Label-Free Electrochemical Aptasensor for the Detection of the 3-O-C12-HSL Quorum-Sensing Molecule in Pseudomonas aeruginosa

 ,
, 
 ,
,  ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Instruments
2.3. Methods
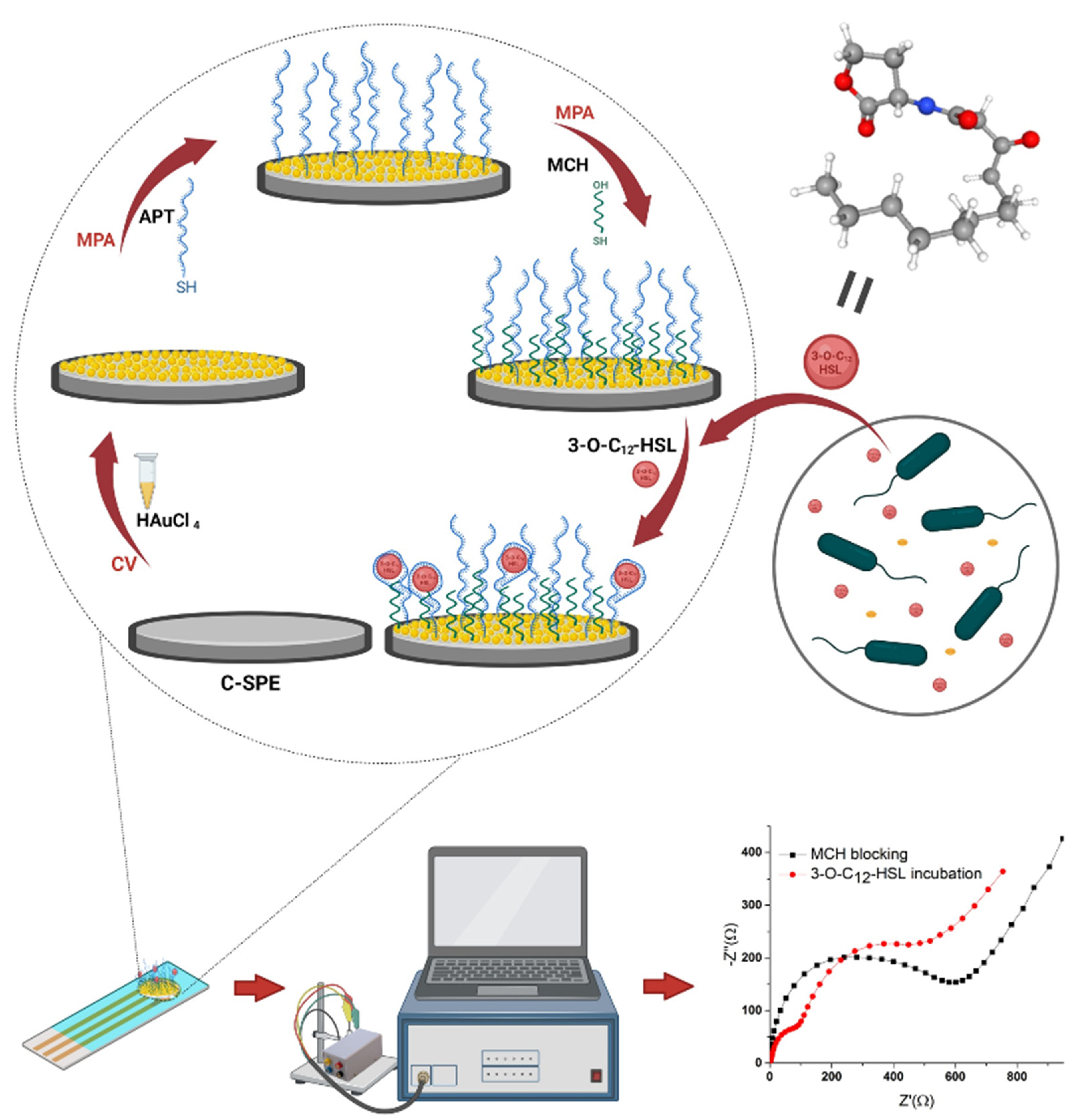
2.3.1. Aptasensor Development
2.3.2. Electrochemical Methods
2.3.3. Analysis Procedure/Quantification Method
2.3.4. SPR Analysis
2.3.5. Interference Studies
2.3.6. Analysis of Real Samples
2.3.7. Estimation of the Number of Microorganisms
- n = the number of colonies developed on a Petri dish;
- d = the dilution from which the respective plate was inoculated;
- N = the number of Petri dishes considered; only plates in which the dilution ratio was observed were considered.
3. Results and Discussion
3.1. Elaboration and Optimization of the Aptasensor
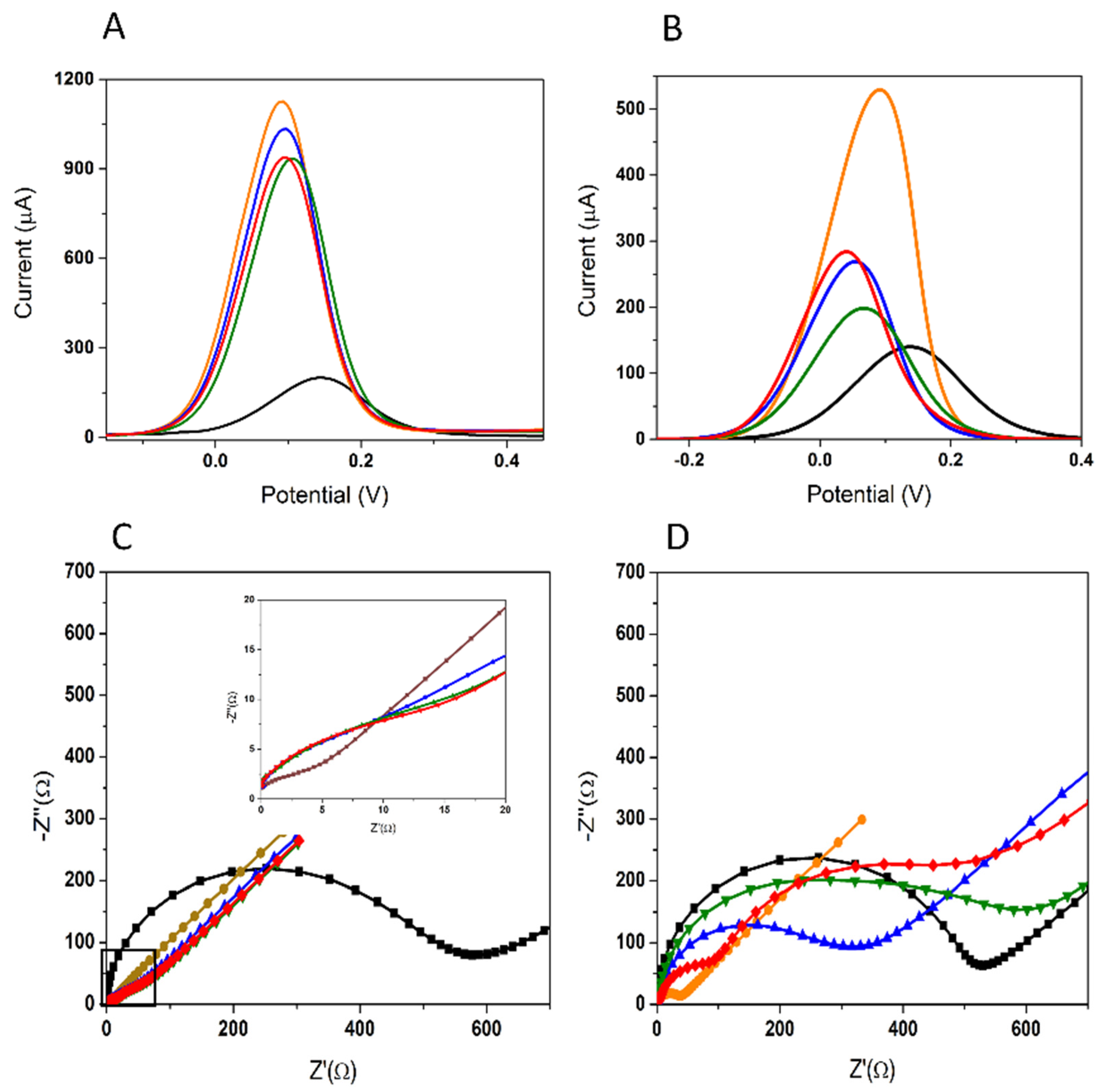
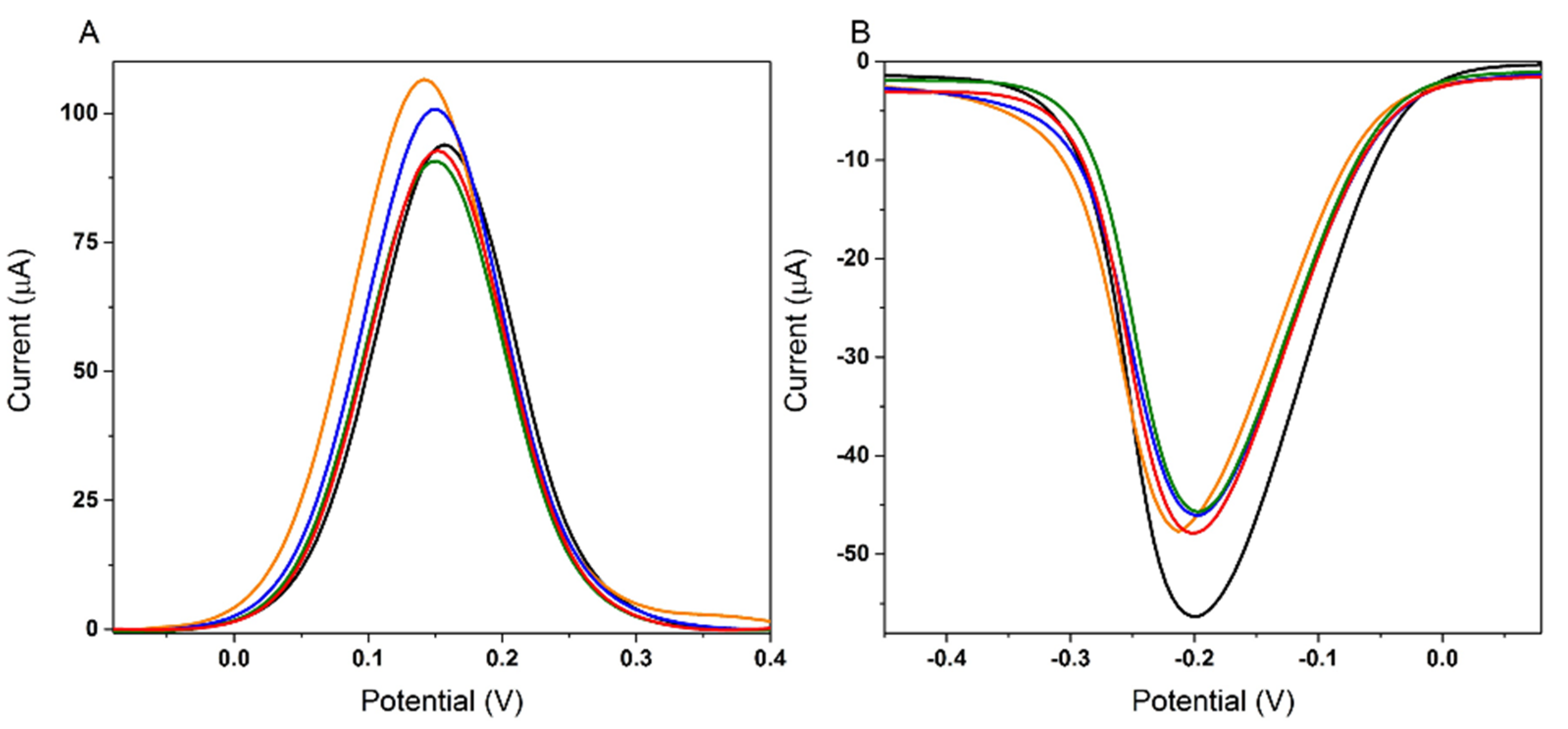
3.1.1. Optimization of the AuNPs Platform
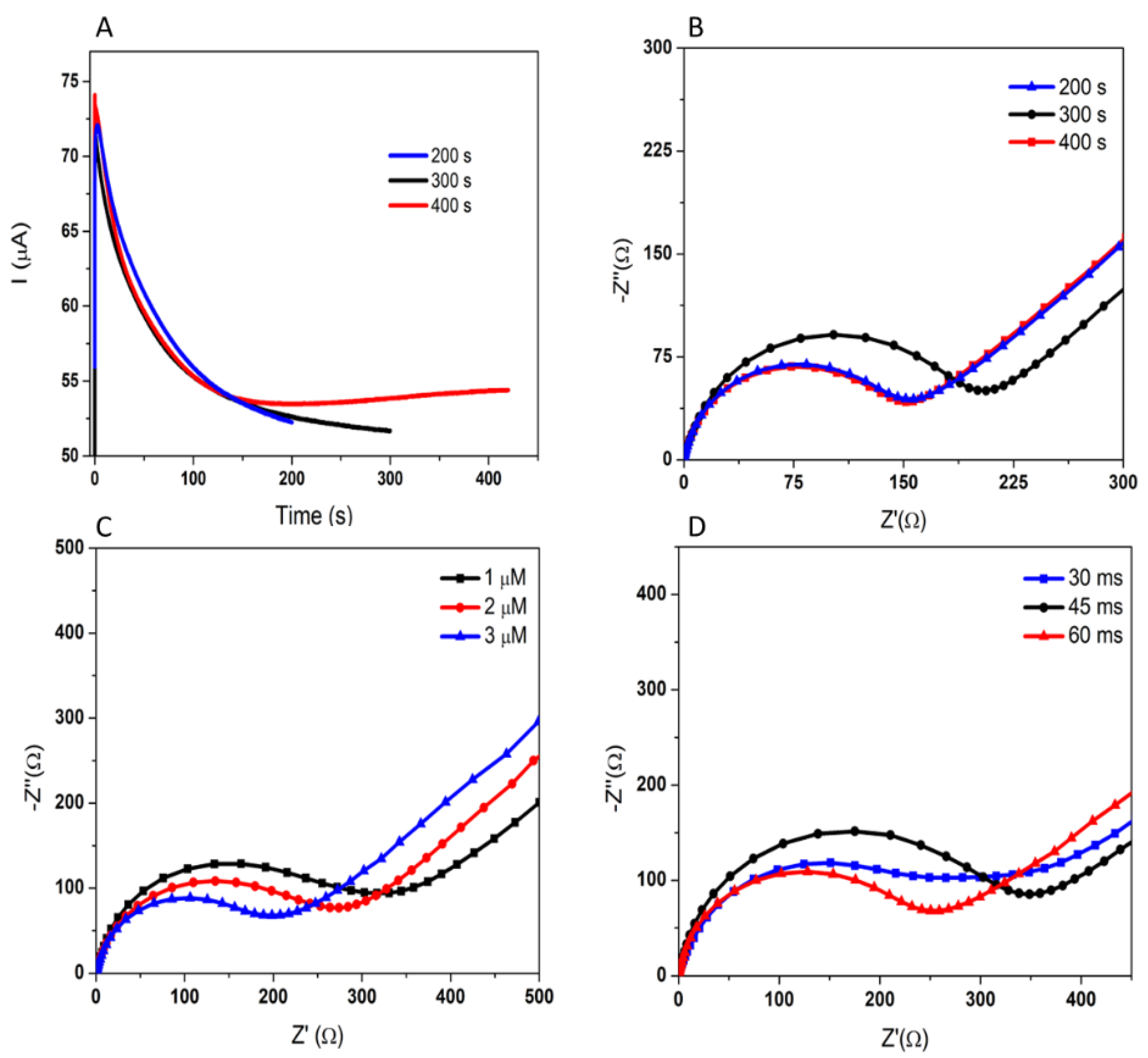
3.1.2. Optimization of the Aptamer Immobilization
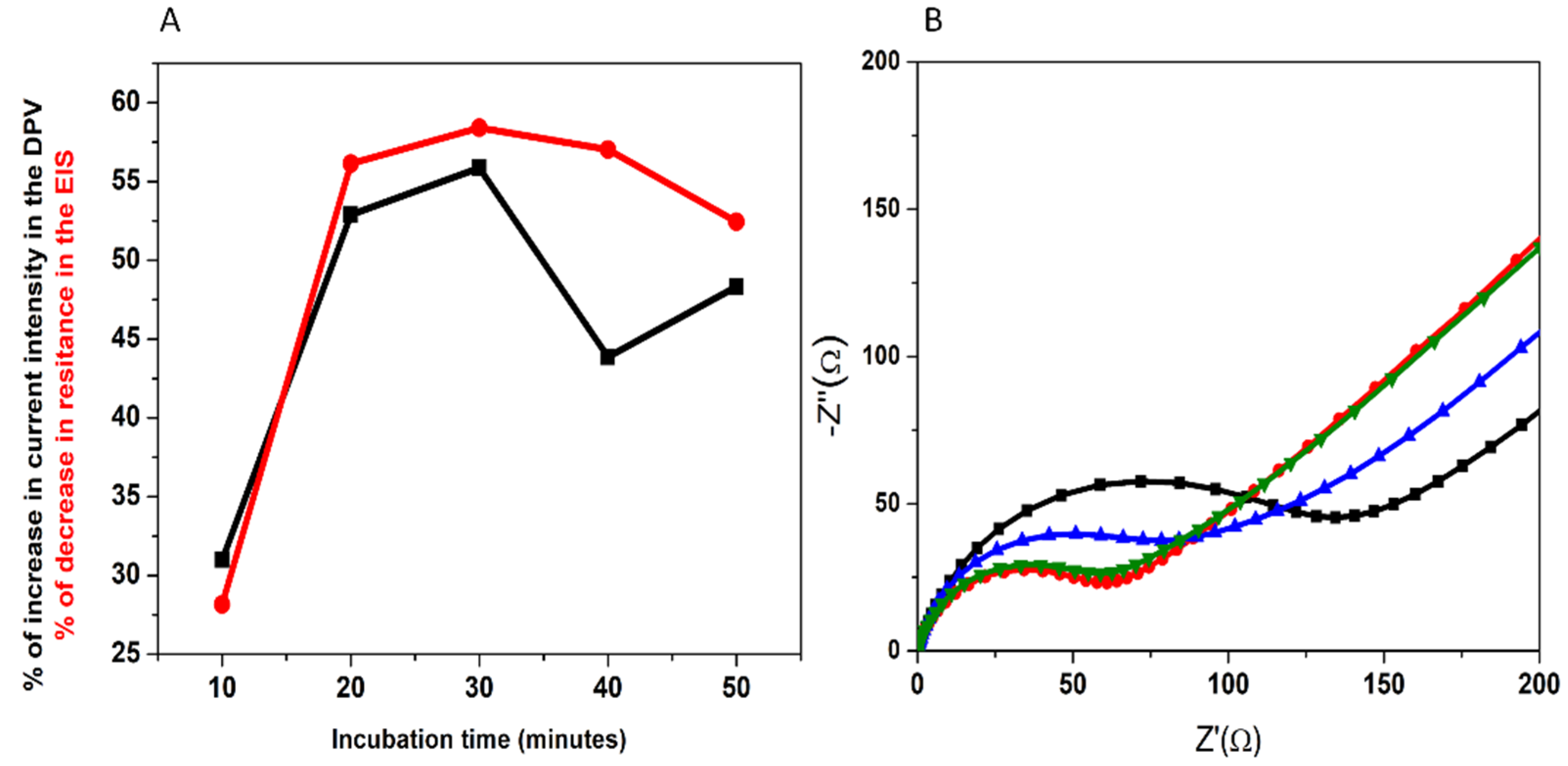
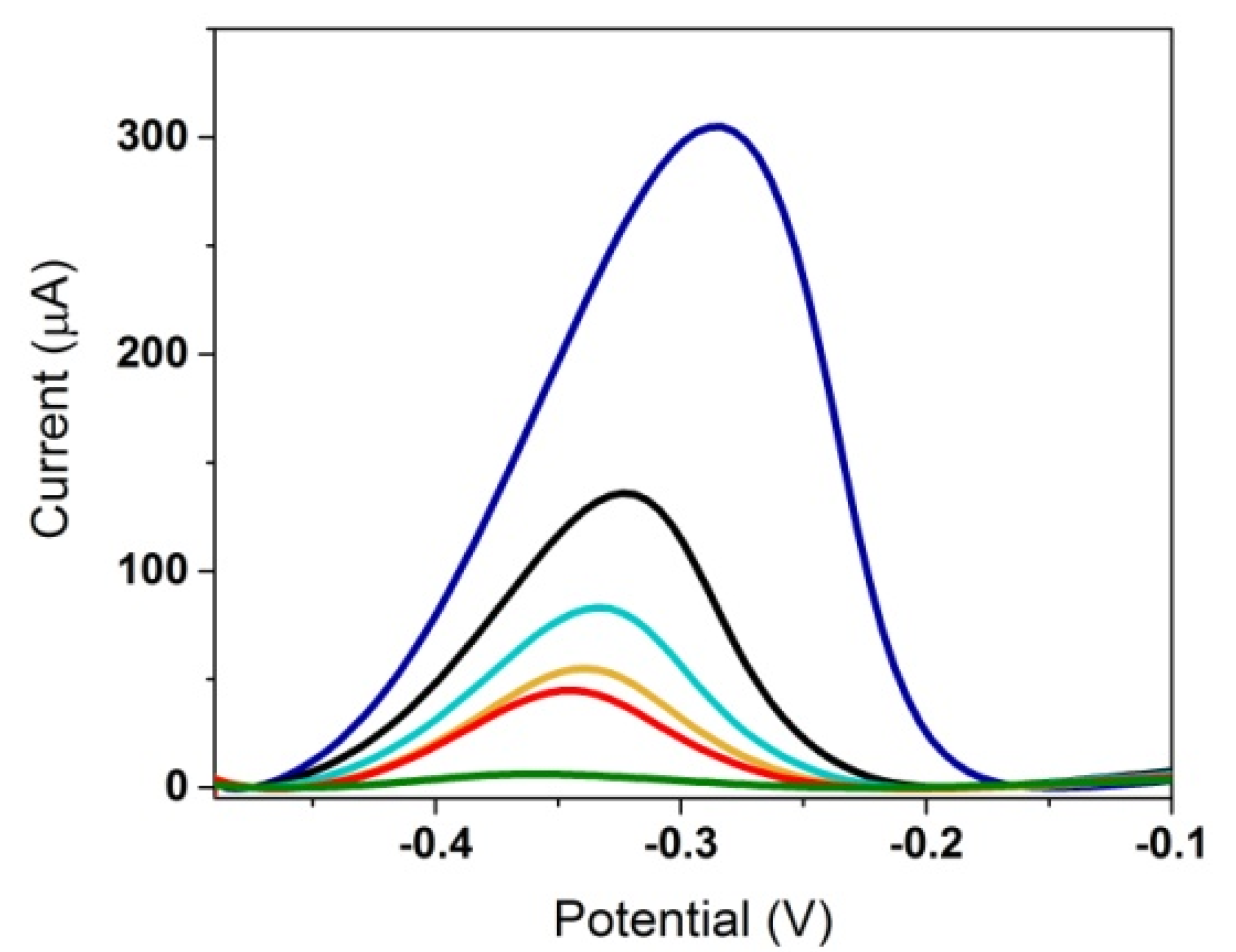
3.1.3. Optimization of the Incubation with 3-O-C12-HSL
3.2. Aptasensor Characterization
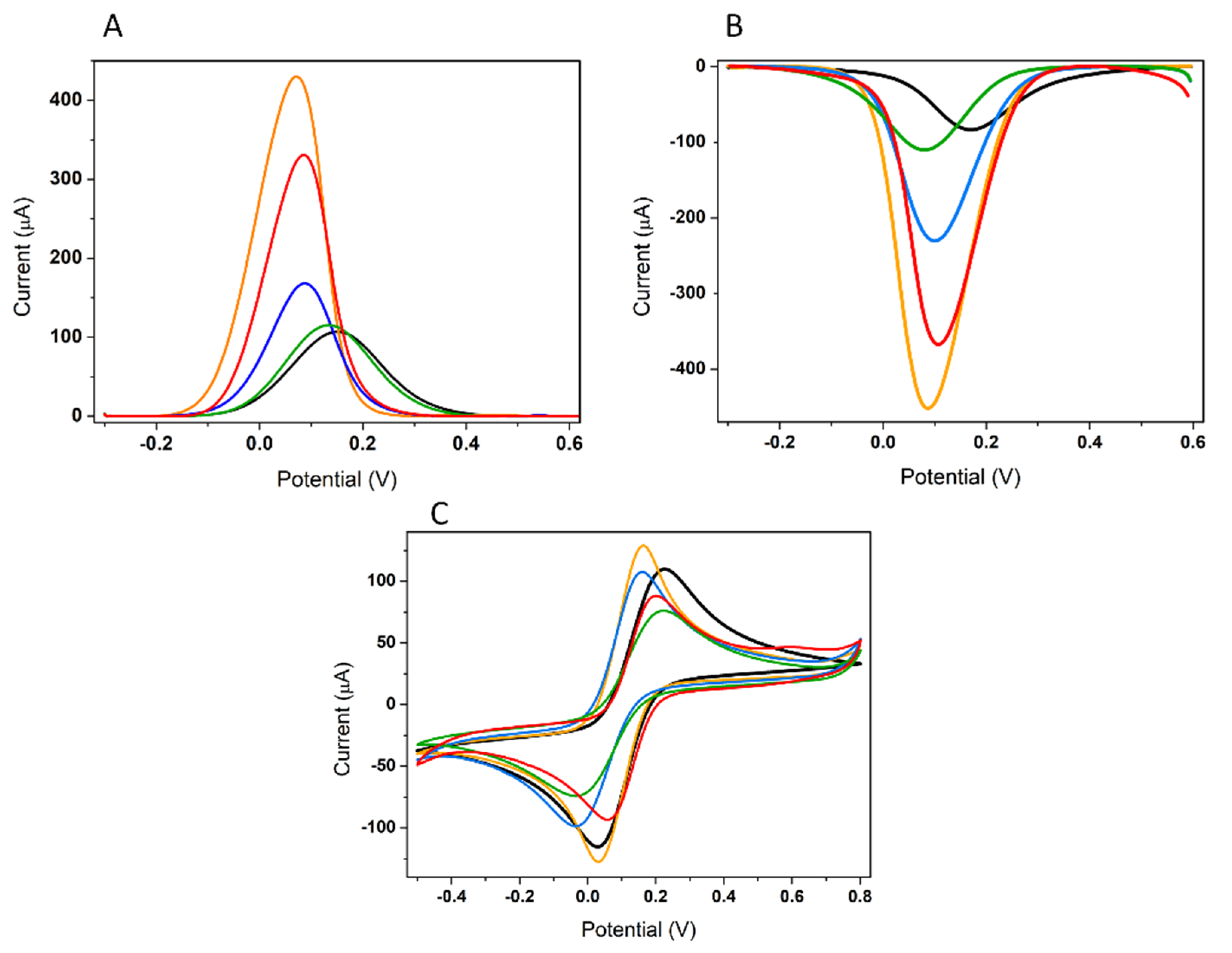
3.2.1. Electrochemical Characterization
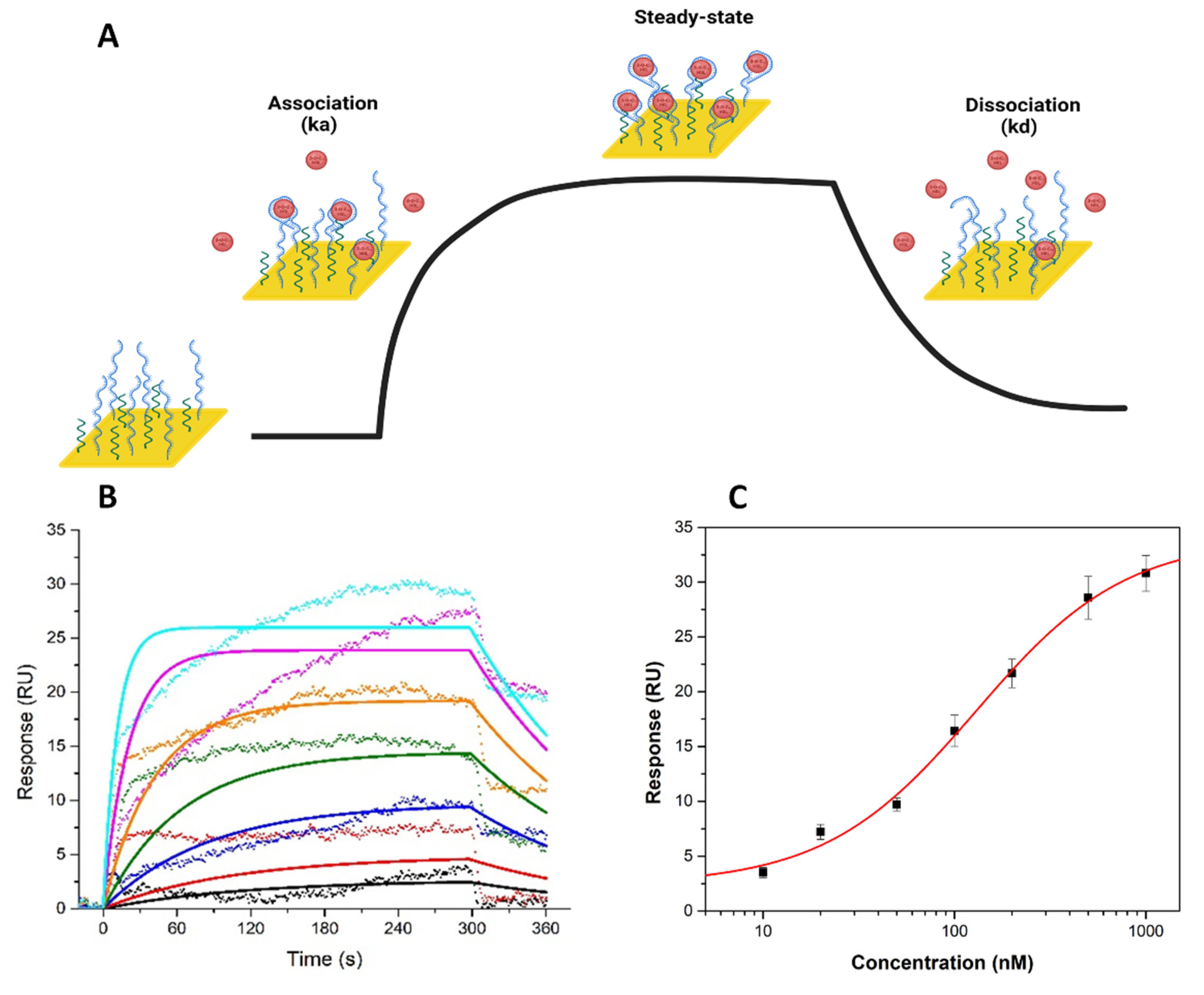
3.2.2. SPR Analysis
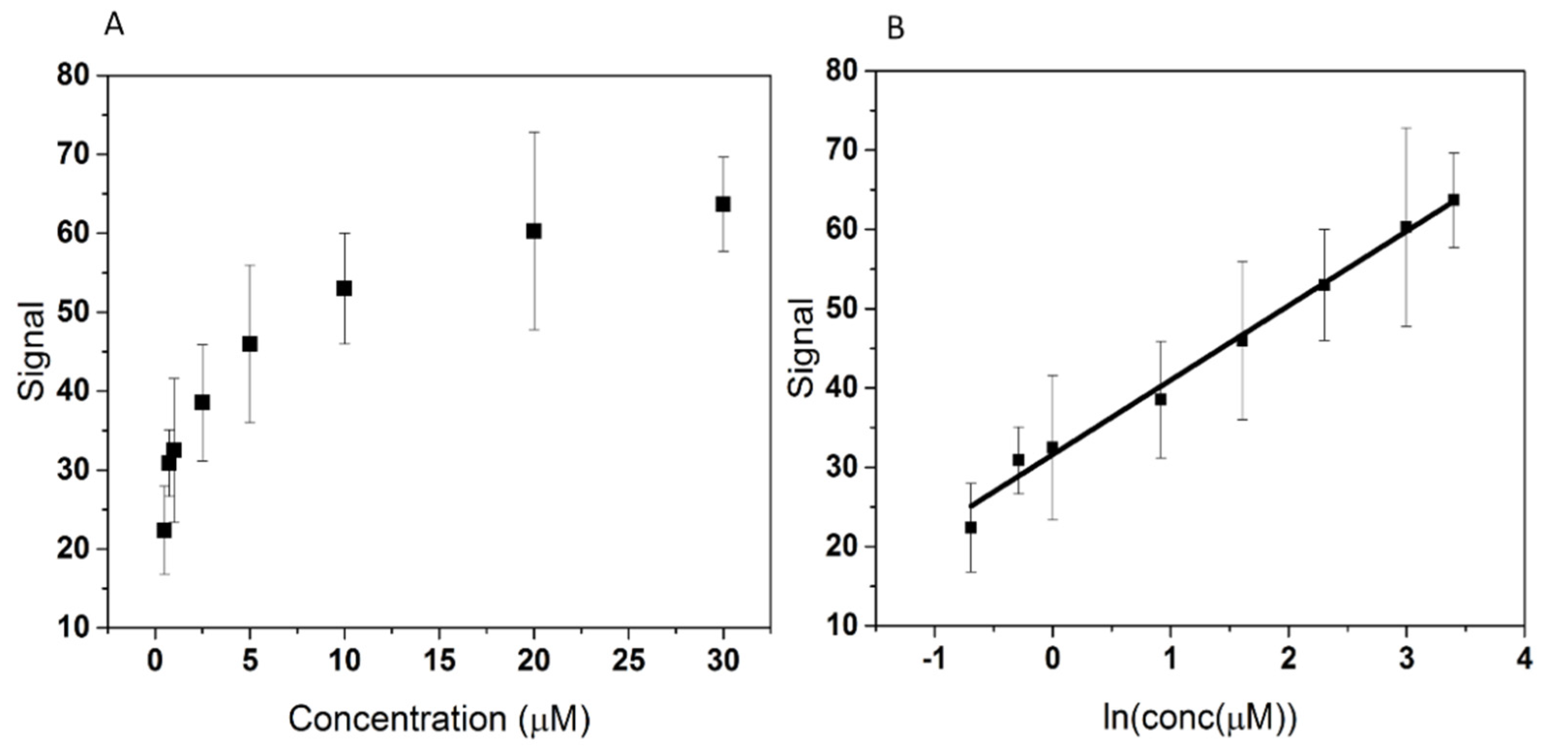
3.3. Calibration Curve and Limit of Detection
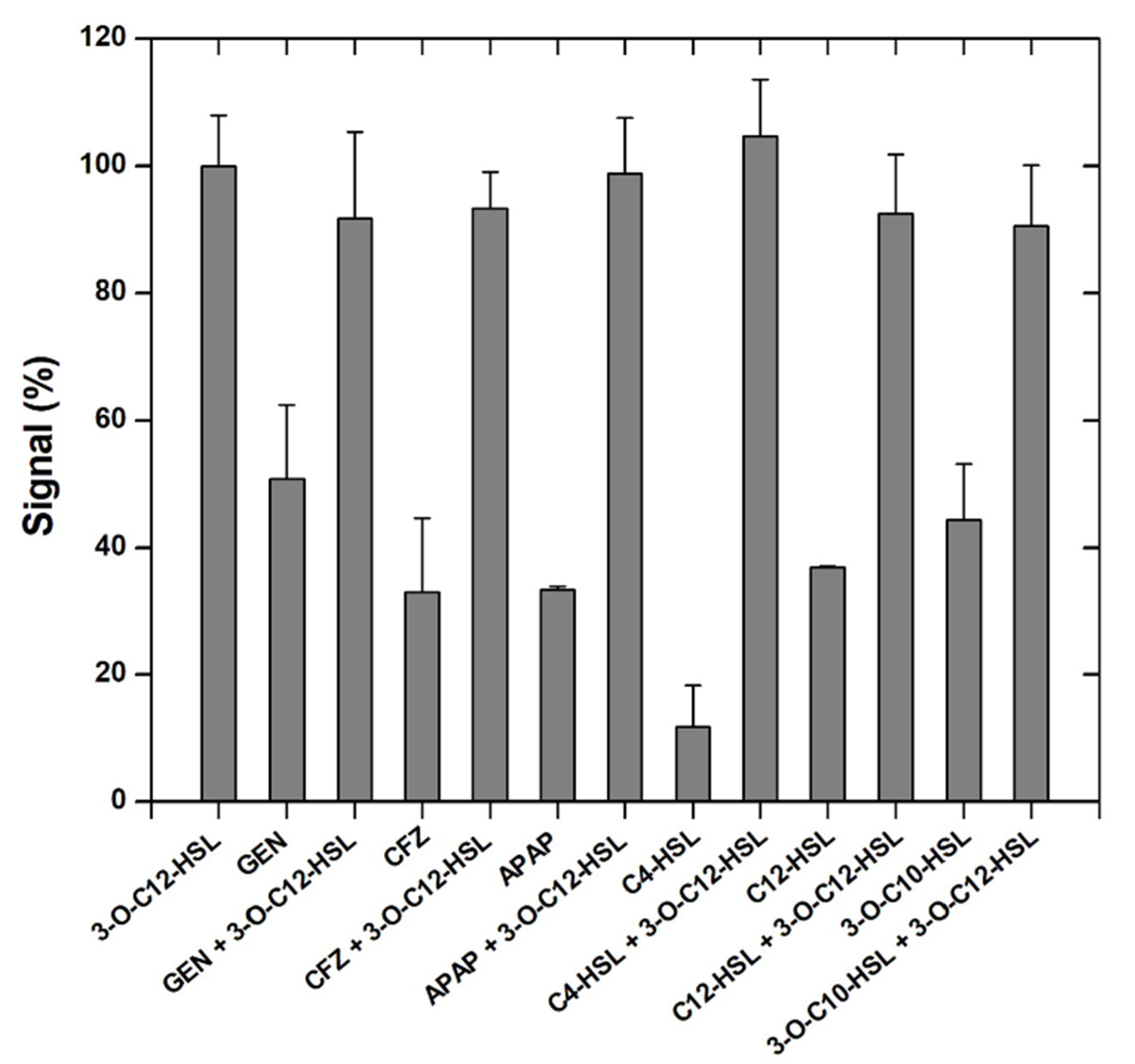
3.4. Interference Study
3.5. Analysis of Real Samples
3.5.1. Urine and Culture Media
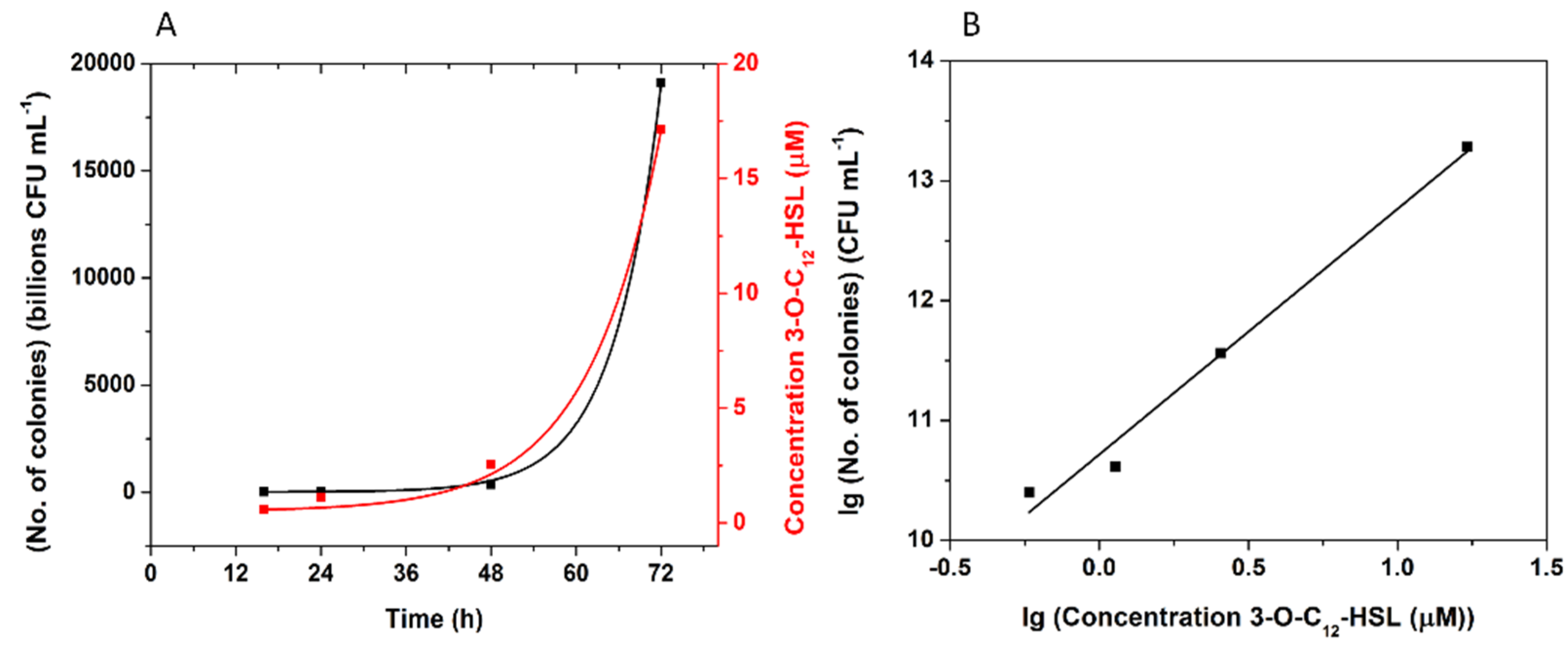
3.5.2. P. aeruginosa Cultures Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Reynolds, D.; Kollef, M. The Epidemiology and Pathogenesis and Treatment of Pseudomonas Aeruginosa Infections: An Update. Drugs 2021, 81, 2117–2131. [Google Scholar] [CrossRef]
- Sindeldecker, D.; Stoodley, P. The Many Antibiotic Resistance and Tolerance Strategies of Pseudomonas Aeruginosa. Biofilm 2021, 3, 100056. [Google Scholar] [CrossRef]
- Saeki, E.K.; Kobayashi, R.K.T.; Nakazato, G. Quorum Sensing System: Target to Control the Spread of Bacterial Infections. Microb. Pathog. 2020, 142, 104068. [Google Scholar] [CrossRef]
- Capatina, D.; Feier, B.; Hosu, O.; Tertis, M.; Cristea, C. Analytical Methods for the Characterization and Diagnosis of Infection with Pseudomonas Aeruginosa: A Critical Review. Anal. Chim. Acta 2022, 1204, 339696. [Google Scholar] [CrossRef]
