Electric Cell-Substrate Impedance Sensing (ECIS) as a Platform for Evaluating Barrier-Function Susceptibility and Damage from Pulmonary Atelectrauma



 , and
, and
Abstract
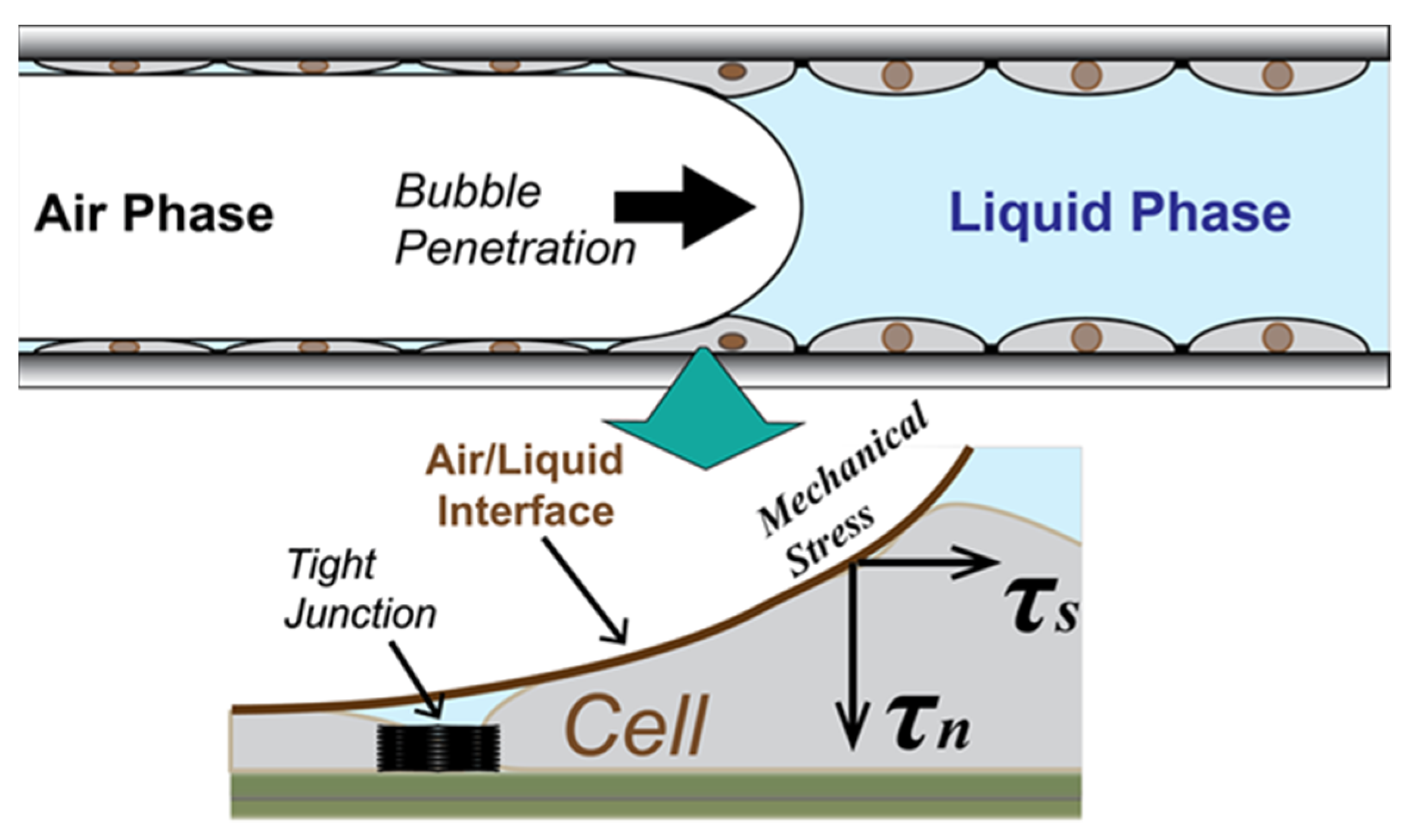
:1. Introduction
- Is there a method to determine the resilience of the epithelium?
