1. Introduction
Cancer metastasis, where malignant cells spread from a primary site to distant organs in the body, is largely responsible for the mortality and morbidity of cancer [
1]. After dissociating from the original tumor mass, metastatic cells migrate through the extracellular matrix (ECM) by reorganizing their attachment to the ECM with altered cell–ECM adhesion dynamics, and then invade through adjacent tissues and intravasate into the blood vessels [
2]. These motile cells journey through the vascular circulation and extravasate via invading the vascular basement membrane and ECM again to ultimately attach at a new location and grow into the secondary tumor [
2]. Therefore, tumor cells experience alterations in cell–cell and cell–ECM adhesion during various stages of cancer invasion and metastasis.
In the tumor microenvironment, integrin receptors mediate cell adhesion with ECM ligands (such as fibronectin or FN, laminin, collagen, and gelatin) [
3,
4]. Such interactions couple the extracellular environment to intracellular signals via the actin cytoskeleton, which enables cell responses to external stimuli in a coordinated manner and enhances cancer cell proliferation, migration, invasion, and metastasis [
2,
4,
5,
6]. The biophysical and biochemical cues from the tumor ECM modulates each one of the ‘hallmarks of cancer’, and defective mechanosensitivity and mechano-responsiveness of the cell–ECM interactions are often associated with metastasis progression [
7,
8,
9]. Therefore, the cell–ECM adhesion strength or avidity has been considered as a general indication for the metastatic potential of tumor cells, which defines the aggressiveness of cancer cells [
6,
10,
11]. Many cell-adhesion proteins have been proposed as potent targets for inhibiting cancer cell invasion and metastasis [
6].
Existing techniques established to measure the cell–ECM avidity include fixed-cell imaging based in vitro adhesion seeding assays [
12], micropipette-based adhesion frequency assays [
13,
14,
15], optical tweezers-based traction force microscopy [
16], as well as the conventional wash assays [
17]. These model systems are able to identify key adhesion components and correlate with metastatic progression [
12] thereafter providing invaluable insights into regulatory mechanisms. However, those methods are either low throughput, not quantitative, or with poor sensitivity and reproducibility [
11,
