization of MSNs
2.2. Loading Lysozyme into MSNs
2.3. PDDMA Coating of MSNs
2.4. SpAcDEX Coating of PDDMA@MSNs
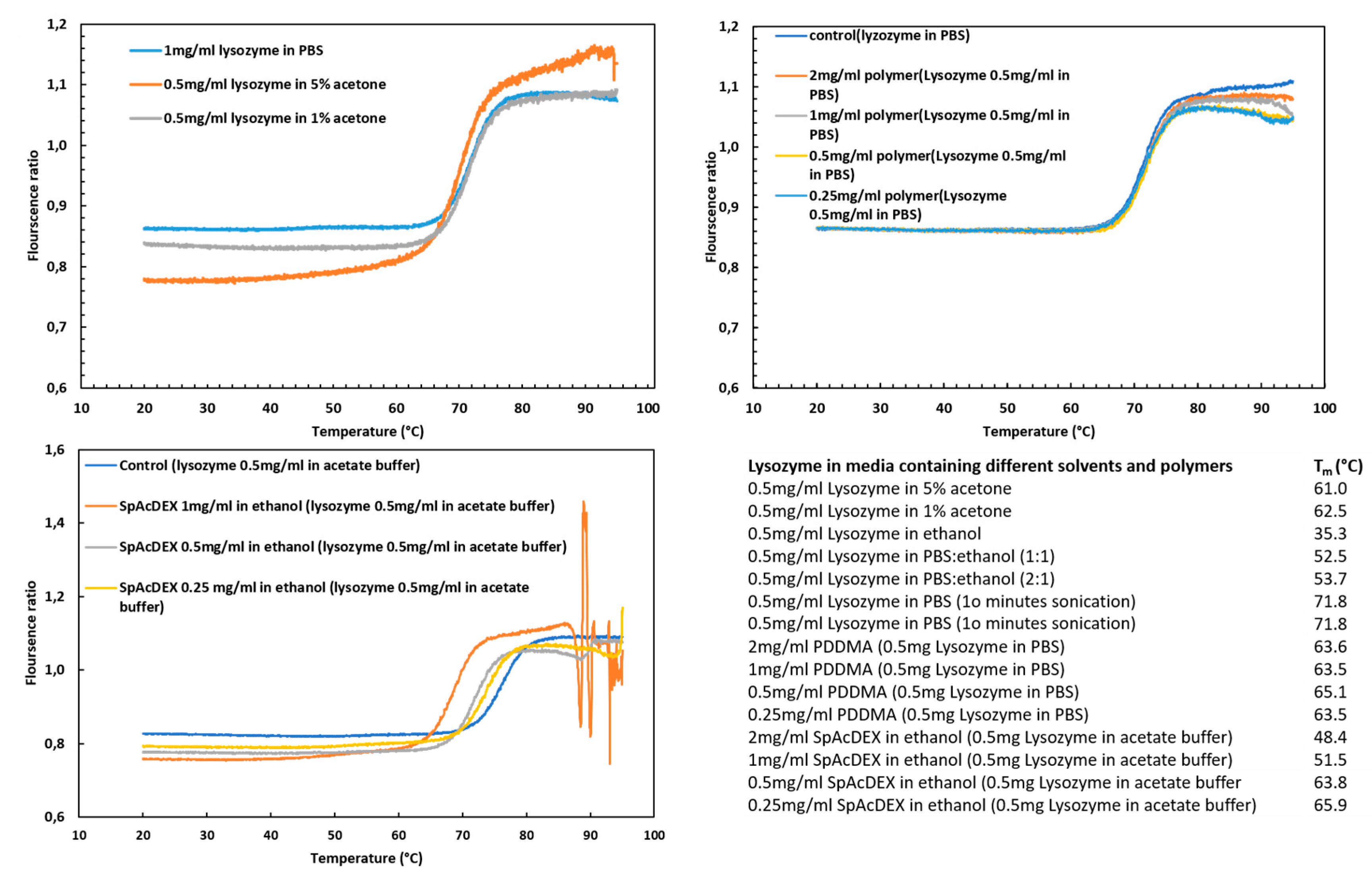
2.5. Stability of Lysozyme
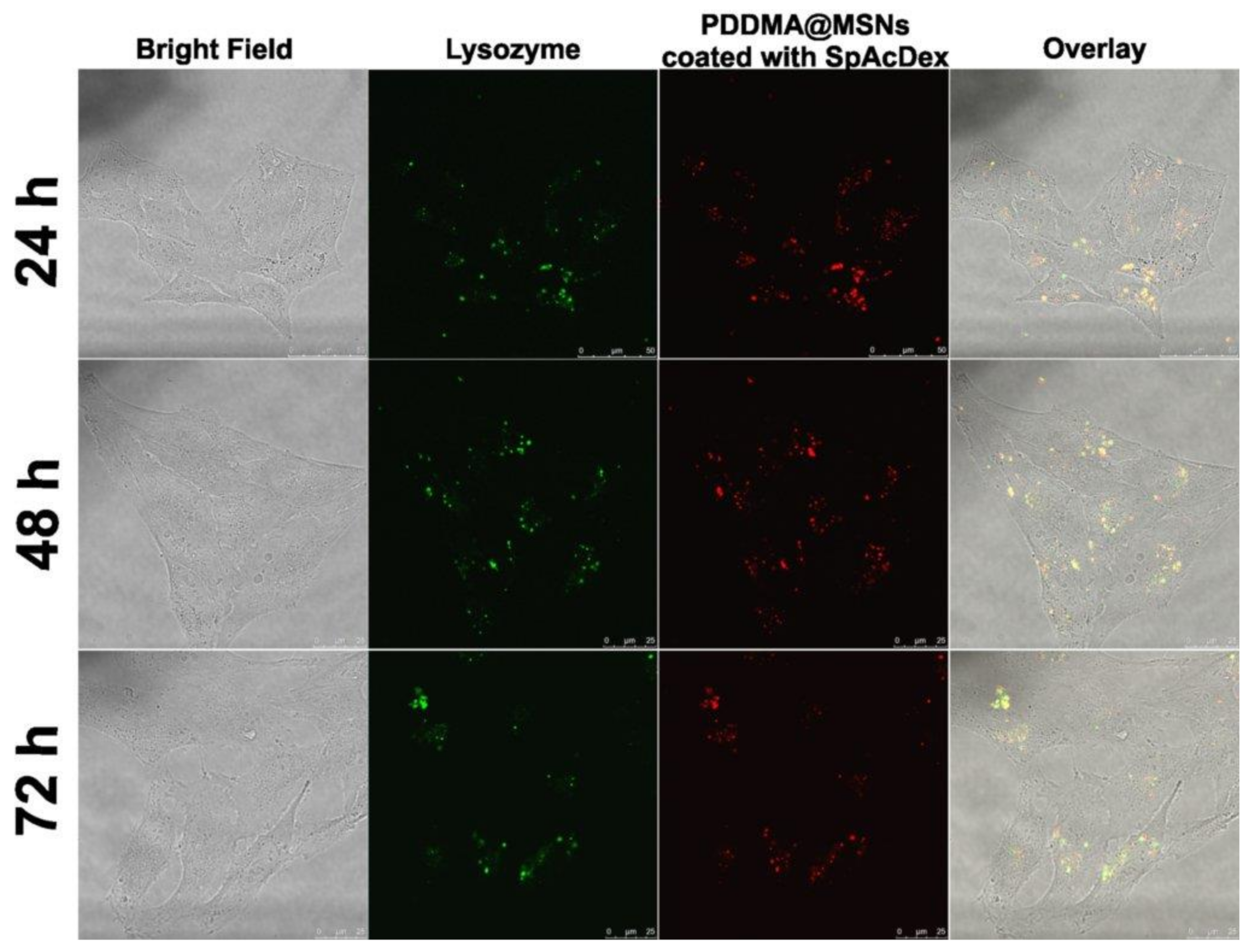
2.6. Cellular Uptake
3. Materials and Methods
3.1. Materials
3.2. Synthesis of SpAcDEX Polymer
3.3. Synthesis of Non-Labelled and Tetramethylrhodamine (TRITC)-Labelled Mesoporous Silica Nanoparticles
3.4. Characterization of Synthesized MSNs
3.5. FITC-Lysozyme Bioconjugation
3.6. Lysozyme Loading
3.7. Fabrication of the Microfluidic Flow-Focusing Glass-Capillary Chip
3.8. PDDMA Coating
3.9. SpAcDEX Coating
3.10. Stability of Lysozyme
3.11. Cellular Uptake
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Whitesides, G.M. The origins and the future of microfluidics. Nature 2006, 442, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Tsui, J.H.; Lee, W.; Pun, S.H.; Kim, J.; Kim, D.-H. Microfluidics-assisted in vitro drug screening and carrier production. Adv. Drug Deliv. Rev. 2013, 65, 1575–1588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Correa, S.; Choi, K.Y.; Dreaden, E.C.; Renggli, K.; Shi, A.; Gu, L.; Shopsowitz, K.E.; Quadir, M.A.; Ben-Akiva, E.; Hammond, P.T. Highly scalable, closed-loop synthesis of drug-loaded, layer-by-layer nanoparticles. Adv. Funct. Mater. 2016, 26, 991–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, D.; Zhang, H.; Fontana, F.; Hirvonen, J.T.; Santos, H.A. Microfluidic-assisted fabrication of carriers for controlled drug delivery. Lab Chip 2017, 17, 1856–1883. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Zhang, H.; Fontana, F.; Hirvonen, J.T.; Santos, H.A. Current developments and applications of microfluidic technology toward clinical translation of nanomedicines. Adv. Drug Deliv. Rev. 2018, 128, 54–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thanh, N.T.; Maclean, N.; Mahiddine, S. Mechanisms of nucleation and growth of nanoparticles in solution. Chem. Rev. 2014, 114, 7610–7630. [Google Scholar] [CrossRef]