- Under which conditions is RD likely to damage the pulmonary epithelium?
- What is the time course and process by which the epithelial layer becomes damaged?
2. Materials and Methods
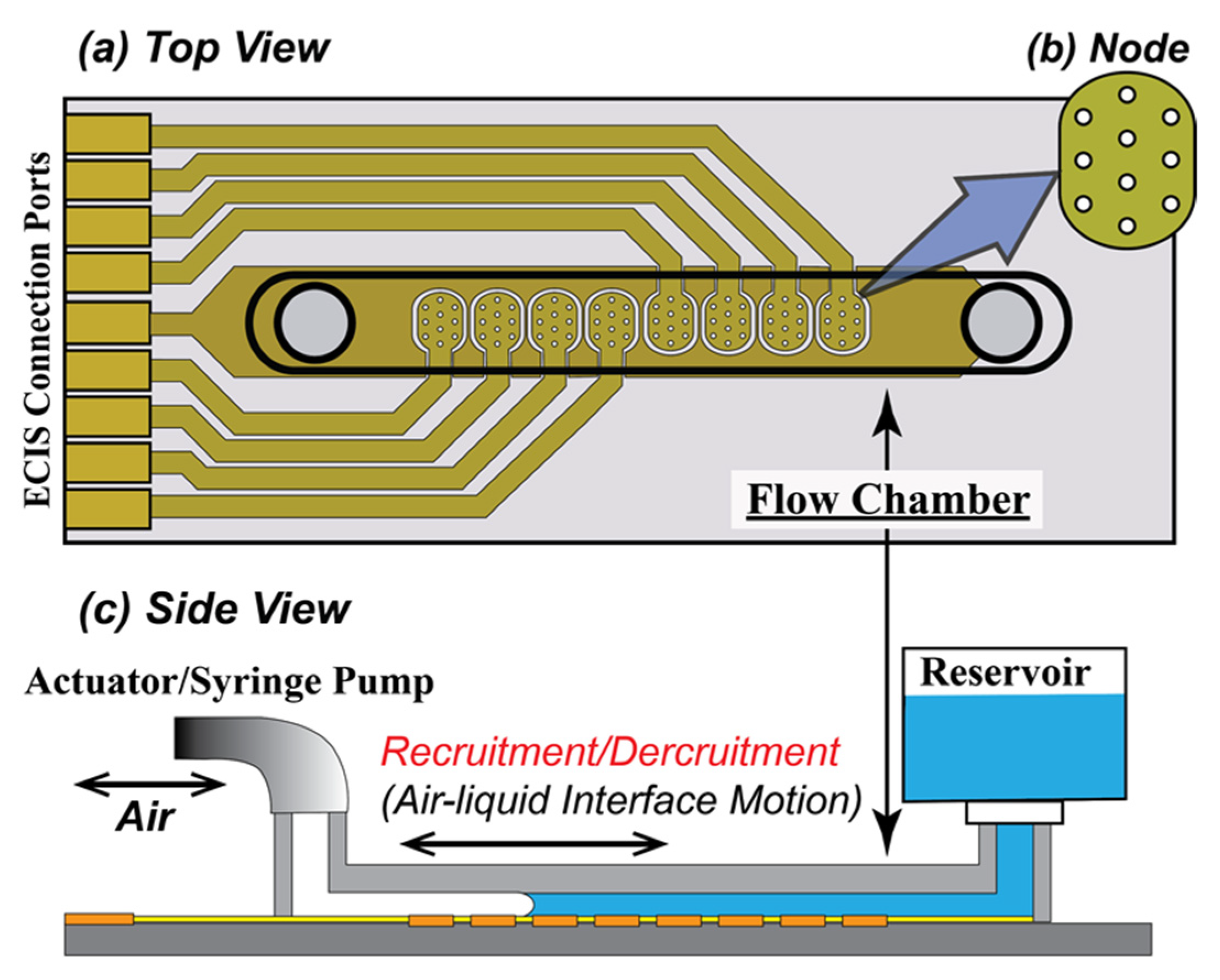
2.1. In Vitro Epithelial Monolayer System
2.1.1. Flow Chamber
2.1.2. Preparation of the Chamber
