
Risk Factors for Antimicrobial Resistance in Turkey Farms: A Cross-Sectional Study in Three European Countries

 , , , , , , , , and
on behalf of the EFFORT-Group
, , , , , , , , and
on behalf of the EFFORT-Group
Abstract
:1. Introduction
2. Results
2.1. Overview of the Sampled Farms and Flocks
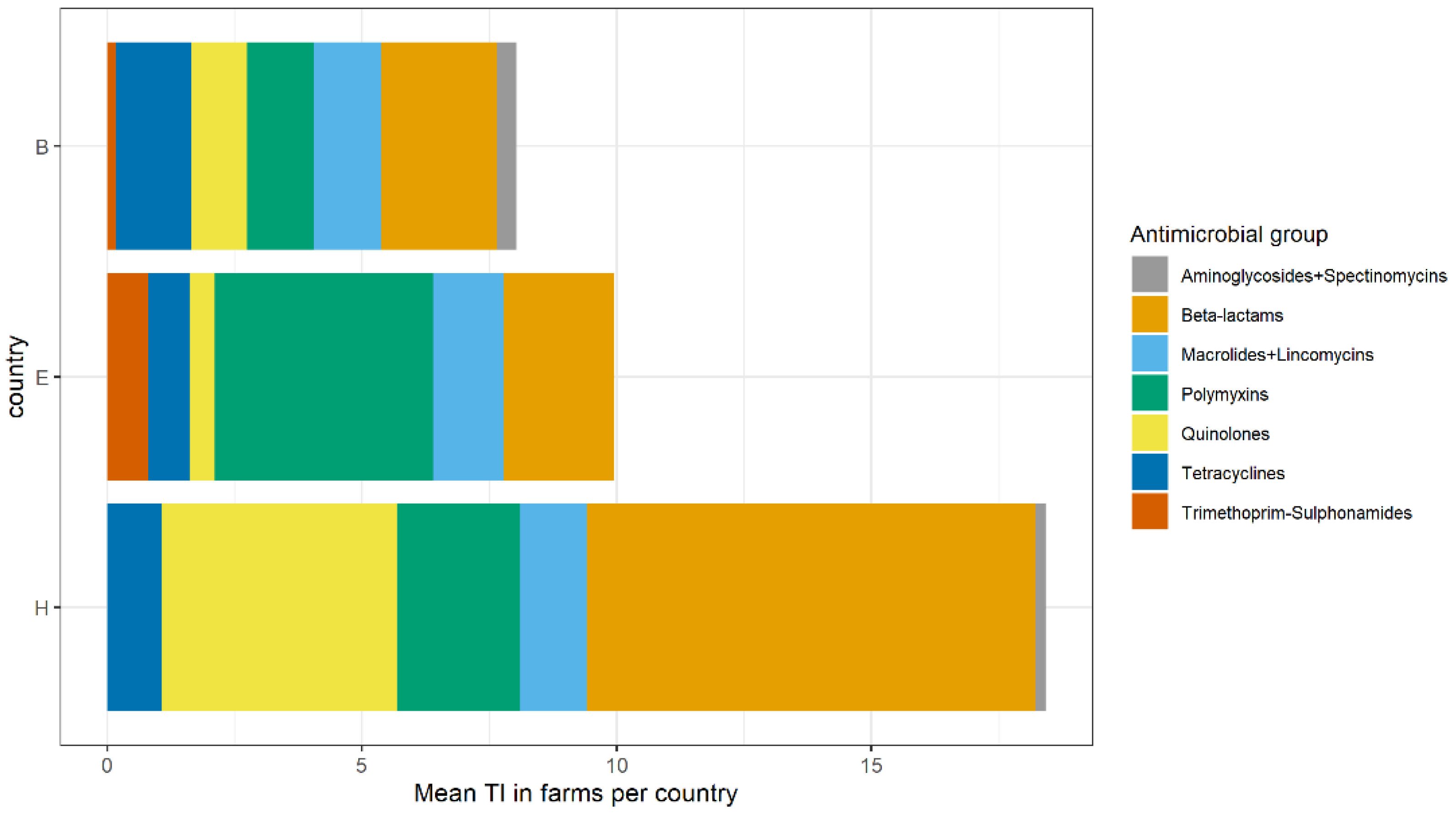
2.2. Antimicrobial Usage
2.3. AMR Genes Identified by Metagenomics
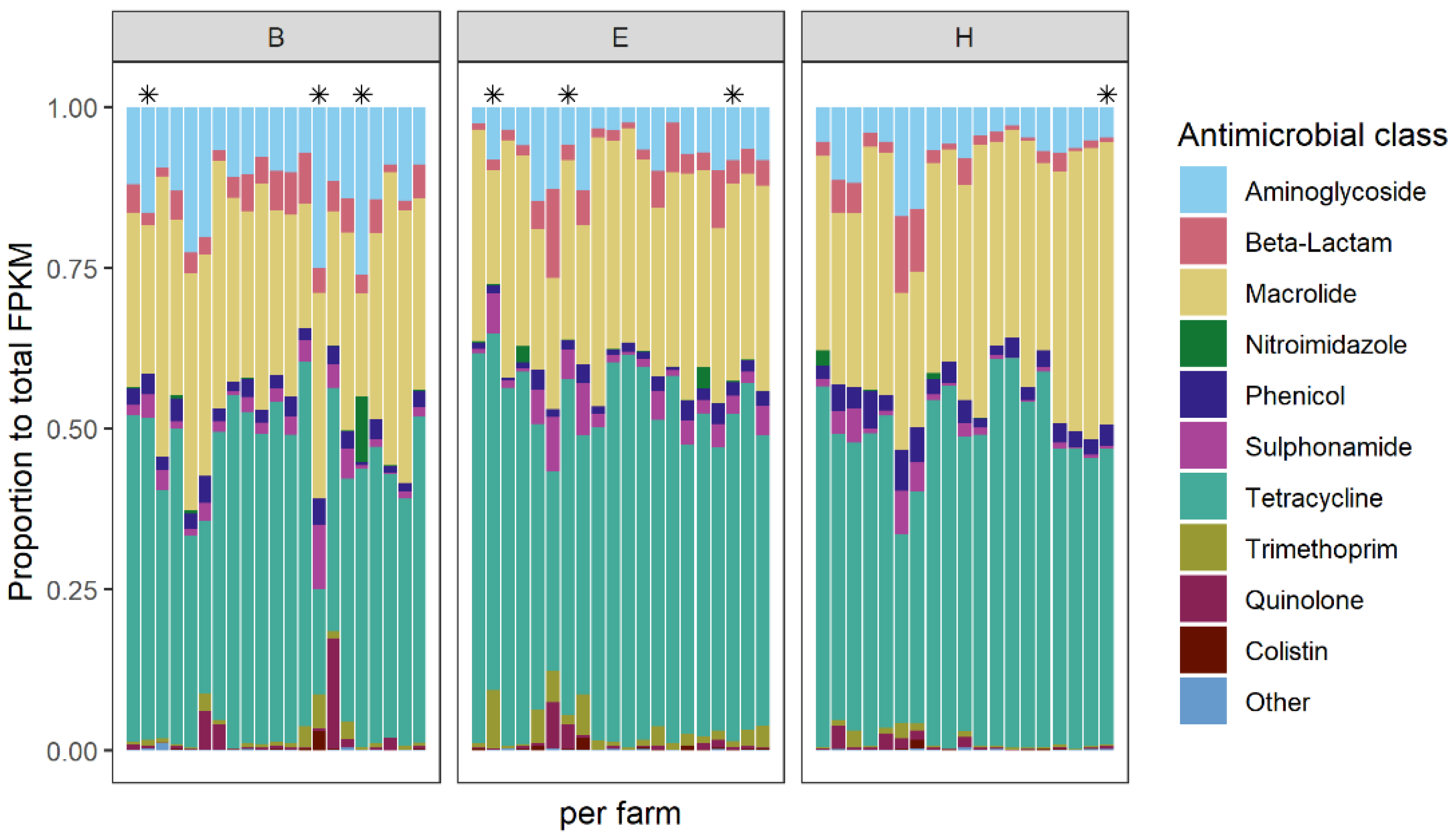
2.3.1. The Abundance and Composition of AMR Genes
2.3.2. Factors Associated with the Abundance of AMR Gene Clusters
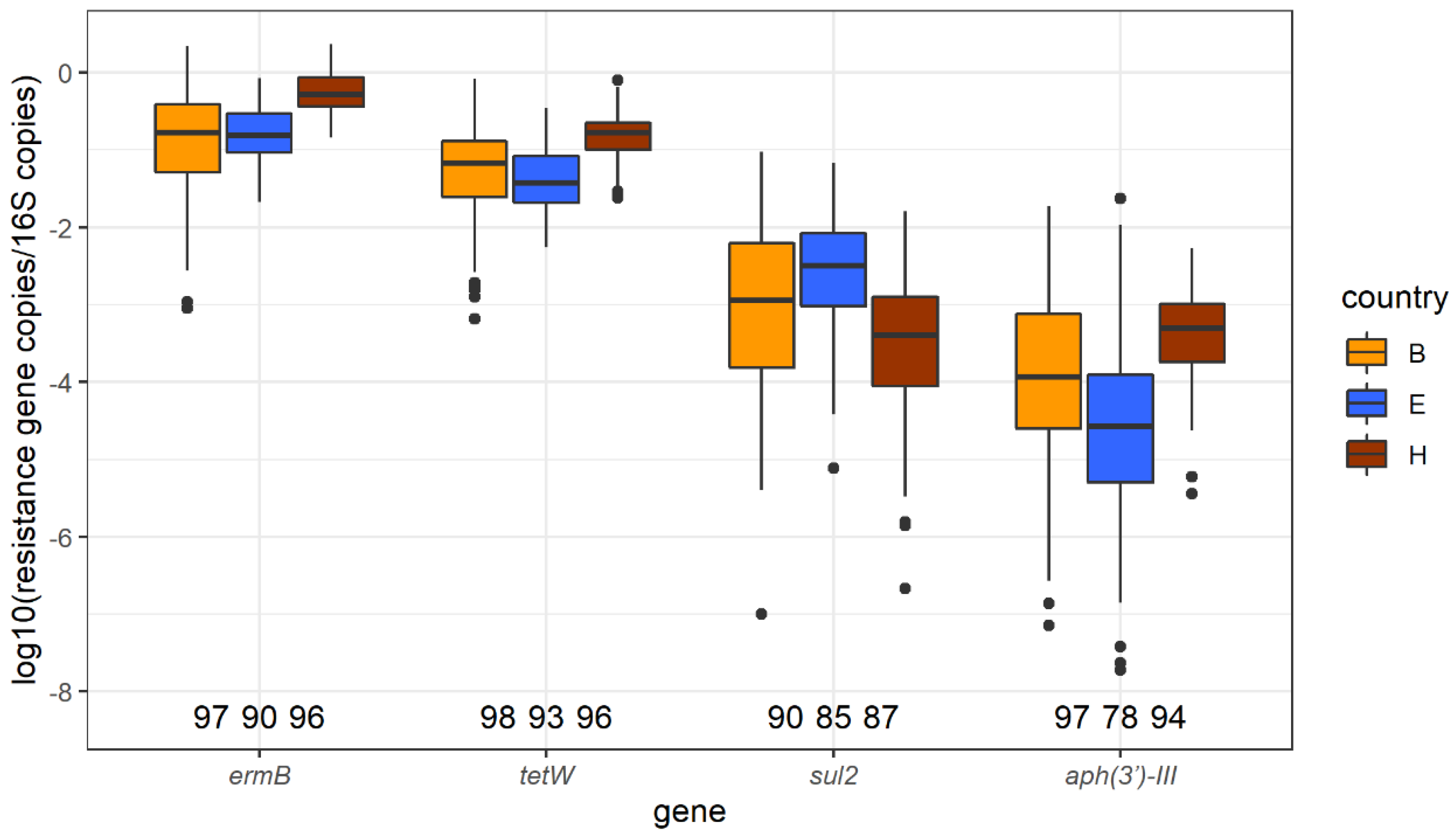
2.4. ermB, tetW, sul2, and aph3′-III Identified by qPCR
2.4.1. Abundance of ermB, tetW, sul2, and aph3′-III