17]. In recent years, Engler et al. established a population-based adhesion assay using the spinning-disk shear to quantify the cell adhesion strength on fibronectin and correlate it with focal adhesion assembly. This method also captured the adhesion heterogeneity within the studied cell population [
10,
11]. However, this measuring system is a custom-built spinning-disk device, and the experimental operation is time-consuming, which requires relatively long sample preparation and processing time with multiple experimental steps. These aspects limit its application as a high-throughput screening system.
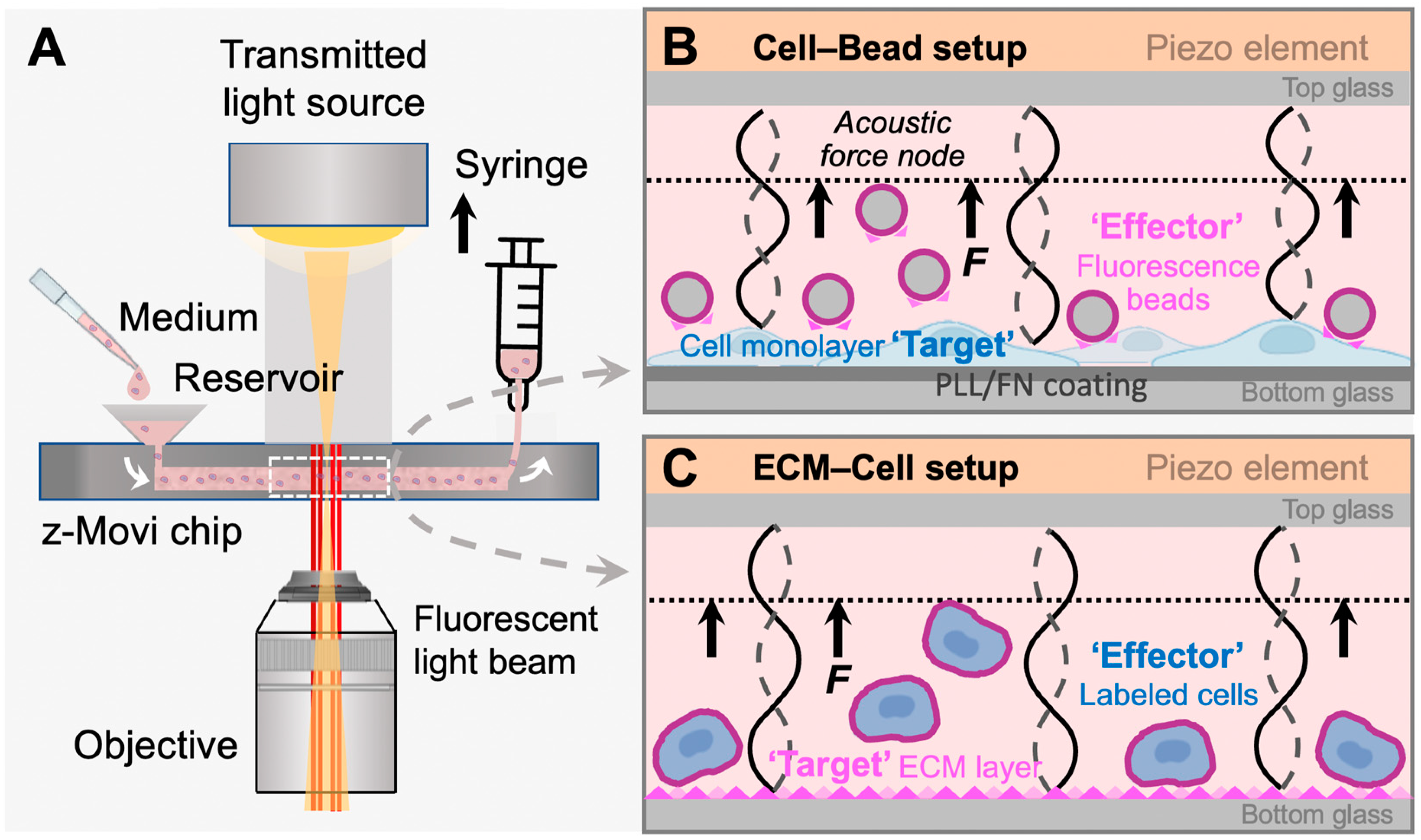
To this end, we established a user-friendly, high-throughput yet effective method to study cell–ECM interaction in vitro using the acoustic force-based z-Movi
® cell–cell avidity analyzer (
Figure 1A). This technology allows sample preparation and avidity testing to be performed within a few hours. It can determine avidities of up to 400 cell pairs in a single run within a few minutes with minimum cell damage and high sensitivity and generate instant statistically relevant data. The z-Movi has rapidly emerged to measure the bona fide avidity between a T cell and its target tumor cell, which determines the adhesive strength of immunological synapse formation and T cell activation [
18,
19,
20]. Assessments of the cell avidity in vitro have been proven to accurately predict cellular responses in vivo and outcomes during immunotherapy and facilitate the selection of optimal immune cells (e.g., CAR-T or TCR-T cells, NK cells) to kill cancer [
18,
19,
20]. Here, we optimized and repurposed the z-Movi technology, for the first time, to measure cell–ECM avidity (
Figure 1B,C), particularly the cell avidity with FN as a showcase. This method can be served as a rapid, simple, and high-throughput screening platform for functional assessment of cell adhesion molecules and their interactions with the tumor microenvironment in a highly predictive and reproducible manner.
2. Materials and Methods
2.1. Cell Culture
The human ovarian cancer cell line A2780 (ECACC 93112519) and human breast cancer cell line MCF-7 (ATCC HTB-22) were cultured in RPMI1640 medium (11875-119, Gibco, Waltham, MA, USA) supplemented with 10% fetal bovine serum (FBS; 10099141, Gibco), and kept in a humidified 37 °C incubator with 5% CO2. All cell lines were negative for mycoplasma using real-time PCR-based screening.