- Johnson, B.K.; Prud’homme, R.K. Mechanism for rapid self-assembly of block copolymer nanoparticles. Phys. Rev. Lett. 2003, 91, 118302. [Google Scholar] [CrossRef]
- Han, J.; Zhu, Z.; Qian, H.; Wohl, A.R.; Beaman, C.J.; Hoye, T.R.; Macosko, C.W. A simple confined impingement jets mixer for flash nanoprecipitation. J. Pharm. Sci. 2012, 101, 4018–4023. [Google Scholar] [CrossRef]
- Feng, Q.; Sun, J.; Jiang, X. Microfluidics-mediated assembly of functional nanoparticles for cancer-related pharmaceutical applications. Nanoscale 2016, 8, 12430–12443. [Google Scholar] [CrossRef]
- Dev, S.; Toster, J.; Vadhan Prasanna, S.; Fitzgerald, M.; Swaminathan Iyer, K.; Raston, C.L. Suppressing regrowth of microfluidic generated drug nanocrystals using polyelectrolyte coatings. RSC Adv. 2013, 3, 695–698. [Google Scholar] [CrossRef]
- Amstad, E.; Spaepen, F.; Weitz, D.A. Stabilization of the Amorphous Structure of Spray-Dried Drug Nanoparticles. J. Phys. Chem. B 2016, 120, 9161–9165. [Google Scholar] [CrossRef] [PubMed]
- Herranz-Blanco, B.; Liu, D.; Mäkilä, E.; Shahbazi, M.-A.; Ginestar, E.; Zhang, H.; Aseyev, V.; Salasubramanian, V.; Salonen, J.; Hirvonen, J.; et al. On-Chip Self-Assembly of a Smart Hybrid Nanocomposite for Antitumoral Applications. Adv. Funct. Mater. 2015, 25, 1488–1497. [Google Scholar] [CrossRef]
- Liu, D.; Zhang, H.; Cito, S.; Fan, J.; Mäkilä, E.; Salonen, J.; Hirvonen, J.; Sikanen, T.M.; Weitz, D.A.; Santos, H.A. Core/Shell Nanocomposites Produced by Superfast Sequential Microfluidic Nanoprecipitation. Nano Lett. 2017, 17, 606–614. [Google Scholar] [CrossRef]