2.4.2. Factors Associated with the Abundance of ermB, tetW, sul2, and aph3′-III
2.5. Phenotypic Resistance Identified by Minimum Inhibitory Concentrations
2.5.1. E. coli Resistance to Antimicrobials
2.5.2. Factors Associated with E. coli Resistance
2.6. Correlations between AMR Genes Abundances Detected by Metagenomics and qPCR
3. Discussion
4. Materials and Methods
4.1. Selection of Farms
4.2. Questionnaire Data: Antimicrobial Usage, Farm Characteristics, and Biosecurity
4.3. Quantification of AMU
4.4. Sampling and Processing of Faecal Samples
4.5. Metagenomic Sequencing and Processing Data
4.6. qPCR Analysis
4.7. E. coli Isolation and MIC Determination
4.8. Variable Selection and Statistical Analysis
4.8.1. Explanatory Variables
4.8.2. Factors Associated with AMR Gene Clusters Identified by Metagenomics Sequencing
4.8.3. Factors Associated with ermB, tetW, sul2, and aph3′-III Identified by qPCR
4.8.4. Factors Associated with E. coli Resistance
4.8.5. Comparisons between Metagenomics and qPCR
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
EFFORT-Group
Conflicts of Interest
References
- O’Neill, J. Antimicrobial Resistance: Tackling a Crisis for the Health and Wealth of Nations the Review on Antimicrobial Resistance Chaired. Available online: http://www.jpiamr.eu/wp-content/uploads/2014/12/AMR-Review-Paper-Tackling-a-crisis-for-the-health-and-wealth-of-nations_1-2.pdf (accessed on 1 May 2021).