2.2. Chip Glass Surface Coating with Poly-L-Lysine (PLL)
The chip needs to be cleaned and dried for at least 1 h at 37 °C before coating. The surface coating was carried out using 0.002% PLL (P4707, Sigma-Aldrich, Macquarie Park, NSW, Australian) in PBS (10010023, Thermo Fisher Scientific, Waltham, MA, USA). For each chip, 50 μL of fresh PLL solution was prepared and pulled into the microfluidic channel using a syringe (SS+03L1, Terumo Medical Canada Inc., Vaughan, ON, Canada) with 10 μL volume left in the reservoir to avoid bubble formation. After 15 min-incubation at room temperature, the PLL solution was completely removed using the syringe. The chip was pulled through with air several times to remove any remaining liquid in the microfluidic channel. The PLL-coated chips were kept in a dry incubator at 37 °C with a cap onto the reservoir (to avoid evaporation) for at least 1 h. The coated chips must be used within 3 days after coating. Upon cell seeding, the chips were rehydrated by pulling in 100 μL warm PBS into the microfluidic channel, leaving about 10 μL in the reservoir to avoid bubble formation. This step was repeated once with 100 μL warm complete cell culture medium.
2.3. Chip Glass Surface Coating with Fibronectin
The chip needs to be cleaned and dried for at least 1 h at 37 °C. For each chip, the glass surface was rehydrated by pulling in the following solutions to the microfluidic channel using a syringe (leave about 10 μL in the reservoir to avoid bubble formation): 200 μL 1M NaOH (incubation for 5 min), 400 μL Milli-Q water for two times, 400 μL 1 M HCl for two times (incubation for 2 min at the second time), 400 μL Milli-Q water for three times, and 400 μL PBS once. For each chip, 50 μL of 100 μg/mL fresh FN (F0895, Sigma-Aldrich) solution in PBS was pulled into the microfluidic channel with about 10 μL left in the reservoir. The inlet was washed with 400 μL PBS three times to prevent fiber or aggregate formation and re-filled with 200 μL PBS. Chips were caped firmly and kept in the 37 °C dry incubator overnight. Before cell seeding, the FN-coated chips were washed 3 times with PBS and once with the complete culture medium. The coated chips need to be used within 3 days after coating.
2.4. Target Cell Monolayer Formation
Target cells with at least 80% confluency were used for z-Movi experiments. Adherent cells were washed once with PBS and trypsinized using TrypLE (12605010, Thermo Fisher Scientific) for 3–5 min at 37 °C. Cells were collected using the complete culture medium, and the cell density was determined using the automatic cell counter. For each chip, 20 μL of MCF-7 or A2780 cells at indicated densities were seeded into the microfluidic channel using a syringe (leave about 10 μL volume in the reservoir to avoid bubble formation). It is important to keep the chips and cells at 37 °C during cell seeding to reduce cell clumping. The inlet was washed with 400 μL complete medium three times and re-filled with 400 μL PBS. Chips were caped firmly and kept in the 37 °C dry incubator for a designated period (1-4 h, cell-line dependent) before z-Movi avidity assay.
2.5. Cell Viability Measurement of the Target Cell Monolayer
The viability of the cell monolayer was evaluated by adding 10 μL Trypan Blue (15250061, Thermo Fisher) in the reservoir containing 50 μL medium. The Trypan Blue dilution was pulled into the microfluidic channel and incubated with the monolayer cells for 30 s to stain the dead cell population, followed by 200 μL complete culture medium to rinse out Trypan Blue. The cell viability was observed by brightfield imaging.
2.6. Effector Cell Staining
Adherent cells were washed once with PBS and trypsinized using TrypLE for 3−5 min at 37 °C. Cells were collected using the complete culture medium, and the cell density was determined using the automatic cell counter. For a single run on the chip, 20 μL of 15 × 106 cells/mL (0.3 × 106 cells) was prepared. Cells were washed once with PBS and stained with the 1× CellTrace Far Red Dye (C34564, Thermo Fisher) dilution in PBS at 1 × 106 cells/mL in the dark at 37 °C for 15 min. Cells were re-suspended by pipetting every 5 min during the staining process. Approximal 5 mL complete culture medium was then added to the cell solution to stop the staining. Stained cells were washed once with PBS, re-suspended in complete culture medium at 15 × 106 cells/mL, and transferred into a 96-well plate with round bottom.
2.7. Effector Beads Coating with Fibronectin (FN)
The red-fluorescent melamine resin particles (MF-FluoRed-L840, microParticles GmbH, Berlin-Adlershof, Germany) were washed with 500 μL PBS three times and once with 500 μL complete culture medium. Beads were then incubated with FN at 10 μg/mL in 200 μL of completed culture medium for 1 h on a rotating stage at room temperature. FN-coated beads were washed three times with 500 μL complete medium and re-suspended in the complete medium (10 μL for a single run).