- Moradali, M.F.; Ghods, S.; Rehm, B.H.A. Pseudomonas Aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence. Front. Cell. Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef] [Green Version]
- Miller, C.; Gilmore, J. Detection of Quorum-Sensing Molecules for Pathogenic Molecules Using Cell-Based and Cell-Free Biosensors. Antibiotics 2020, 9, 259. [Google Scholar] [CrossRef]
- Abdalla, M.Y.; Hoke, T.; Seravalli, J.; Switzer, B.L.; Bavitz, M.; Fliege, J.D.; Murphy, P.J.; Britigan, B.E. Pseudomonas Quinolone Signal Induces Oxidative Stress and Inhibits Heme Oxygenase-1 Expression in Lung Epithelial Cells. Infect. Immun. 2017, 85, e00176-17. [Google Scholar] [CrossRef] [Green Version]
- Barr, H.L.; Halliday, N.; Barrett, D.A.; Williams, P.; Forrester, D.L.; Peckham, D.; Williams, K.; Smyth, A.R.; Honeybourne, D.; Whitehouse, J.L.; et al. Diagnostic and Prognostic Significance of Systemic Alkyl Quinolones for P. Aeruginosa in Cystic Fibrosis: A Longitudinal Study. J. Cyst. Fibros. 2017, 16, 230–238. [Google Scholar] [CrossRef] [Green Version]
- Das, S.; Sarkar, H.S.; Uddin, M.R.; Mandal, S.; Sahoo, P. A Chemosensor to Recognize N-Acyl Homoserine Lactone in Bacterial Biofilm. Sens. Actuators B Chem. 2018, 259, 332–338. [Google Scholar] [CrossRef]
- Montagut, E.J.; Vilaplana, L.; Martin-Gomez, M.T.; Marco, M.P. High-Throughput Immunochemical Method to Assess the 2-Heptyl-4-Quinolone Quorum Sensing Molecule as a Potential Biomarker of Pseudomonas Aeruginosa Infections. ACS Infect. Dis. 2020, 6, 3237–3246. [Google Scholar] [CrossRef]
- Patel, N.M.; Moore, J.D.; Blackwell, H.E.; Amador-Noguez, D. Identification of Unanticipatedand Novel N-Acyl L-Homoserine Lactones (AHLs) Using a Sensitive Non-Targeted LC-MS/MS Method. PLoS ONE 2016, 11, e0163469. [Google Scholar] [CrossRef] [PubMed]
- Lepine, F.; Milot, S.; Groleau, M.-C.; Deziel, E. Liquid Chromatography/Mass Spectrometry (LC/MS) for the Detection and Quantification of N-Acyl-L-Homoserine Lactones (AHLs) and 4-Hydroxy-2-Alkylquinolines (HAQs). In Quorum Sensing. Methods in Molecular Biology; Leoni, L., Rampion, G., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1673, pp. 49–59. ISBN 9781493973095. [Google Scholar]