- Liu, X.; Wu, F.; Ji, Y.; Yin, L. Recent Advances in Anti-cancer Protein/Peptide Delivery. Bioconjugate Chem. 2019, 30, 305–324. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Deng, S.; You, Y.; Chan, H.F. The role of microfluidics in protein formulations with pre-programmed functional characteristics. Biologics 2018, 12, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.J.; Xu, P. Smart Mesoporous Silica Nanoparticles for Protein Delivery. Nanomaterials 2019, 9, 511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, L.; Bi, J.; Tang, Y.; Qiao, S.Z. Magnetic Core-Shell Silica Nanoparticles with Large Radial Mesopores for siRNA Delivery. Small 2016, 12, 4735–4742. [Google Scholar] [CrossRef]
- Kao, K.-C.; Lin, T.-S.; Mou, C.-Y. Enhanced Activity and Stability of Lysozyme by Immobilization in the Matching Nanochannels of Mesoporous Silica Nanoparticles. J. Phys. Chem. C 2014, 118, 6734–6743. [Google Scholar] [CrossRef]
- Watermann, A.; Brieger, J. Mesoporous Silica Nanoparticles as Drug Delivery Vehicles in Cancer. Nanomaterials 2017, 7, 189. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Goel, S.; Shi, S.; Barnhart, T.E.; Lan, X.; Cai, W. General synthesis of silica-based yolk/shell hybrid nanomaterials and in vivo tumor vasculature targeting. Nano Res. 2018, 11, 4890–4904. [Google Scholar] [CrossRef]
- Marcelo, G.; Tarazona, M.P.; Saiz, E. Solution properties of poly(diallyldimethylammonium chloride) (PDDA). Polymer 2005, 46, 2584–2594. [Google Scholar] [CrossRef]
- Butzbach, K.; Konhäuser, M.; Fach, M.; Bamberger, D.N.; Breitenbach, B.; Epe, B.; Wich, P.R. Receptor-mediated Uptake of Folic Acid-functionalized Dextran Nanoparticles for Applications in Photodynamic Therapy. Polymers 2019, 11, 896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blanco, E.; Shen, H.; Ferrari, M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat. Biotechnol. 2015, 33, 941–951. [Google Scholar] [CrossRef]