- World Health Organization. Critically Important Antimicrobials for Human Medicine. Available online: https://apps.who.int/iris/bitstream/handle/10665/312266/9789241515528-eng.pdf (accessed on 1 May 2021).
- World Health Organization. Global Action Plan on Antimicrobial Resistance. Available online: https://apps.who.int/iris/bitstream/handle/10665/193736/9789241509763_eng.pdf?sequence=1 (accessed on 30 January 2021).
- European Comission. A European One Health Action Plan against Antimicrobial Resistance (AMR). Available online: https://ec.europa.eu/health/sites/health/files/antimicrobial_resistance/docs/amr_2017_action-plan.pdf (accessed on 30 March 2021).
- Mughini-Gras, L.; Dorado-García, A.; van Duijkeren, E.; van den Bunt, G.; Dierikx, C.M.; Bonten, M.J.M.; Bootsma, M.C.J.; Schmitt, H.; Hald, T.; Evers, E.G.; et al. Articles Attributable Sources of Community-Acquired Carriage of Echerichia coli Containing β-Lactam Antibiotic Resistance Genes: A Population-Based Modelling Study. Lancet 2019, 8, 357–369. [Google Scholar] [CrossRef] [Green Version]
- DG AGRI DASHBOARD: POULTRY MEAT. Available online: https://ec.europa.eu/info/sites/info/files/food-farming-fisheries/farming/documents/poultry-meat-dashboard_en.pdf (accessed on 29 March 2021).
- European Food Safety Authority and European Centre for Disease Prevention and Control. The European Union Summary Report on Antimicrobial Resistance in Zoonotic and Indicator Bacteria from Humans, Animals and Food in 2018/2019. EFSA J. 2021, 19, e06007. [Google Scholar] [CrossRef]
- Jones, E.M.; Snow, L.C.; Carrique-Mas, J.J.; Gosling, R.J.; Clouting, C.; Davies, R.H. Risk Factors for Antimicrobial Resistance in Echerichia coli Found in GB Turkey Flocks. Vet. Rec. 2013, 173, 422. [Google Scholar] [CrossRef] [PubMed]
- Taylor, N.M.; Wales, A.D.; Ridley, A.M.; Davies, R.H. Farm Level Risk Factors for Fluoroquinolone Resistance in E. coli and Thermophilic Campylobacter spp. on Poultry Farms. Avian Pathol. 2016, 45, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Agunos, A.; Gow, S.P.; Leger, D.F.; Carson, C.A.; Deckert, A.E.; Bosman, A.L.; Loest, D.; Irwin, R.J.; Reid-Smith, R.J. Antimicrobial Use and Antimicrobial Resistance Indicators—Integration of Farm-Level Surveillance Data from Broiler Chickens and Turkeys in British Columbia, Canada. Front. Vet. Sci. 2019, 6, 131. [Google Scholar] [CrossRef] [PubMed]
- Boulianne, M.; Arsenault, J.; Daignault, D.; Archambault, M.; Letellier, A.; Dutil, L. Drug Use and Antimicrobial Resistance among Echerichia coli and Enterococcus spp. Isolates from Chicken and Turkey Flocks Slaughtered in Quebec, Canada. Can. J. Vet. Res. 2016, 80, 49–59. [Google Scholar] [PubMed]
- Chuppava, B.; Keller, B.; Meißner, J.; Kietzmann, M.; Visscher, C. Effects of Different Types of Flooring Design on the Development of Antimicrobial Resistance in Commensal Echerichia coli in Fattening Turkeys. Vet. Microbiol. 2018, 217, 18–24. [Google Scholar] [CrossRef]
- Chuppava, B.; Keller, B.; El-Wahab, A.; Meißner, J.; Kietzmann, M.; Visscher, C. Resistance of Echerichia coli in Turkeys after Therapeutic or Environmental Exposition with Enrofloxacin Depending on Flooring. Int. J. Environ. Res. Public Health 2018, 15, 1993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- European Centre for Disease Prevention and Control, European Food Safety Authority and European Medicines Agency. ECDC/EFSA/EMA Second Joint Report on the Integrated Analysis of the Consumption of Antimicrobial Agents and Occurrence of Antimicrobial Resistance in Bacteria from Humans and Food-Producing Animals: Joint Interagency Antimicrobial Consumption and Resistan. EFSA J. 2017, 15, e05017. [Google Scholar] [CrossRef]