- Yang, Y.; Zhou, M.; Hardwidge, P.R.; Cui, H.; Zhu, G. Isolation and Characterization of N-Acyl Homoserine Lactone-Producing Bacteria from Cattle Rumen and Swine Intestines. Front. Cell. Infect. Microbiol. 2018, 8, 155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montagut, E.J.; Martin-Gomez, M.T.; Marco, M.P. An Immunochemical Approach to Quantify and Assess the Potential Value of the Pseudomonas Quinolone Signal as a Biomarker of Infection. Anal. Chem. 2021, 93, 4859–4866. [Google Scholar] [CrossRef] [PubMed]
- Rothballer, M.; Uhl, J.; Kunze, J.; Schmitt-Kopplin, P.; Hartmann, A. Detection of the Bacterial Quorum-Sensing Signaling Molecules N-Acyl-Homoserine Lactones (HSL) and N-Acyl-Homoserine (HS) with an Enzyme-Linked Immunosorbent Assay (ELISA) and via Ultrahigh-Performance Liquid Chromatography Coupled to Mass Spectrometry (UHPLC-MS). Methods Mol. Biol. 2018, 1673, 61–72. [Google Scholar] [CrossRef]
- Massai, F.; Imperi, F.; Quattrucci, S.; Zennaro, E.; Visca, P.; Leoni, L. A Multitask Biosensor for Micro-Volumetric Detection of N-3-Oxo-Dodecanoyl-Homoserine Lactone Quorum Sensing Signal. Biosens. Bioelectron. 2011, 26, 3444–3449. [Google Scholar] [CrossRef]
- O’Connor, G.; Knecht, L.D.; Salgado, N.; Strobel, S.; Pasini, P.; Daunert, S. Whole-Cell Biosensors as Tools for the Detection of Quorum-Sensing Molecules: Uses in Diagnostics and the Investigation of the Quorum-Sensing Mechanism. Adv. Biochem. Eng. Biotechnol. 2016, 154, 181–200. [Google Scholar] [CrossRef]
- Baldrich, E.; Muñoz, F.X.; García-Aljaro, C. Electrochemical Detection of Quorum Sensing Signaling Molecules by Dual Signal Confirmation at Microelectrode Arrays. Anal. Chem. 2011, 83, 2097–2103. [Google Scholar] [CrossRef]
- Li, F.; Yu, Z.; Han, X.; Lai, R.Y. Electrochemical Aptamer-Based Sensors for Food and Water Analysis: A Review. Anal. Chim. Acta 2019, 1051, 1–23. [Google Scholar] [CrossRef]
- Forouzanfar, S.; Alam, F.; Pala, N.; Wang, C. Review—A Review of Electrochemical Aptasensors for Label-Free Cancer Diagnosis. J. Electrochem. Soc. 2020, 167, 067511. [Google Scholar] [CrossRef]
- Hernandez-Vargas, G.; Sosa-Hernández, J.E.; Saldarriaga-Hernandez, S.; Villalba-Rodríguez, A.M.; Parra-Saldivar, R.; Iqbal, H.M.N. Electrochemical Biosensors: A Solution to Pollution Detection with Reference to Environmental Contaminants. Biosensors 2018, 8, 29. [Google Scholar] [CrossRef] [Green Version]