2.1.3. Seeding Density
2.1.4. Feeding Condition
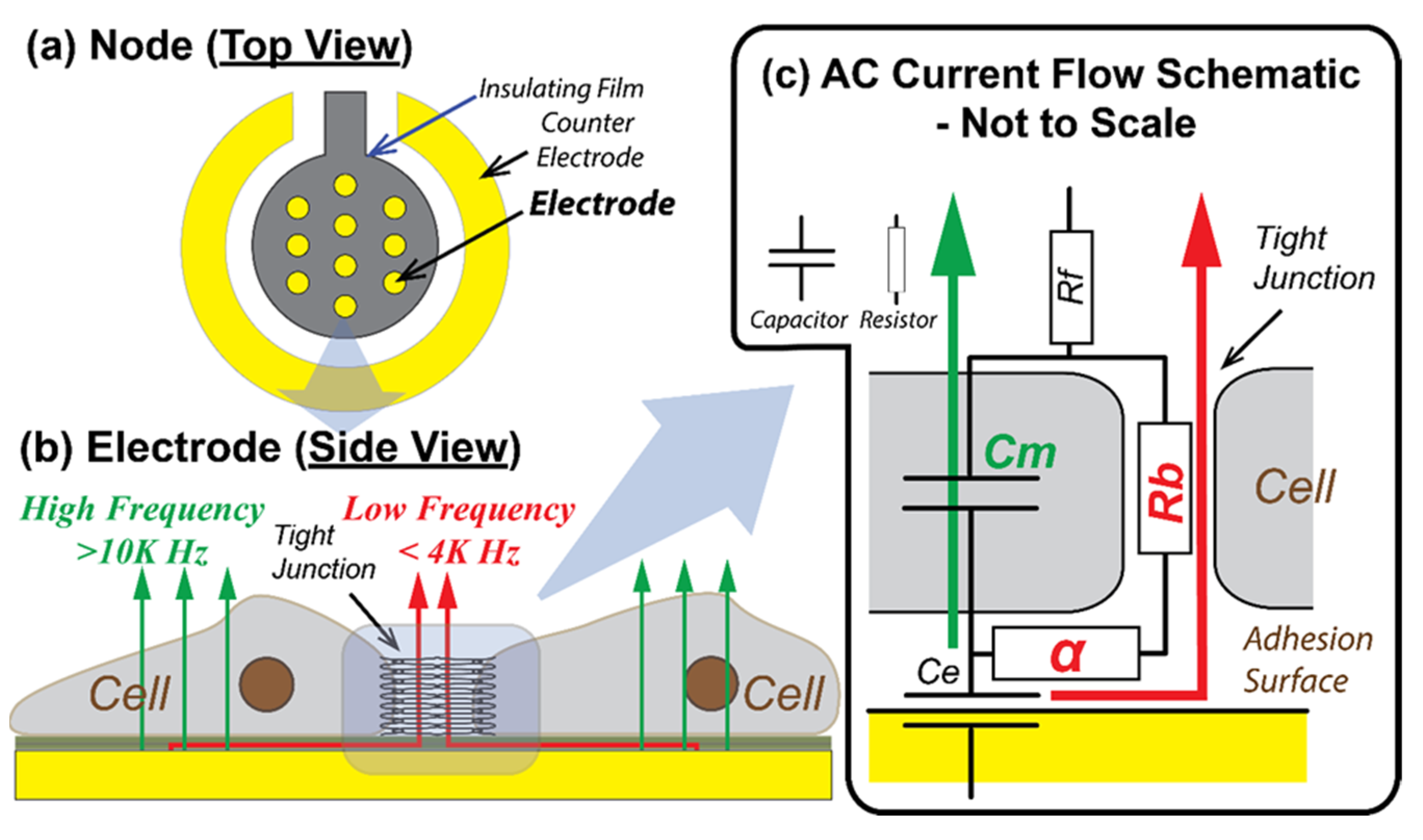
2.2. Electrical Cell-Substrate Impedance Sensing
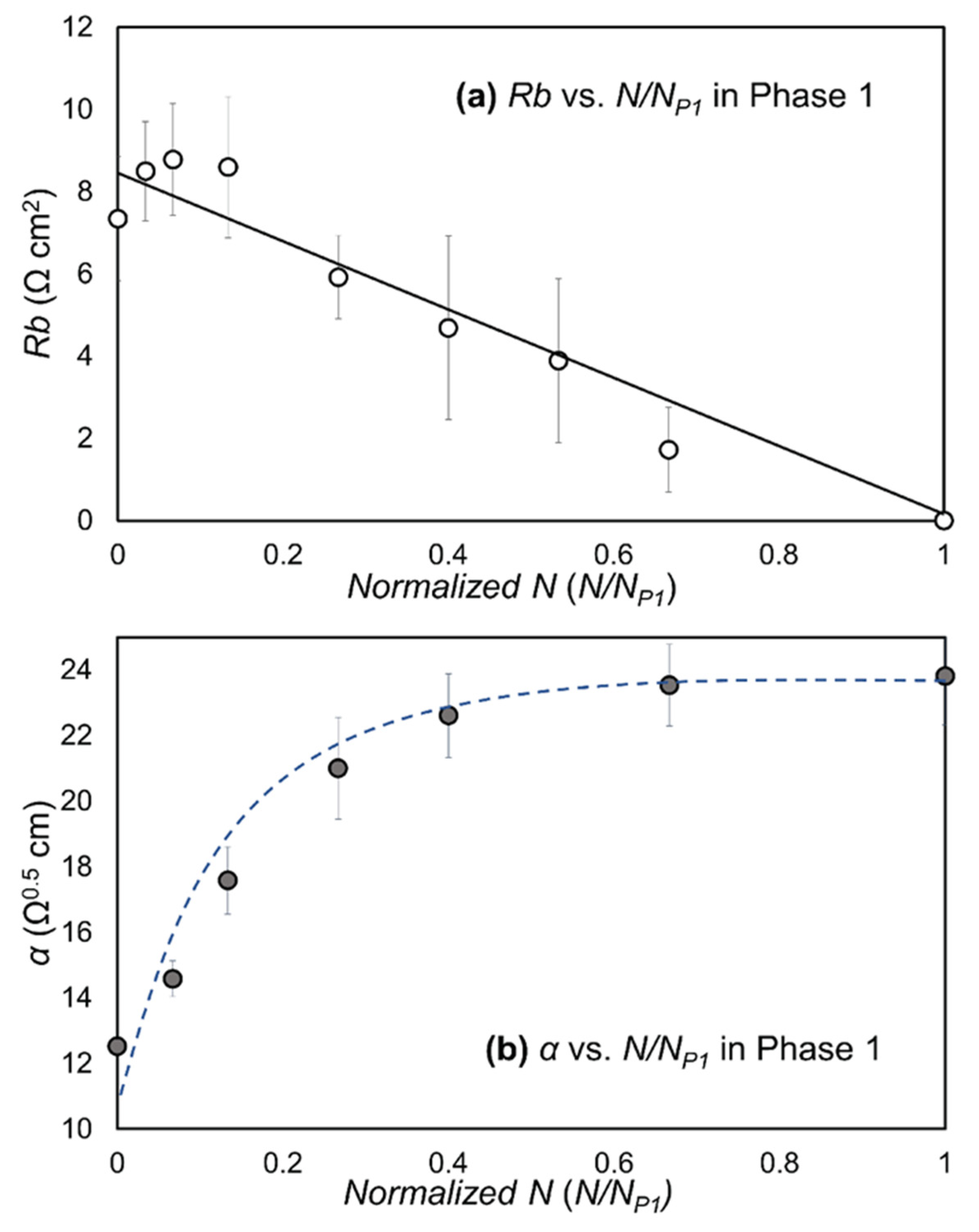
- Rb (Ω cm2) that quantifies the integrity of cell–cell tight junctions,
- Cm (µF/cm2) that quantifies the confluency of the cell membrane, and