- European Food Safety Authority and European Centre for Disease Prevention and Control. The European Union Summary Report on Antimicrobial Resistance in Zoonotic and Indicator Bacteria from Humans, Animals and Food in 2017/2018. EFSA J. 2020, 18, e06007. [Google Scholar] [CrossRef] [Green Version]
- Dierikx, C.M.; van der Goot, J.A.; Smith, H.E.; Kant, A.; Mevius, D.J. Presence of ESBL/AmpC -Producing Echerichia coli in the Broiler Production Pyramid: A Descriptive Study. PLoS ONE 2013, 8, e79005. [Google Scholar] [CrossRef]
- Van Gompel, L.; Luiken, R.E.C.; Sarrazin, S.; Munk, P.; Knudsen, B.E.; Hansen, R.B.; Bossers, A.; Aarestrup, F.M.; Dewulf, J.; Wagenaar, J.A.; et al. The Antimicrobial Resistome in Relation to Antimicrobial Use and Biosecurity in Pig Farming, a Metagenome-Wide Association Study in Nine European Countries. J. Antimicrob. Chemother. 2019, 74, 865–876. [Google Scholar] [CrossRef] [PubMed]
- Luiken, R.E.C.; Van Gompel, L.; Munk, P.; Sarrazin, S.; Joosten, P.; Dorado-García, A.; Borup Hansen, R.; Knudsen, B.E.; Bossers, A.; Wagenaar, J.A.; et al. Associations between Antimicrobial Use and the Faecal Resistome on Broiler Farms from Nine European Countries. J. Antimicrob. Chemother. 2019, 74, 2596–2604. [Google Scholar] [CrossRef]
- Yang, D.; Van Gompel, L.; Luiken, R.E.C.; Sanders, P.; Joosten, P.; van Heijnsbergen, E.; Wouters, I.M.; Scherpenisse, P.; Chauvin, C.; Wadepohl, K.; et al. Association of Antimicrobial Usage with Faecal Abundance of aph(3’)-III, ermB, sul2 and tetW Resistance Genes in Veal Calves in Three European Countries. Int. J. Antimicrob. Agents 2020, 56, 106131. [Google Scholar] [CrossRef] [PubMed]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of Acquired Antimicrobial Resistance Genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef] [PubMed]
- Ceccarelli, D.; Hesp, A.; van der Goot, J.; Joosten, P.; Sarrazin, S.; Wagenaar, J.A.; Dewulf, J.; Mevius, D.J. Antimicrobial Resistance Prevalence in Commensal Echerichia coli from Broilers, Fattening Turkeys, Fattening Pigs and Veal Calves in European Countries and Association with Antimicrobial Usage at Country Level. J. Med. Microbiol. 2020, 69, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Caucci, C.; Di Martino, G.; Dalla Costa, A.; Santagiuliana, M.; Lorenzetto, M.; Capello, K.; Mughini-Gras, L.; Gavazzi, L.; Bonfanti, L. Trends and Correlates of Antimicrobial Use in Broiler and Turkey Farms: A Poultry Company Registry-Based Study in Italy. J. Antimicrob. Chemother. 2019, 74, 2784–2787. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Vanderhaeghen, W.; Fertner, M.; Fuchs, K.; Obritzhauser, W.; Agunos, A.; Carson, C.; Borck Høg, B.; Dalhoff Andersen, V.; Chauvin, C.; et al. Monitoring of Farm-Level Antimicrobial Use to Guide Stewardship: Overview of Existing Systems and Analysis of Key Components and Processes. Front. Vet. Sci. 2020, 7, 540. [Google Scholar] [CrossRef]
- Joosten, P.; Sarrazin, S.; Van Gompel, L.; Luiken, R.E.C.; Mevius, D.J.; Wagenaar, J.A.; Heederik, D.J.J.; Dewulf, J.; Graveland, H.; Schmitt, H.; et al. Quantitative and Qualitative Analysis of Antimicrobial Usage at Farm and Flock Level on 181 Broiler Farms in Nine European Countries. J. Antimicrob. Chemother. 2019, 74, 798–806. [Google Scholar] [CrossRef]