- Beluomini, M.A.; da Silva, J.L.; de Sá, A.C.; Buffon, E.; Pereira, T.C.; Stradiotto, N.R. Electrochemical Sensors Based on Molecularly Imprinted Polymer on Nanostructured Carbon Materials: A Review. J. Electroanal. Chem. 2019, 840, 343–366. [Google Scholar] [CrossRef]
- Zhou, L.; Glennon, J.D.; Luong, J.H.T.; Reen, F.J.; O’Gara, F.; McSweeney, C.; McGlacken, G.P. Detection of the Pseudomonas Quinolone Signal (PQS) by Cyclic Voltammetry and Amperometry Using a Boron Doped Diamond Electrode. Chem. Commun. 2011, 47, 10347–10349. [Google Scholar] [CrossRef] [PubMed]
- Buzid, A.; Shang, F.; Reen, F.J.; Muimhneacháin, E.; Clarke, S.L.; Zhou, L.; Luong, J.H.T.; O’Gara, F.; McGlacken, G.P.; Glennon, J.D. Molecular Signature of Pseudomonas Aeruginosa with Simultaneous Nanomolar Detection of Quorum Sensing Signaling Molecules at a Boron-Doped Diamond Electrode. Sci. Rep. 2016, 6, 30001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buzid, A.; Luong, J.H.T.; Reen, F.J.; O’Gara, F.; Glennon, J.D.; McGlacken, G.P.; Rev, N.; Discov, D.; Care, D.; Res, C. Rapid Electrochemical Detection of Pseudomonas Aeruginosa Signaling Molecules by Boron-Doped Diamond Electrode. In Quorum Sensing: Methods and Protocols, Methods in Molecular Biology; Leoni, L., Rampioni, G., Eds.; Humana Press: New York, NY, USA, 2018; Volume 1673, pp. 107–116. ISBN 978-1-4939-7309-5. [Google Scholar]
- Özcan, M.; Sesal, N.C.; Şener, M.K.; Koca, A. An Alternative Strategy to Detect Bacterial Contamination in Milk and Water: A Newly Designed Electrochemical Biosensor. Eur. Food Res. Technol. 2020, 246, 1317–1324. [Google Scholar] [CrossRef]
- Jiang, H.; Jiang, D.; Shao, J.; Sun, X. Magnetic Molecularly Imprinted Polymer Nanoparticles Based Electrochemical Sensor for the Measurement of Gram-Negative Bacterial Quorum Signaling Molecules (N-Acyl-Homoserine-Lactones). Biosens. Bioelectron. 2016, 75, 411–419. [Google Scholar] [CrossRef]
- Ștefan, G.; Hosu, O.; de Wael, K.; Lobo-Castañón, M.J.; Cristea, C. Aptamers in Biomedicine: Selection Strategies and Recent Advances. Electrochim. Acta 2021, 376, 137994. [Google Scholar] [CrossRef]
- Sang, S.; Wang, Y.; Feng, Q.; Wei, Y.; Ji, J.; Zhang, W. Progress of New Label-Free Techniques for Biosensors: A Review. Crit. Rev. Biotechnol. 2016, 36, 465–481. [Google Scholar] [CrossRef]