- α (Ω0.5 cm) that quantifies cell–surface adhesion [12].
2.3. RD Experimental Protocol
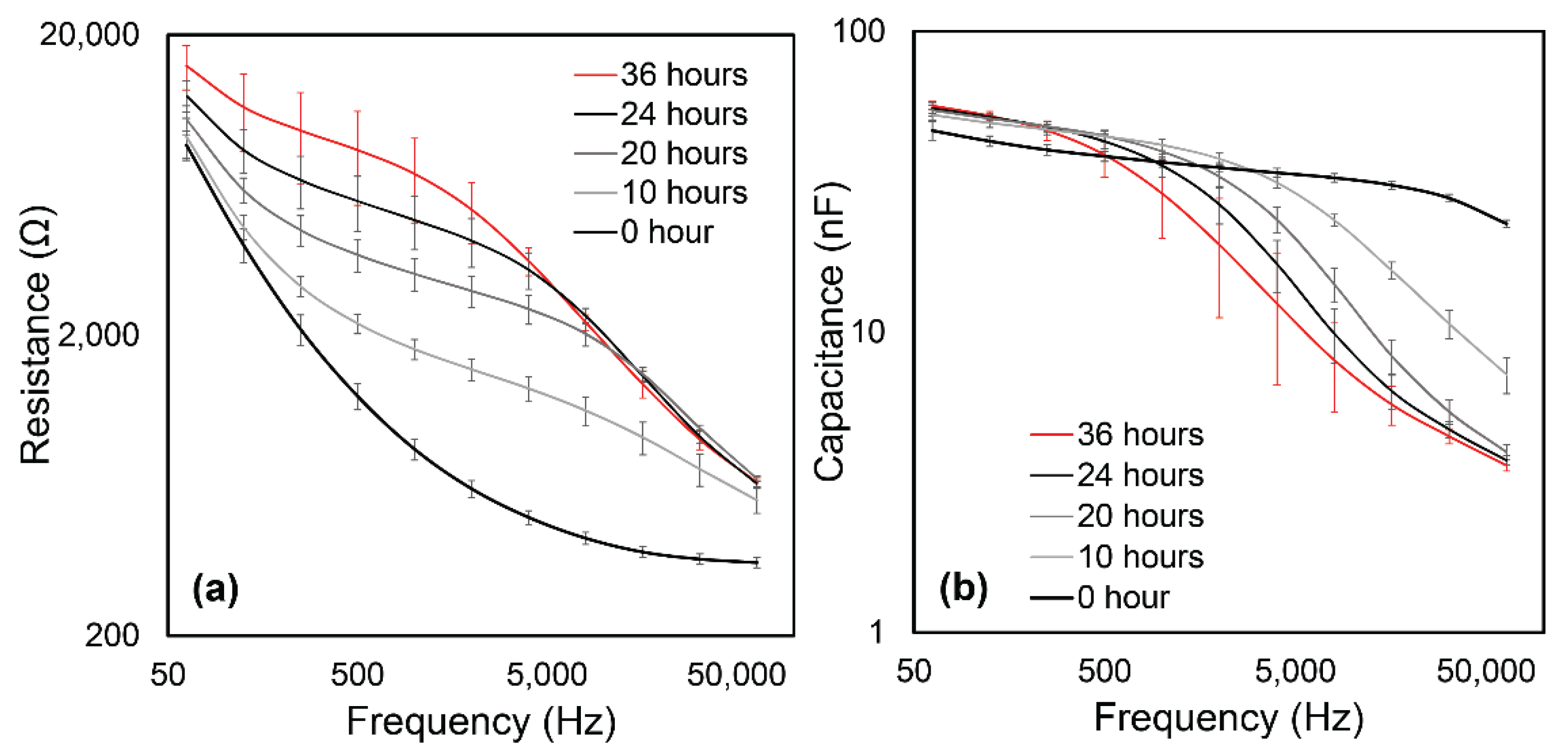
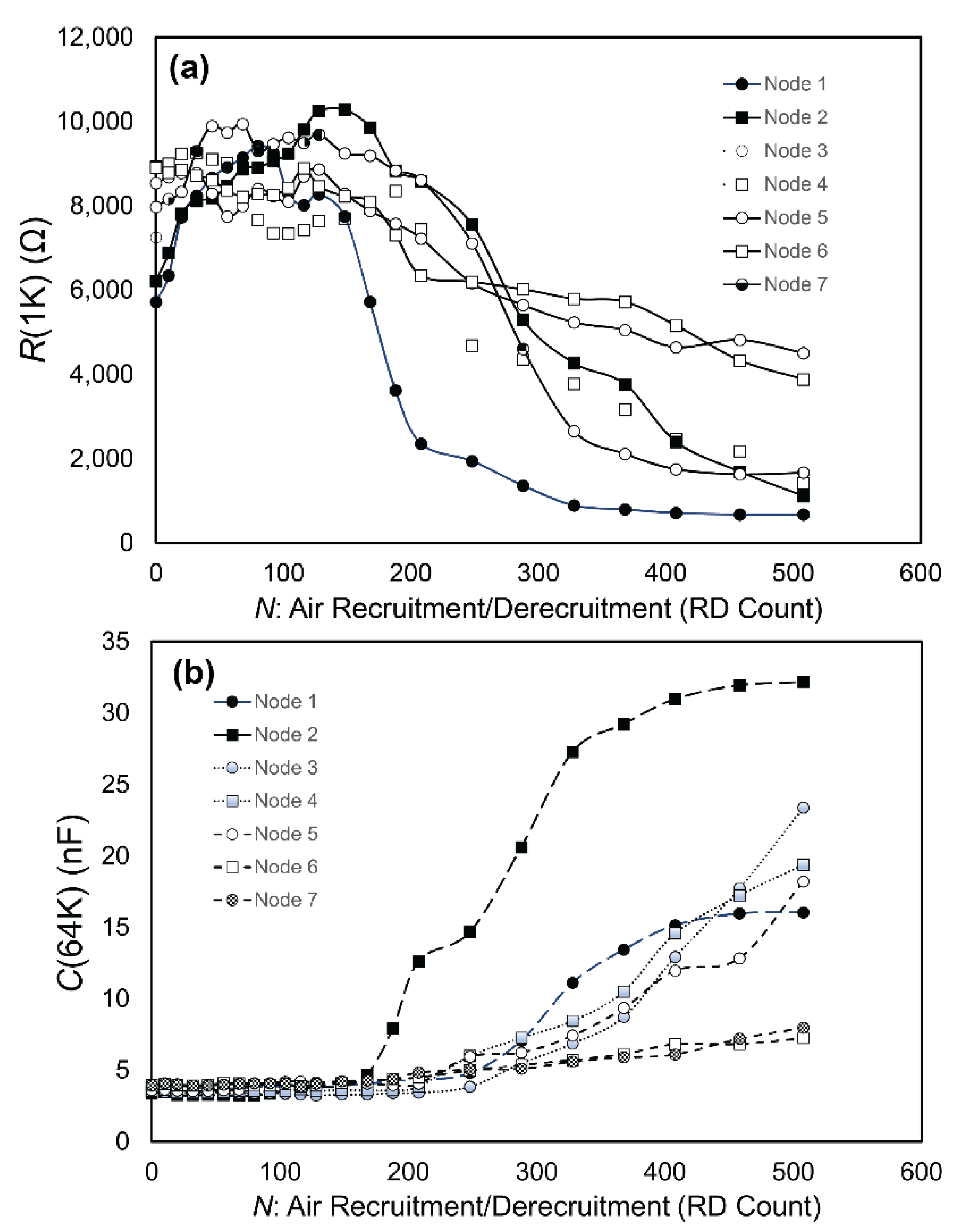
3. Results
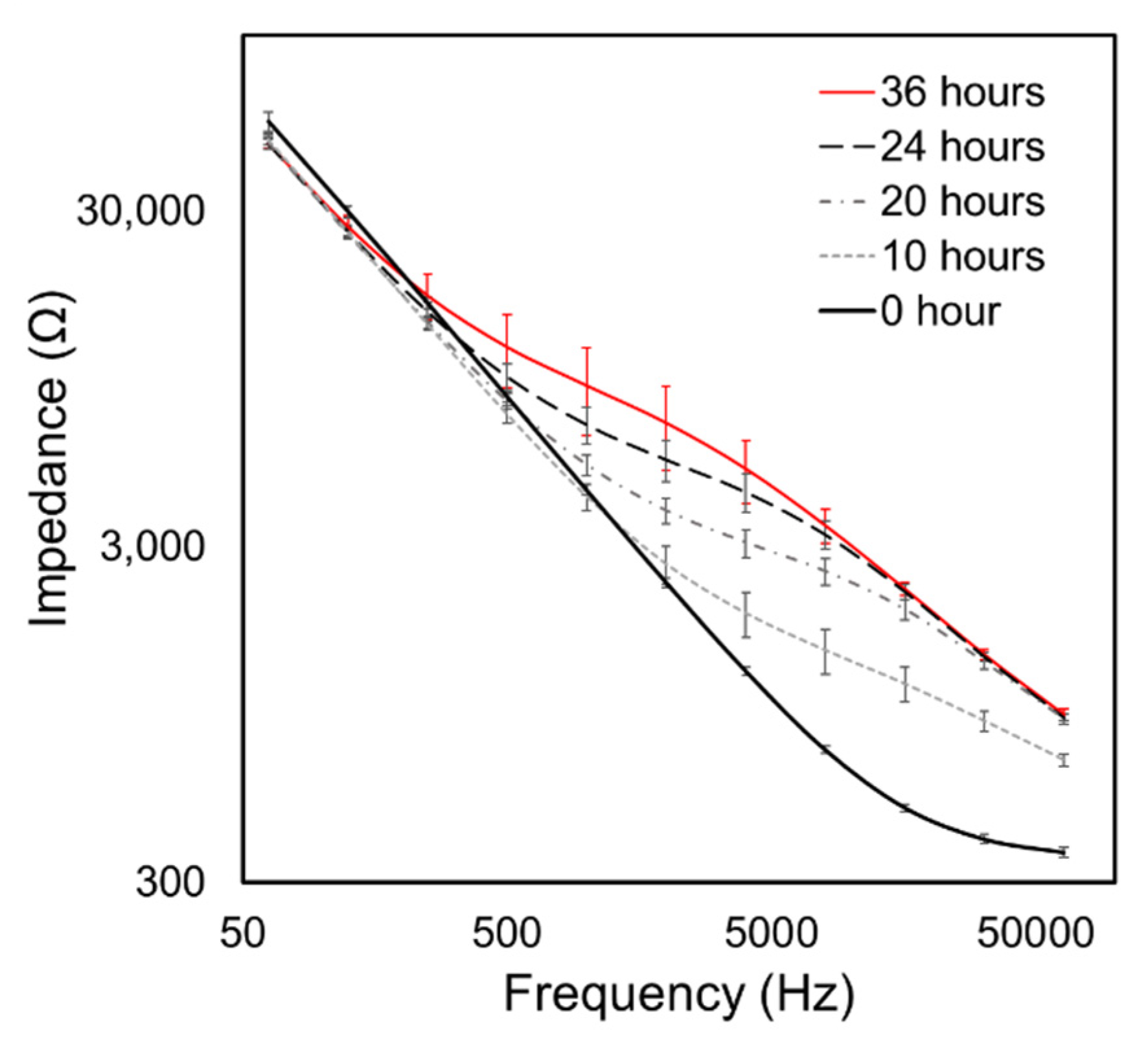
3.1. Experimental Data
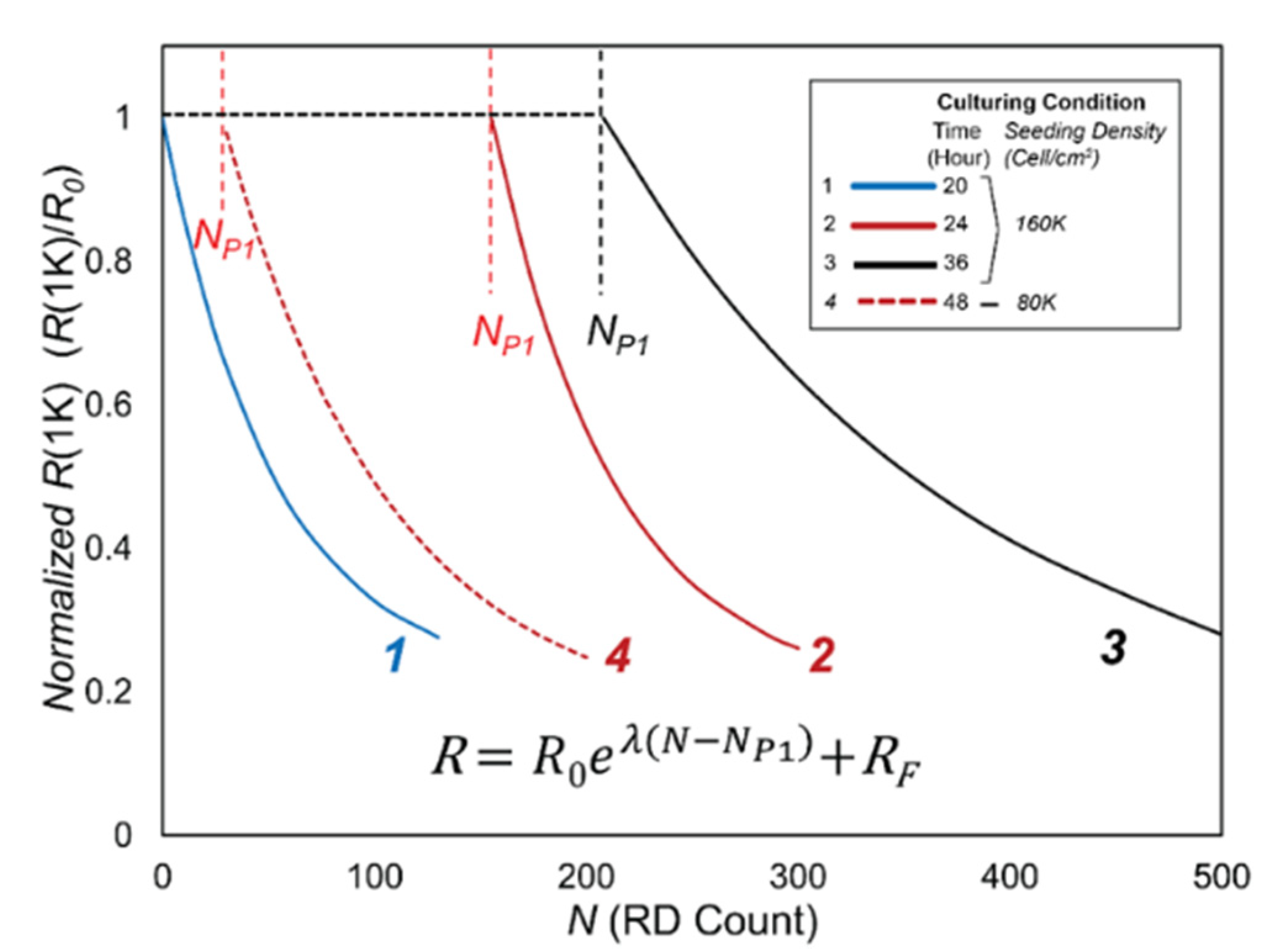
3.2. Data Analysis
- R0, the initial value of R(1K) prior to RD insults,
- NP1, the length of Phase 1, during which confluency is sustained as determined by C(64K),
- λ, the exponential decay constant of the monotonic reduction of R(1K) in Phase 2, and
- RF, the cell-free value of R(1K).