- Li, Y.; Lian, Y.; Zhang, L.T.; Aldousari, S.M.; Hedia, H.S.; Asiri, S.A.; Liu, W.K. Cell and nanoparticle transport in tumour microvasculature: The role of size, shape and surface functionality of nanoparticles. Interface Focus 2016, 6, 20150086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yadav, I.; Kumar, S.; Aswal, V.K.; Kohlbrecher, J. Structure and Interaction in the pH-Dependent Phase Behavior of Nanoparticle-Protein Systems. Langmuir 2017, 33, 1227–1238. [Google Scholar] [CrossRef] [PubMed]
- Henry, N.; Clouet, J.; Le Visage, C.; Weiss, P.; Gautron, E.; Renard, D.; Cordonnier, D.; Boury, F.; Humbert, B.; Terrisse, H.; et al. Silica nanofibers as a new drug delivery system: A study of the protein–silica interactions. J. Mater. Chem. B 2017, 5, 2908–2920. [Google Scholar] [CrossRef]
- Mora-Huertas, C.E.; Fessi, H.; Elaissari, A. Polymer-based nanocapsules for drug delivery. Int. J. Pharm. 2010, 385, 113–142. [Google Scholar] [CrossRef]
- Danaei, M.; Dehghankhold, M.; Ataei, S.; Hasanzadeh Davarani, F.; Javanmard, R.; Dokhani, A.; Knorasani, S.; Mozafari, M.R. Impact of Particle Size and Polydispersity Index on the Clinical Applications of Lipidic Nanocarrier Systems. Pharmaceutics 2018, 10, 57. [Google Scholar] [CrossRef] [Green Version]
- Chiesa, E.; Greco, A.; Riva, F.; Dorati, R.; Conti, B.; Modena, T.; Genta, I. Hyaluronic Acid-Based Nanoparticles for Protein Delivery: Systematic Examination of Microfluidic Production Conditions. Pharmaceutics 2021, 13, 1565. [Google Scholar] [CrossRef]
- Shen, J.; Ma, M.; Zhang, H.; Yu, H.; Xue, F.; Hao, N.; Chen, H. Microfluidics-Assisted Surface Trifunctionalization of a Zeolitic Imidazolate Framework Nanocarrier for Targeted and Controllable Multitherapies of Tumors. ACS Appl. Mater. Interfaces 2020, 12, 45838–45849. [Google Scholar] [CrossRef]