- Koyappayil, A.; Lee, M.H. Ultrasensitive Materials for Electrochemical Biosensor Labels. Sensors 2021, 21, 89. [Google Scholar] [CrossRef]
- Zhao, Z.G.; Yu, Y.M.; Xu, B.Y.; Yan, S.S.; Xu, J.F.; Liu, F.; Li, G.M.; Ding, Y.L.; Wu, S.Q. Screening and Anti-Virulent Study of N-Acyl Homoserine Lactones DNA Aptamers against Pseudomonas Aeruginosa Quorum Sensing. Biotechnol. Bioprocess Eng. 2013, 18, 406–412. [Google Scholar] [CrossRef]
- Tertiş, M.; Ciui, B.; Suciu, M.; Săndulescu, R.; Cristea, C. Label-Free Electrochemical Aptasensor Based on Gold and Polypyrrole Nanoparticles for Interleukin 6 Detection. Electrochim. Acta 2017, 258, 1208–1218. [Google Scholar] [CrossRef]
- Chang, A.L.; McKeague, M.; Liang, J.C.; Smolke, C.D. Kinetic and Equilibrium Binding Characterization of Aptamers to Small Molecules Using a Label-Free, Sensitive, and Scalable Platform. Anal. Chem. 2014, 86, 3273–3278. [Google Scholar] [CrossRef] [PubMed]
- Muhamad, N.B.; Khairul, W.M.; Yusoff, F. Synthesis and Characterization of Poly(3,4-Ethylenedioxythiophene) Functionalized Graphene with Gold Nanoparticles as a Potential Oxygen Reduction Electrocatalyst. J. Solid State Chem. 2019, 275, 30–37. [Google Scholar] [CrossRef]
- Ranjbar, S.; Shahrokhian, S. Design and Fabrication of an Electrochemical Aptasensor Using Au Nanoparticles/Carbon Nanoparticles/Cellulose Nanofibers Nanocomposite for Rapid and Sensitive Detection of Staphylococcus Aureus. Bioelectrochemistry 2018, 123, 70–76. [Google Scholar] [CrossRef] [PubMed]
- Cesiulis, H.; Tsyntsaru, N.; Ramanavicius, A.; Ragoisha, G. The Study of Thin Films by Electrochemical Impedance Spectroscopy. In Nanostructures and Thin Films for Multifunctional Applications. NanoScience and Technology; Tiginyanu, I., Topala, P., Ursaki, V., Eds.; Springer: Cham, Switzerland, 2016; pp. 3–42. [Google Scholar]
- Jambrec, D.; Conzuelo, F.; Estrada-Vargas, A.; Schuhmann, W. Potential-Pulse-Assisted Formation of Thiol Monolayers within Minutes for Fast and Controlled Electrode Surface Modification. ChemElectroChem 2016, 3, 1484–1489. [Google Scholar] [CrossRef]
- Xue, Y.; Li, X.; Li, H.; Zhang, W. Quantifying Thiol-Gold Interactions towards the Efficient Strength Control. Nat. Commun. 2014, 5, 4348. [Google Scholar] [CrossRef] [Green Version]
- Ge, C.; Liao, J.; Yu, W.; Gu, N. Electric Potential Control of DNA Immobilization on Gold Electrode. Biosens. Bioelectron. 2003, 18, 53–58. [Google Scholar] [CrossRef]