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviation
| Abbreviation | Meaning |
| R(1K) | Resistance at a 1 kHz input frequency. (Ω) |
| R0 | The initial value of R(1K) prior to RD insults. (Ω) |
| RF | The cell-free value of R(1K). (Ω) |
| C(64K) | Capacitance at input frequency at 64 kHz. (nF) |
| Rb | Barrier function (resistance between cells) defined by the Giaever–Keese model. (Ω·cm2) |
| RbInit | The initial Rb prior to RD insults. (Ω·cm2) |
| α | Current flow resistance parameter in the cell adhesion surface in the Giaever–Keese model. (Ω0.5·cm) |
| Cm | The cell membrane capacitance in the Giaever–Keese model. (µF/cm2) |
| RD | The Recruitment/Derecruitment of air–liquid interface. |
| N | Number of RD cycle. (N) |
| NP1 | The length of Phase 1 (N) |
| λ | The exponential decay constant of R(1K) in Phase 2. (N−1) |
References
- Rubenfeld, G.D.; Caldwell, E.; Peabody, E.; Weaver, J.R.R.T.; Martin, D.P.; Neff, M.; Stern, E.J.; Hudson, L.D. Incidence and Outcomes of Acute Lung Injury. N. Engl. J. Med. 2005, 353, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Nieman, G.F.; Satalin, J.; Andrews, P.; Aiash, H.; Habashi, N.M.; Gatto, L.A. Personalizing mechanical ventilation according to physiologic parameters to stabilize alveoli and minimize ventilator induced lung injury (VILI). Intensive Care Med. Exp. 2017, 5, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seah, A.S.; Grant, K.A.; Aliyeva, M.; Allen, G.B.; Bates, J.H. Quantifying the roles of tidal volume and PEEP in the pathogenesis of ventilator-induced lung injury. Ann. Biomed. Eng. 2011, 39, 1505–1516. [Google Scholar] [CrossRef] [PubMed]
- Moloney, E.D.; Griffiths, M.J.D. Protective ventilation of patients with acute respiratory distress syndrome. Br. J. Anaesth. 2004, 92, 261–270. [Google Scholar] [CrossRef] [Green Version]
- Bilek, A.M.; Dee, K.C.; Gaver, D.P., III. Mechanisms of surface-tension-induced epithelial cell damage in a model of pulmonary airway reopening. J. Appl. Physiol. 2003, 94, 770–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacob, A.-M.; Gaver, D.P. Atelectrauma disrupts pulmonary epithelial barrier integrity and alters the distribution of tight junction proteins ZO-1 and claudin 4. J. Appl. Physiol. 2012, 113, 1377–1387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaver, D.P.; Nieman, G.F.; Gatto, L.A.; Cereda, M.; Habashi, N.M.; Bates, J.H.T. The POOR Get POORer: A Hypothesis for the Pathogenesis of Ventilator-induced Lung Injury. Am. J. Respir. Crit. Care Med. 2020, 202, 1081–1087. [Google Scholar] [CrossRef]
- Salomon, J.J.; Muchitsch, V.E.; Gausterer, J.C.; Schwagerus, E.; Huwer, H.; Daum, N.; Lehr, C.-M.; Ehrhardt, C. The Cell Line NCl-H441 Is a Useful in Vitro Model for Transport Studies of Human Distal Lung Epithelial Barrier. Mol. Pharm. 2014, 11, 995–1006. [Google Scholar] [CrossRef]
- Hermanns, M.I.; Unger, R.E.; Kehe, K.; Peters, K.; Kirkpatrick, C.J. Lung epithelial cell lines in coculture with human pulmonary microvascular endothelial cells: Development of an alveolo-capillary barrier in vitro. Lab. Investig. 2004, 84, 736–752. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Birch, N.P.; Suresh, V. An Optimised Human Cell Culture Model for Alveolar Epithelial Transport. PLoS ONE 2016, 11, e0165225. [Google Scholar] [CrossRef]
- Weibel, E.R.; Gomez, D.M. Architecture of the Human Lung. Science 1962, 137, 577–585. [Google Scholar] [CrossRef] [PubMed]
- Giaever, I.; Keese, C.R. Micromotion of mammalian cells measured electrically. Proc. Natl. Acad. Sci. USA 1991, 88, 7896–7900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hucklesby, J.J.W.; Anchan, A.; O’Carroll, S.J.; Unsworth, C.P.; Graham, E.S.; Angel, C.E. Comparison of Leading Biosensor Technologies to Detect Changes in Human Endothelial Barrier Properties in Response to Pro-Inflammatory TNFα and IL1β in Real-Time. Biosensors 2021, 11, 159. [Google Scholar] [CrossRef] [PubMed]
- Gu, A.Y.; Kho, D.T.; Johnson, R.H.; Graham, E.S.; O’Carroll, S.J. In Vitro Wounding Models Using the Electric Cell-Substrate Impedance Sensing (ECIS)-Zθ Technology. Biosensors 2018, 8, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DePaola, N.; Phelps, J.E.; Florez, L.; Keese, C.R.; Minnear, F.L.; Giaever, I.; Vincent, P. Electrical impedance of cultured endothelium under fluid flow. Ann. Biomed. Eng. 2001, 29, 648–656. [Google Scholar] [CrossRef]