- Deng, J.; Davies, D.R.; Wisedchaisri, G.; Wu, M.; Hol, W.G.; Mehlin, C. An improved protocol for rapid freezing of protein samples for long-term storage. Acta. Crystallogr. D. Biol. Crystallogr. 2004, 60, 203–204. [Google Scholar] [CrossRef] [PubMed]
- Inoue, M.; Yamada, H.; Yasukochi, T.; Kuroki, R.; Miki, T.; Horiuchi, T.; Imoto, T. Multiple role of hydrophobicity of tryptophan-108 in chicken lysozyme: Structural stability, saccharide binding ability, and abnormal pKa of glutamic acid-35. Biochemistry 1992, 31, 5545–5553. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Thulstrup, P.W.; Mu, H.; Hansen, S.H.; van de Weert, M.; Rantanen, J.; Yang, M. Effect of ethanol as a co-solvent on the aerosol performance and stability of spray-dried lysozyme. Int. J. Pharm. 2016, 513, 175–182. [Google Scholar] [CrossRef] [PubMed]
- van de Weert, M.; Hoechstetter, J.; Hennink, W.E.; Crommelin, D.J.A. The effect of a water/organic solvent interface on the structural stability of lysozyme. J. Control. Release 2000, 68, 351–359. [Google Scholar] [CrossRef]
- Cohen, J.L.; Schubert, S.; Wich, P.R.; Cui, L.; Cohen, J.A.; Mynar, J.L.; Fréchet, J.M.J. Acid-degradable cationic dextran particles for the delivery of siRNA therapeutics. Bioconjug. Chem. 2011, 22, 1056–1065. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhang, Y.; Weisensee, K. Conducting Polymeric Nanocomposites with a Three-Dimensional Co-flow Microfluidics Platform. Micromachines 2019, 10, 383. [Google Scholar] [CrossRef] [Green Version]