- Jambrec, D.; Gebala, M.; la Mantia, F.; Schuhmann, W. Potential-Assisted DNA Immobilization as a Prerequisite for Fast and Controlled Formation of DNA Monolayers. Angew. Chem. Int. Ed. Engl. 2015, 54, 15064–15068. [Google Scholar] [CrossRef]
- Baraket, A.; Lee, M.; Zine, N.; Sigaud, M.; Bausells, J.; Errachid, A. A Fully Integrated Electrochemical Biosensor Platform Fabrication Process for Cytokines Detection. Biosens. Bioelectron. 2017, 93, 170–175. [Google Scholar] [CrossRef]
- Lukács, Z.; Kristóf, T. A Generalized Model of the Equivalent Circuits in the Electrochemical Impedance Spectroscopy. Electrochim. Acta 2020, 363, 137199. [Google Scholar] [CrossRef]
- Tertis, M.; Leva, P.I.; Bogdan, D.; Suciu, M.; Graur, F.; Cristea, C. Impedimetric Aptasensor for the Label-Free and Selective Detection of Interleukin-6 for Colorectal Cancer Screening. Biosens. Bioelectron. 2019, 137, 123–132. [Google Scholar] [CrossRef]
- Chang, A.L.; McKeague, M.; Smolke, C.D. Facile Characterization of Aptamer Kinetic and Equilibrium Binding Properties Using Surface Plasmon Resonance. Methods Enzymol. 2014, 549, 451–466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spiga, F.M.; Maietta, P.; Guiducci, C. More DNA-Aptamers for Small Drugs: A Capture-SELEX Coupled with Surface Plasmon Resonance and High-Throughput Sequencing. ACS Comb. Sci. 2015, 17, 326–333. [Google Scholar] [CrossRef] [Green Version]
- Wen, K.Y.; Cameron, L.; Chappell, J.; Jensen, K.; Bell, D.J.; Kelwick, R.; Kopniczky, M.; Davies, J.C.; Filloux, A.; Freemont, P.S. A Cell-Free Biosensor for Detecting Quorum Sensing Molecules in P. Aeruginosa-Infected Respiratory Samples. ACS Synth. Biol. 2017, 6, 2293–2301. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, S.; Srinivasan, P.; Neelakantan, P.; Rayappan, J.B.B.; Solomon, A.P. Photoluminescence-Based Bioassay With Cysteamine-Capped TiO2 Nanoparticles for the Selective Recognition of N-Acyl Homoserine Lactones. Front. Bioeng. Biotechnol. 2021, 9, 750933. [Google Scholar] [CrossRef] [PubMed]
- Fujitani, S.; Moffett, K.S.; Yu, V.L. Pseudomonas Aeruginosa. Antimicrobe Infect. Dis. Antimicrob. Agents 2019, 2, 1–25. [Google Scholar] [CrossRef]