2.8. Cell Binding Avidity Measurement
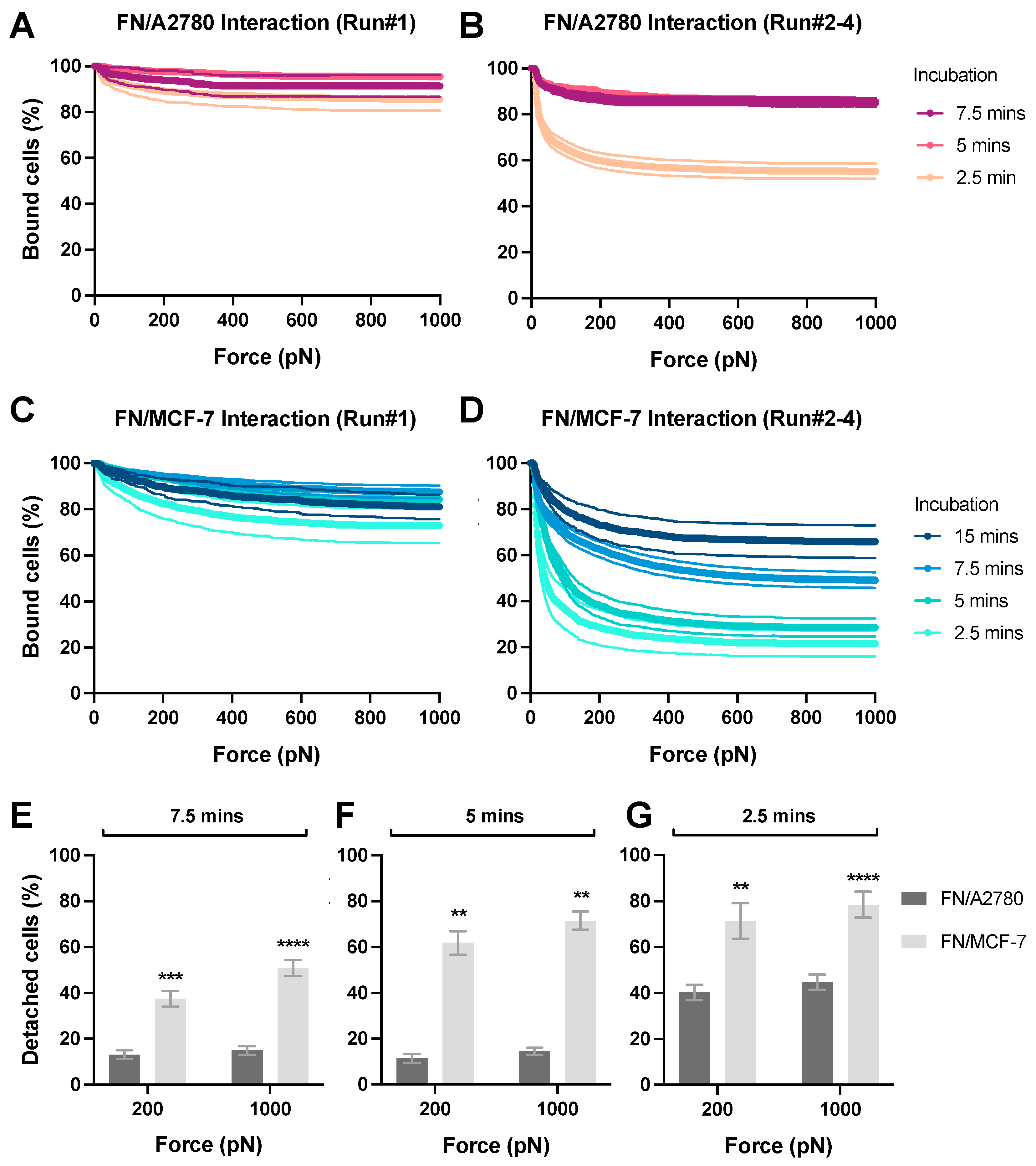
The chip was placed on the z-Movi (LUMICKS B.V., Amsterdam, The Netherlands) stage, and the in-chip cell monolayer quality was evaluated and validated by applying force at 1000 pN for 10 s. Once the monolayer passed the validation, the medium level in the inlet was brought down to about 10 μL. 20 μL of stained effector cells were added into the reservoir and pulled into the microfluidic channel to interact with the target cell monolayer for a designated period. Depending on the effector density, up to 400 effectors that being considered as individual events will be incubated in the field of view (FOV) for the avidity measurement. During the incubation, the inlet was gently washed with complete culture medium three times to remove the remaining cells and refilled with 100 μL complete medium. After interaction, the acoustic force was applied with a linear force ramp from 0 to 1000 pN over 2.5 min. The percentage of bound cells to the monolayer under different levels of applied force was calculated simultaneously. To block FN binding with cancer cells, a FN antibody (MA5-11981, Thermo Fisher) was used. To block non-specific bindings, 2% BSA (A3311, Sigma-Aldrich) in complete culture medium was used.