- Cavallini, F.; Tarantola, M. ECIS based wounding and reorganization of cardiomyocytes and fibroblasts in co-cultures. Prog. Biophys. Mol. Biol. 2019, 144, 116–127. [Google Scholar] [CrossRef]
- Stolwijk, J.A.; Matrougui, K.; Renken, C.W.; Trebak, M. Impedance analysis of GPCR-mediated changes in endothelial barrier function: Overview and fundamental considerations for stable and reproducible measurements. Pflug. Arch. 2015, 467, 2193–2218. [Google Scholar] [CrossRef] [Green Version]
- Szulcek, R.; Bogaard, H.J.; van Nieuw Amerongen, G.P. Electric cell-substrate impedance sensing for the quantification of endothelial proliferation, barrier function, and motility. J. Vis. Exp. 2014, 85, e51300. [Google Scholar] [CrossRef] [Green Version]
- Anchan, A.; Kalogirou-Baldwin, P.; Johnson, R.; Kho, D.T.; Joseph, W.; Hucklesby, J.; Finlay, G.J.; O’Carroll, S.J.; Angel, C.E.; Graham, E.S. Real-Time Measurement of Melanoma Cell-Mediated Human Brain Endothelial Barrier Disruption Using Electric Cell-Substrate Impedance Sensing Technology. Biosensors 2019, 9, 56. [Google Scholar] [CrossRef] [Green Version]
- Kho, D.T.; Johnson, R.H.; O’Carroll, S.J.; Angel, C.E.; Graham, E.S. Biosensor Technology Reveals the Disruption of the Endothelial Barrier Function and the Subsequent Death of Blood Brain Barrier Endothelial Cells to Sodium Azide and Its Gaseous Products. Biosensors 2017, 7, 41. [Google Scholar] [CrossRef] [Green Version]
- Robilliard, L.D.; Yu, J.; Jun, S.M.; Anchan, A.; Finlay, G.; Angel, C.E.; Graham, E.S. Can ECIS Biosensor Technology Be Used to Measure the Cellular Responses of Glioblastoma Stem Cells? Biosensors 2021, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Robilliard, L.D.; Kho, D.T.; Johnson, R.H.; Anchan, A.; O’Carroll, S.J.; Graham, E.S. The Importance of Multifrequency Impedance Sensing of Endothelial Barrier Formation Using ECIS Technology for the Generation of a Strong and Durable Paracellular Barrier. Biosensors 2018, 8, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, K.; Cramer, S.; Galla, H.-J. Impedance-based cell monitoring: Barrier properties and beyond. Fluids Barriers CNS 2013, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lo, C.M.; Keese, C.R.; Giaever, I. Cell–Substrate Contact: Another Factor May Influence Transepithelial Electrical Resistance of Cell Layers Cultured on Permeable Filters. Exp. Cell Res. 1999, 250, 576–580. [Google Scholar] [CrossRef]
- Kataoka, N.; Iwaki, K.; Hashimoto, K.; Mochizuki, S.; Ogasawara, Y.; Sato, M.; Tsujioka, K.; Kajiya, F. Measurements of endothelial cell-to-cell and cell-to-substrate gaps and micromechanical properties of endothelial cells during monocyte adhesion. Proc. Natl. Acad. Sci. USA 2002, 99, 15638–15643. [Google Scholar] [CrossRef] [Green Version]
- Jacob, A.-M.; Gaver, D.P., III. An investigation of the influence of cell topography on epithelial mechanical stresses during pulmonary airway reopening. Phys. Fluids 2005, 17, 031502. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, E.; Giannetti, M.J.; Van Houten, M.J.; Forouzan, O.; Shevkoplyas, S.S.; Gaver, D.P. The unusual symmetric reopening effect induced by pulmonary surfactant. J. Appl. Physiol. 2014, 116, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Gattinoni, L.; Quintel, M.; Marini, J.J. Volutrauma and atelectrauma: Which is worse? Crit. Care 2018, 22, 264. [Google Scholar] [CrossRef] [Green Version]
- Modrykamien, A.M.; Gupta, P. The acute respiratory distress syndrome. Bayl. Univ. Med Cent. Proc. 2015, 28, 163–171. [Google Scholar] [CrossRef]
- Bates, J.H.T.; Gaver, D.P.; Habashi, N.M.; Nieman, G.F. Atelectrauma Versus Volutrauma: A Tale of Two Time-Constants. Crit. Care Explor. 2020, 2, e0299. [Google Scholar] [CrossRef]
- Cressoni, M.; Cadringher, P.; Chiurazzi, C.; Amini, M.; Gallazzi, E.; Marino, A.; Brioni, M.; Carlesso, E.; Chiumello, D.; Quintel, M.; et al. Lung inhomogeneity in patients with acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2014, 189, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Gelsinger, M.L.; Tupper, L.L.; Matteson, D.S. Cell Line Classification Using Electric Cell-Substrate Impedance Sensing (ECIS). Int. J. Biostat. 2020, 16, 20180083. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bagnaninchi, P.O.; Drummond, N. Real-time label-free monitoring of adipose-derived stem cell differentiation with electric cell-substrate impedance sensing. Proc. Natl. Acad. Sci. USA 2011, 108, 6462–6467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matthews, B.D.; Overby, D.R.; Mannix, R.; Ingber, D.E. Cellular adaptation to mechanical stress: Role of integrins, Rho, cytoskeletal tension and mechanosensitive ion channels. J. Cell Sci. 2006, 119, 508–518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nestor-Bergmann, A.; Stooke-Vaughan, G.A.; Goddard, G.K.; Starborg, T.; Jensen, O.E.; Woolner, S. Decoupling the Roles of Cell Shape and Mechanical Stress in Orienting and Cueing Epithelial Mitosis. Cell Rep. 2019, 26, 2088–2100.e4. [Google Scholar] [CrossRef] [Green Version]