- Ferreira, M.P.A.; Talman, V.; Torrieri, G.; Liu, D.; Marques, G.; Moslova, K.; Liu, Z.; Pinto, J.F.; Hirvonen, J.; Ruskoaho, H. Dual-Drug Delivery Using Dextran-Functionalized Nanoparticles Targeting Cardiac Fibroblasts for Cellular Reprogramming. Adv. Funct. Mater. 2018, 28, 1705134. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, D.; Wang, L.; Liu, Z.; Wu, R.; Janoniene, A.; Ma, M.; Pan, G.; Baranauskiene, L.; Zhang, L.; et al. Microfluidic Encapsulation of Prickly Zinc-Doped Copper Oxide Nanoparticles with VD1142 Modified Spermine Acetalated Dextran for Efficient Cancer Therapy. Adv. Healthc. Mater. 2017, 6, 1601406. [Google Scholar] [CrossRef]
- Desai, D.; Karaman, D.S.; Prabhakar, N.; Tadayon, S.; Duchanoy, A.; Toivola, D.M.; Rajput, S.; Näreoja, T.; Rosenholm, J. Design considerations for mesoporous silica nanoparticulate systems in facilitating biomedical applications. Open Mater. Sci. 2014, 1, 16–43. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
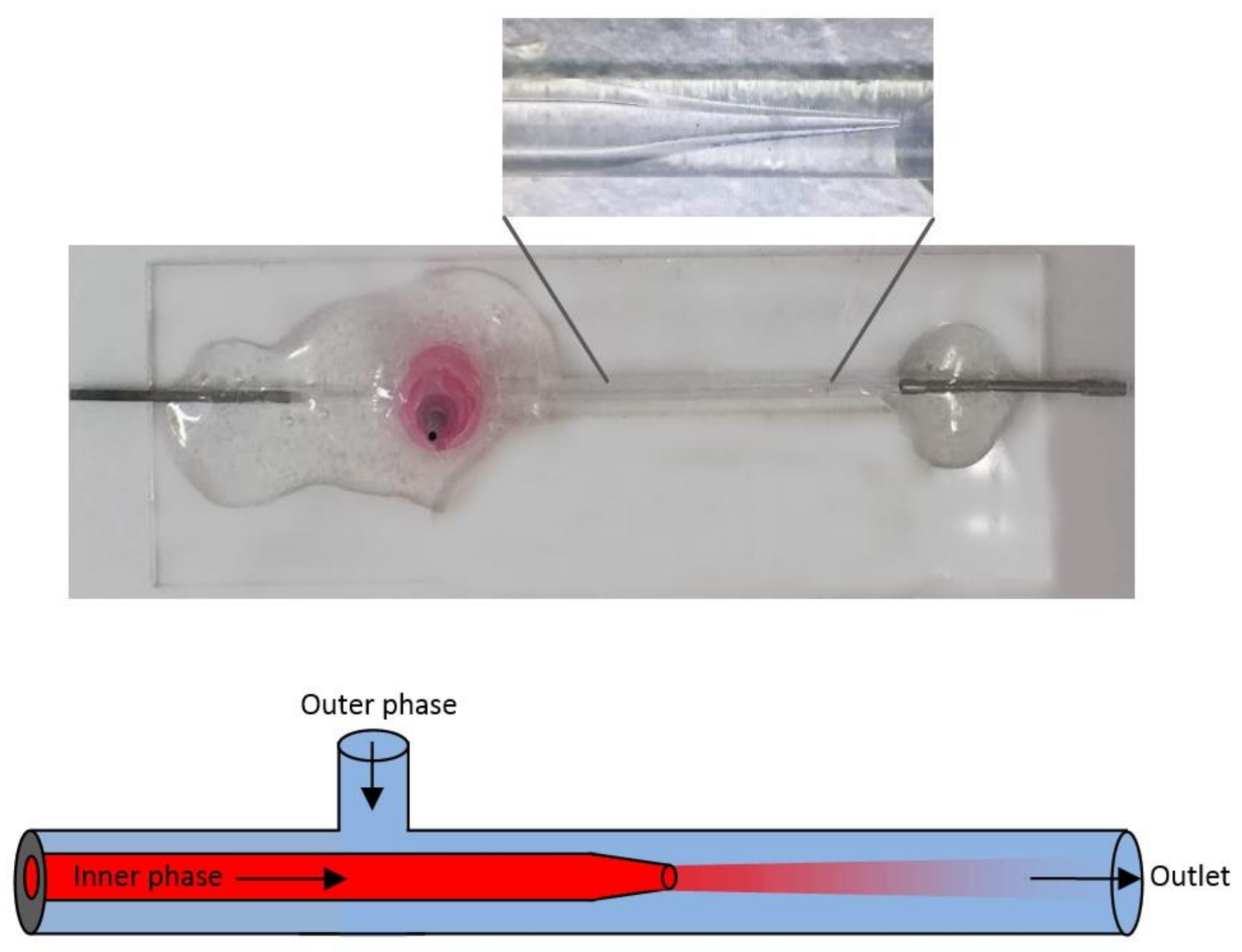{kind=link}
| Code | PDDMA (mg/mL) | Flow Rate (mL/h) (PDDMA: Acetone) | MSN (mg/mL) | Size (nm) | PDI | Zeta Potential (mV) |
|---|---|---|---|---|---|---|
| F1 | 5 | 2:40 | 0.25 | 96.16 ± 4.09 | 0.401 ± 0.064 | −2.02 ± 0.77 |
| F2 | 10 | 2:40 | 0.25 | 146.5 ± 4.17 | 0.197 ± 0.025 | 17.0 ± 0.90 |
| F3 | 10 | 2:20 | 0.25 | 140.1 ± 3.60 | 0.385 ± 0.021 | 9.49 ± 0.90 |
| F4 | 15 | 2:40 | 0.25 | 105.2 ± 9.45 | 0.466 ± 0.049 | 11.1 ± 1.17 |
| F5 | 20 | 2:20 | 0.5 | 792.5 ± 152.0 | 0.436 ± 0.238 | 17.7 ± 0.43 |
| F6 | 20 | 2:40 | 0.5 | 276.7 ± 2.85 | 0.151 ± 0.054 | 33.5 ± 1.30 |
| F7 | 20 | 2:60 | 0.5 | 146.5 ± 2.12 | 0.262 ± 0.008 | 14.5 ± 0.32 |
| F8 | 20 | 2:80 | 0.5 | 218.1 ± 1.50 | 0.187 ± 0.012 | 24.9 ± 0.75 |
| F9 | 22 | 2:20 | 0.5 | 463.5 ± 59.62 | 0.384 ± 0.539 | 22.9 ± 0.43 |
| F10 | 22 | 2:40 | 0.5 | 396.9 ± 22.16 | 0.521 ± 0.268 | 7.63 ± 1.81 |
| F11 | 22 | 2:60 | 0.5 | 282.2 ± 17.16 | 0.698 ± 0.365 | 24.9 ± 0.50 |