2.9. Chip Cleaning
The cleaning solution containing 5% bleach (A1727, Sigma-Aldrich) was pulled into the microfluidic channel, and the chip was incubated at room temperature for at least 20 min and up to 4 h. The channel was then washed with 400 μL Milli-Q water and the glass surface was scrubbed by introducing air and moving the bubbles back and forth several times. The chip surface was scrubbed with 200 μL bleach twice, followed by pulling through (1) 400 μL bleach, (2) 400 μL Milli-Q water twice, (3) 100 μL 12M HCl (H1758, Sigma-Aldrich), and (4) 200 μL Milli-Q water twice. The surface was then scrubbed with 400 μL 1 M NaOH twice and incubated with 1 M NaOH for 1 h. After pulling through the NaOH solution, the surface was scrubbed with 200 μL 1 M NaOH twice, followed by washing with 400 μL Milli-Q water twice. Cleaned chips were stored in the dry incubator.
2.10. Avidity Data Analysis
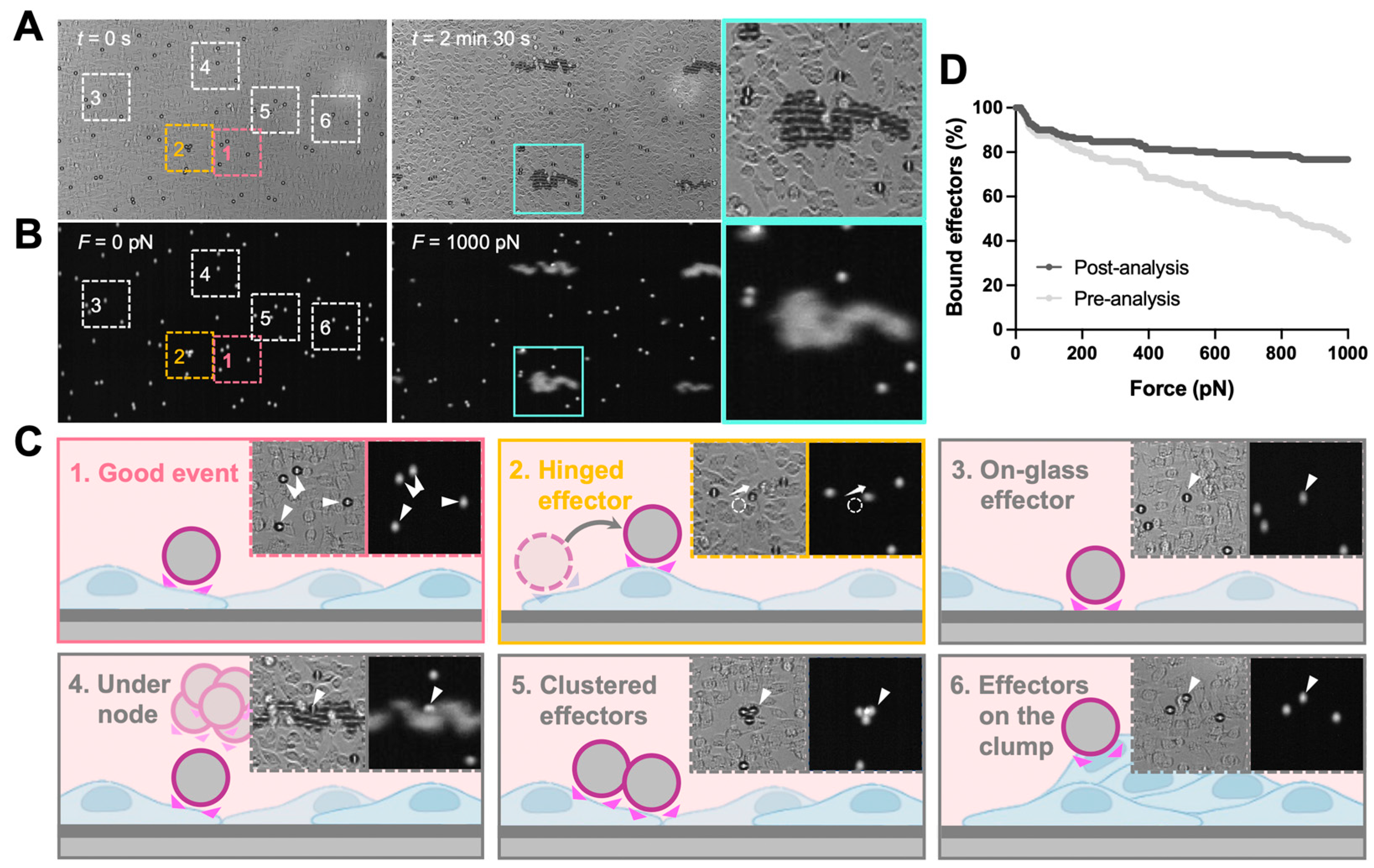
The analysis of z-Movi data was performed offline by using Oceon 1.4.1 (LUMICKS B.V., Amsterdam, The Netherlands). Two-channel images were loaded into the software: (1) brightfield images which were used to examine the monolayer confluency and screen the effector events (
Figure 2A,
left); (2) fluorescence images were used to track the position of effectors (
Figure 2A,
right) and, thus, judge whether the effector was lifted. During the z-Movi application, the control software will move the field of view (FOV) to the force-calibrated region of the chip based on the information stored in the chip, while the software will capture both brightfield (monolayer tracking) and fluorescence (effectors tracking) signals within the FOV for the whole force application. The region of interest (ROI) for each effector is a 12 pixel-wide circle (with the cell as the center) by default. ROIs of up to 400 effectors within the FOV were identified by the software. Effectors left the ROIs while force application would be considered as ‘lifted’ and the software tracked all ROIs automatically. Manual selections were performed after software ROI selections (