- Crosby, L.M.; Waters, C.M. Epithelial repair mechanisms in the lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 298, 715–731. [Google Scholar] [CrossRef] [Green Version]
- White, S.R.; Tse, R.; Marroquin, B.A. Stress-activated protein kinases mediate cell migration in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2005, 32, 301–310. [Google Scholar] [CrossRef]
- Marini, J.J.; Gattinoni, L. Time Course of Evolving Ventilator-Induced Lung Injury: The “Shrinking Baby Lung”. Crit. Care Med. 2020, 48, 1203–1209. [Google Scholar] [CrossRef]
- Gaver, D.P., III; Samsel, R.W.; Solway, J. Effects of surface tension and viscosity on airway reopening. J. Appl. Physiol. 1990, 69, 74–85. [Google Scholar] [CrossRef]
- Kay, S.S.; Bilek, A.M.; Dee, K.C.; Gaver, D.P., III. Pressure gradient, not exposure duration, determines the extent of epithelial cell damage in a model of pulmonary reopening. J. Appl. Physiol. 2004, 97, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Novak, C.; Ballinger, M.N.; Ghadiali, S. Mechanobiology of Pulmonary Diseases: A Review of Engineering Tools to Understand Lung Mechanotransduction. J. Biomech. Eng. 2021, 143, 110801. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Fujioka, H.; Tung, Y.-C.; Futai, N.; Paine, R., III; Grotberg, J.B. Acoustically detectable cellular-level lung injury induced by fluid mechanical stress in microfluidic airway systems. Proc. Natl. Acad. Sci. USA 2007, 104, 18886–18891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wray, C.; Mao, Y.; Pan, J.; Chandrasena, A.; Piasta, F.; Frank, J.A. Claudin-4 augments alveolar epithelial barrier function and is induced in acute lung injury. Am. J. Physiol. Lung Cell. Mol. Physiol. 2009, 297, L219–L227. [Google Scholar] [CrossRef] [PubMed] [Green Version]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
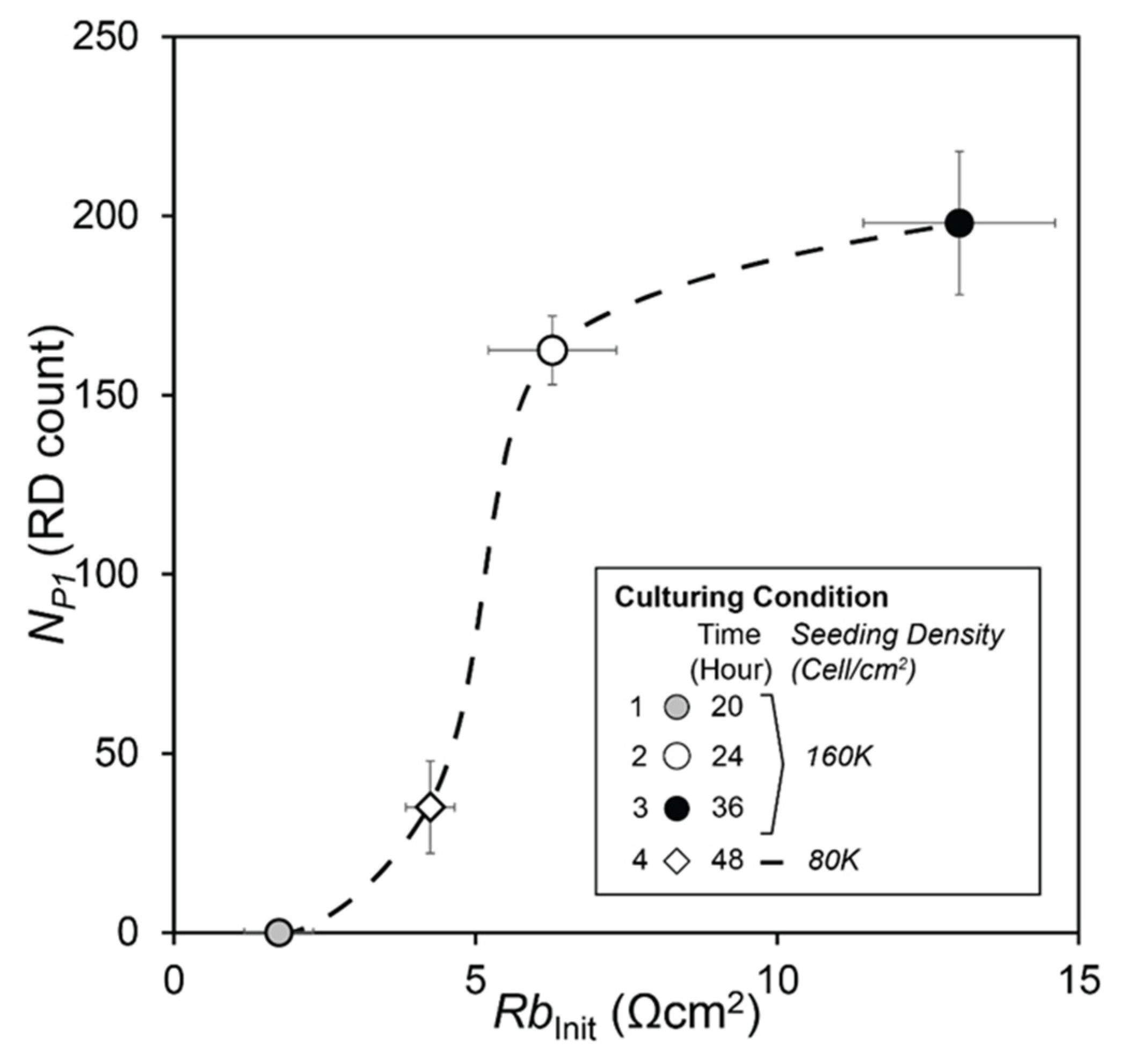
| (a) | Case | S.Density | C.Time | NP1 | λ | |||
|---|---|---|---|---|---|---|---|---|
| (Cell/cm2) | (hour) | (N) | std | (N −1) | std | |||
| 1 | 160K | 20 | 0 | 0 | −0.0192 | 0.0154 | ||
| 2 | 160K | 24 | 143 | 9.6 | −0.0173 | 0.0120 | ||
| 3 | 160K | 36 | 198 | 20 | −0.0059 | 0.0051 | ||
| 4 | 80K | 48 | 35 | 12.9 | −0.0127 | 0.0054 | ||
| (b) | NP1 | λ | ||||||
| Case | 1 | 2 | 3 | Case | 1 | 2 | 3 | |
| 2 | 1.66 × 10−5 | 2 | 1.42 × 10−1 | |||||
| 3 | 7.78 × 10−6 | 5.62 × 10−3 | 3 | 1.95 × 10−1 | 2.44 × 10−2 | |||
| 4 | 6.18 × 10−4 | 1.15× 10−4 | 1.18 × 10−4 | 4 | 7.88 × 10−2 | 1.31 × 10−2 | 4.40 × 10−1 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yamaguchi, E.; Yao, J.; Aymond, A.; Chrisey, D.B.; Nieman, G.F.; Bates, J.H.T.; Gaver, D.P. Electric Cell-Substrate Impedance Sensing (ECIS) as a Platform for Evaluating Barrier-Function Susceptibility and Damage from Pulmonary Atelectrauma. Biosensors 2022, 12, 390. https://doi.org/10.3390/bios12060390
Yamaguchi E, Yao J, Aymond A, Chrisey DB, Nieman GF, Bates JHT, Gaver DP. Electric Cell-Substrate Impedance Sensing (ECIS) as a Platform for Evaluating Barrier-Function Susceptibility and Damage from Pulmonary Atelectrauma. Biosensors. 2022; 12(6):390. https://doi.org/10.3390/bios12060390
Chicago/Turabian StyleYamaguchi, Eiichiro, Joshua Yao, Allison Aymond, Douglas B. Chrisey, Gary F. Nieman, Jason H. T. Bates, and Donald P. Gaver. 2022. "Electric Cell-Substrate Impedance Sensing (ECIS) as a Platform for Evaluating Barrier-Function Susceptibility and Damage from Pulmonary Atelectrauma" Biosensors 12, no. 6: 390. https://doi.org/10.3390/bios12060390
APA StyleYamaguchi, E., Yao, J., Aymond, A., Chrisey, D. B., Nieman, G. F., Bates, J. H. T., & Gaver, D. P. (2022). Electric Cell-Substrate Impedance Sensing (ECIS) as a Platform for Evaluating Barrier-Function Susceptibility and Damage from Pulmonary Atelectrauma. Biosensors, 12(6), 390. https://doi.org/10.3390/bios12060390