- Blidar, A.; Feier, B.; Tertis, M.; Galatus, R.; Cristea, C. Electrochemical Surface Plasmon Resonance (EC-SPR) Aptasensor for Ampicillin Detection. Anal. Bioanal. Chem. 2019, 411, 1053–1065. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Platform | Au-SPE | C-SPE/AuNPs | ||
|---|---|---|---|---|
| DPV (%) | EIS (%) | DPV (%) | EIS (%) | |
| APT immobilization | −8.40 | +207.18 | −49.11 | +532.8 |
| MCH blocking | −9.58 | +51.85 | −26.14 | +55.50 |
| Incubation with 50 μM 3-O-C12-HSL | +0.21 | −13.28 | +42.92 | −68.32 |
| Aptasensor Elaboration Step | Optimized Parameters | Signal Modification in EIS (%) | Signal Modification in DPV (%) | |
|---|---|---|---|---|
| AuNP generation | HAuCl4 Conc. (μM) | 1.5 | −91 | +205.68 |
| 3 | −88.42 | +190.61 | ||
| 5 | −83.90 | +168.38 | ||
| Number of CV cycles (Conc. HAuCl4 = 1.5 μM) | 25 | −82 | +95.03 | |
| 35 | −91 | +197.64 | ||
| APT immobilization | Immobilization technique | Overnight | - | −33.88 |
| MPA | - | −52.68 | ||
| APT solution concentration (μM) (MPA) | 1 | +407.06 | −31.33 | |
| 2 | +366.50 | −22.08 | ||
| 3 | +341.62 | −27.17 | ||
| Total immobilization time (s) (Conc. APT 1 µM) | 200 | +316.27 | −23.10 | |
| 300 | +433.26 | −36.82 | ||
| 400 | +295.83 | −23.62 | ||
| Pulse duration (s) (Conc. APT 1 µM, total immobilization time 300 s) | 0.030 | +616.30 | −29.02 | |
| 0.045 | +885.75 | −43.06 | ||
| 0.060 | +565.47 | −14.97 | ||
| Incubation with 3-O-C12-HSL | Incubation time (min) | 10 | −3.41 | +0.15 |
| 20 | −52.88 | +56.12 | ||
| 30 | −55.85 | +58.40 | ||
| 40 | −43.86 | +57.04 | ||
| 50 | −48.32 | +52.44 | ||
| Kinetic conditions (30 min) | Static | −52.41 | +10.26 | |
| Dynamic | −20.29 | +6 |
| RS (Ω) | Ret (Ω) | W (mMho) | CPE (µMho) | N | C (µF) | χ2 | |
|---|---|---|---|---|---|---|---|
| C-SPE | 135 | 279 | 3.09 | - | - | 1.00 | 0.003 |
| C-SPE/AuNPs | 133 | 27.20 | 3.25 | - | - | 5.79 | 0.001 |
| C-SPE/AuNPs/APT | 133 | 186 | 2.75 | 4.52 | 0.93 | - | 0.003 |
| C-SPE/AuNPs/APT/MCH | 133 | 647 | 2.58 | 4.34 | 0.91 | - | 0.006 |
| C-SPE/AuNPs/APT/MCH/3-O-C12-HSL | 134 | 222; 228 | 3.02 | - | - | 37.90; 5.27 | 0.01 |
| Method | Linear Range (nM) | LOD (nM) | Sample | Ref. |
|---|---|---|---|---|
| HPLC-MS/MS | 2.6–350 | 2.6 | Culture media | [11] |
| Whole-cell biosensor (PA14-R3) | 0.152–12 | 0.01 | Culture media and CF sputum | [16] |
| Genetically engineered (electrochemical) biosensor | 0.01–10 | 0.002 | Liquid cultures and artificial saliva | [18] |