| F12 | 25 | 2:60 | 0.5 | 570.2 ± 108.0 | 0.827 ± 0.300 | 14.8 ± 0.60 |
| F13 | 30 | 2:60 | 0.5 | 517.4 ± 41.25 | 0.743 ± 0.371 | 16.5 ± 2.21 |
| SpAcDEX (mg/mL) | Flow Rate (mL/h) (SpAcDEX: 0.1% Pluronic F127) | PDDMA@MSN (mg/mL) | Size (nm) | PDI | Zeta Potential (mV) | |
|---|---|---|---|---|---|---|
| S1 | 2 | 2:40 | 2 | 225.2 ± 2.90 | 0.316 ± 0.032 | 26.4 ± 1.11 |
| S2 | 1 | 2:40 | 2 | 166.2 ± 2.40 | 0.204 ± 0.014 | 14.5 ± 0.90 |
| S3 | 1 | 2:20 | 2 | 173.4 ± 3.77 | 0.225 ± 0.006 | 16.5 ± 1.61 |
| S4 | 1 | 2:20 | 1 | 168.3 ± 3.69 | 0.184 ± 0.012 | 20.1 ± 1.33 |
| S5 | 0.5 | 2:20 | 1 | 166.3 ± 14.06 | 0.226 ± 0.026 | 14.1 ± 0.43 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Küçüktürkmen, B.; Inam, W.; Howaili, F.; Gouda, M.; Prabhakar, N.; Zhang, H.; Rosenholm, J.M. Microfluidic-Assisted Fabrication of Dual-Coated pH-Sensitive Mesoporous Silica Nanoparticles for Protein Delivery. Biosensors 2022, 12, 181. https://doi.org/10.3390/bios12030181
Küçüktürkmen B, Inam W, Howaili F, Gouda M, Prabhakar N, Zhang H, Rosenholm JM. Microfluidic-Assisted Fabrication of Dual-Coated pH-Sensitive Mesoporous Silica Nanoparticles for Protein Delivery. Biosensors. 2022; 12(3):181. https://doi.org/10.3390/bios12030181
Chicago/Turabian StyleKüçüktürkmen, Berrin, Wali Inam, Fadak Howaili, Mariam Gouda, Neeraj Prabhakar, Hongbo Zhang, and Jessica M. Rosenholm. 2022. "Microfluidic-Assisted Fabrication of Dual-Coated pH-Sensitive Mesoporous Silica Nanoparticles for Protein Delivery" Biosensors 12, no. 3: 181. https://doi.org/10.3390/bios12030181
APA StyleKüçüktürkmen, B., Inam, W., Howaili, F., Gouda, M., Prabhakar, N., Zhang, H., & Rosenholm, J. M. (2022). Microfluidic-Assisted Fabrication of Dual-Coated pH-Sensitive Mesoporous Silica Nanoparticles for Protein Delivery. Biosensors, 12(3), 181. https://doi.org/10.3390/bios12030181