Figure 2B). Clustered effectors, effectors stuck on the glass, on the target cell clump, and under the acoustic force nodes were excluded from the analysis. In addition, effectors that escaped from the ROI but were not lifted (hinged effectors) were considered as attached effectors for the whole force application. After the manual selections, the software ran the automatic detection on lifted cells over the force ramp from 0–1000 pN (
Figure 2C). Since the avidity curve would be dramatically different if no manual selection were performed (
Figure 2D), we strongly recommended consistent manual selection over different runs of experiments.
2.11. Statistical Analysis
Unpaired two-tailed Student’s t test was performed in Prism 9 for the measurement of statistical significance. p < 0.05 was considered statistically significant. All data points with error bars are presented as mean ± standard error of the mean (S.E.M).
4. Conclusions
In brief, we have established a novel high-throughput measuring platform using the emerging acoustic force-dependent z-Movi technology to determine the adhesion strength of different types of cancer cells on ECM in vitro. This robust and highly maneuverable method will offer a rapid and simple solution to perform predictive, reproducible, and fast characterization of bona fide interactions between tumor cells and ECM components. More importantly, z-Movi allows us to capture early adhesion events and quantitate the adhesion strength from the initial cell attachment, flattening, to fully spreading on ECM, which cannot be achieved by traditional methods. Furthermore, given that acoustic force causes minimal damage to live cells, this system is suitable for studying fragile cells like primary patient samples and cancer stem cells, regardless of the tumor types. Although the experimental setup is designed for in vitro testing, the adhesion strength quantified by z-Movi serves as a general marker to predict the metastatic potential of the cancer cells in vivo. Moreover, by measuring the avidity of hundreds of ‘Target–Effector’ pairs in parallel with minimum cell damage with high sensitivity, this method is of great potential for preclinical drug screening to identify new candidates with potential anti-metastatic properties in a high-throughput manner.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