| Magnetic MIP-based electrochemical sensor | 2.5–100 | 0.8 | Bacteria supernatant samples | [27] |
| LasRV cell-free biosensor | 5–100 | 4.9 | CF sputum | [46] |
| Photoluminescence-based assay using cysteamine-capped TiO2 nanoparticles | 10–160 | 10 | Artificial urine | [47] |
| AuNPs-based electrochemical aptasensor | 500–30,000 | 500 | Spiked culture media, urine samples, culture media with bacteria | This work |
| Sample | Signal (%) | Spiked Conc. of 3-O-C12-HSL (μM) | Found Conc. of 3-O-C12-HSL (μM) | Recovery (%) |
|---|---|---|---|---|
| Urine | 53.18 | 10 | 10.01 | 100.1 ± 8.59 |
| NB | 55.05 | 10 | 12.06 | 120.61 ± 7.84 |
| Time | P. aeruginosa ATCC 27853 | P. aeruginosa Clinical Isolate | ||
|---|---|---|---|---|
| No. of Colonies (CFU mL−1) | Conc. of 3-O-C12-HSL (µM) | No. of Colonies (CFU mL−1) | Conc. of 3-O-C12-HSL (µM) | |
| 16 h | 2507 × 107 | 0.582 | 1522 × 107 | 0.383 |
| 24 h | 4097 × 107 | 1.131 | 2935 × 107 | 0.675 |
| 48 h | 36,455 × 107 | 2.55 | 28,385 × 107 | 3.06 |
| 72 h | 1911 × 1010 | 17.14 | 1386 × 1010 | 7.76 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Capatina, D.; Lupoi, T.; Feier, B.; Blidar, A.; Hosu, O.; Tertis, M.; Olah, D.; Cristea, C.; Oprean, R. Label-Free Electrochemical Aptasensor for the Detection of the 3-O-C12-HSL Quorum-Sensing Molecule in Pseudomonas aeruginosa. Biosensors 2022, 12, 440. https://doi.org/10.3390/bios12070440
Capatina D, Lupoi T, Feier B, Blidar A, Hosu O, Tertis M, Olah D, Cristea C, Oprean R. Label-Free Electrochemical Aptasensor for the Detection of the 3-O-C12-HSL Quorum-Sensing Molecule in Pseudomonas aeruginosa. Biosensors. 2022; 12(7):440. https://doi.org/10.3390/bios12070440
Chicago/Turabian StyleCapatina, Denisa, Teodora Lupoi, Bogdan Feier, Adrian Blidar, Oana Hosu, Mihaela Tertis, Diana Olah, Cecilia Cristea, and Radu Oprean. 2022. "Label-Free Electrochemical Aptasensor for the Detection of the 3-O-C12-HSL Quorum-Sensing Molecule in Pseudomonas aeruginosa" Biosensors 12, no. 7: 440. https://doi.org/10.3390/bios12070440
APA StyleCapatina, D., Lupoi, T., Feier, B., Blidar, A., Hosu, O., Tertis, M., Olah, D., Cristea, C., & Oprean, R. (2022). Label-Free Electrochemical Aptasensor for the Detection of the 3-O-C12-HSL Quorum-Sensing Molecule in Pseudomonas aeruginosa. Biosensors, 12(7), 440. https://doi.org/10.3390/bios12070440