- Munk, P.; Knudsen, B.E.; Lukjacenko, O.; Duarte, A.S.R.; Van Gompel, L.; Luiken, R.E.C.; Smit, L.A.M.; Schmitt, H.; Garcia, A.D.; Hansen, R.B.; et al. Abundance and Diversity of the Faecal Resistome in Slaughter Pigs and Broilers in Nine European Countries. Nat. Microbiol. 2018, 3, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Skarżyńska, M.; Leekitcharoenphon, P.; Hendriksen, R.S.; Aarestrup, F.M.; Wasyl, D. A Metagenomic Glimpse into the Gut of Wild and Domestic Animals: Quantification of Antimicrobial Resistance and More. PLoS ONE 2020, 15, e0242987. [Google Scholar] [CrossRef] [PubMed]
- Joyce, A.; McCarthy, C.G.P.; Murphy, S.; Walsh, F. Antibiotic Resistomes of Healthy Pig Faecal Metagenomes. Microb. Genom. 2019, 5, e000272. [Google Scholar] [CrossRef]
- Davies, R.; Wales, A. Antimicrobial Resistance on Farms: A Review Including Biosecurity and the Potential Role of Disinfectants in Resistance Selection. Compr. Rev. Food Sci. Food Saf. 2019, 18, 753–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alba, P.; Leekitcharoenphon, P.; Franco, A.; Feltrin, F.; Ianzano, A.; Caprioli, A.; Stravino, F.; Hendriksen, R.S.; Bortolaia, V.; Battisti, A. Molecular Epidemiology of mcr-Encoded Colistin Resistance in Enterobacteriaceae from Food-Producing Animals in Italy Revealed through the EU Harmonized Antimicrobial Resistance Monitoring. Front. Microbiol. 2018, 9, 1217. [Google Scholar] [CrossRef] [PubMed]
- Saliu, E.M.; Vahjen, W.; Zentek, J. Types and Prevalence of Extended-Spectrum Beta-Lactamase Producing Enterobacteriaceae in Poultry. Anim. Health Res. Rev. 2017, 18, 46–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andersen, V.D.; Jensen, M.S.; Munk, P.; Vigre, H. Robustness in Quantifying the Abundance of Antimicrobial Resistance Genes in Pooled Faeces Samples from Batches of Slaughter Pigs Using Metagenomics Analysis. J. Glob. Antimicrob. Resist. 2021, 24, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Andersen, S.C.; Fachmann, M.S.R.; Kiil, K.; Nielsen, E.M.; Hoorfar, J. Gene-Based Pathogen Detection: Can We Use qPCR to Predict the Outcome of Diagnostic Metagenomics? Genes 2017, 8, 332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bortolaia, V.; Kaas, R.S.; Ruppe, E.; Roberts, M.C.; Schwarz, S.; Cattoir, V.; Philippon, A.; Allesoe, R.L.; Rebelo, A.R.; Florensa, A.F.; et al. ResFinder 4.0 for Predictions of Phenotypes from Genotypes. J. Antimicrob. Chemother. 2020, 75, 3491–3500. [Google Scholar] [CrossRef] [PubMed]
- Starke, R.; Pylro, V.S.; Morais, D.K. 16S rRNA Gene Copy Number Normalization Does Not Provide More Reliable Conclusions in Metataxonomic Surveys. Microb. Ecol. 2021, 81, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Doebeli, M.; Parfrey, L.W. Correcting for 16S rRNA Gene Copy Numbers in Microbiome Surveys Remains an Unsolved Problem. Microbiome 2018, 6, 41. [Google Scholar] [CrossRef]
- European Medicines Agency. Revised ESVAC Reflection Paper on Collecting Data on Consumption of Antimicrobial Agents per Animal Species, on Technical Units of Measurement and Indicators for Reporting Consumption of Antimicrobial Agents in Animals. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/revised-european-surveillance-veterinary-antimicrobial-consumption-esvac-reflection-paper-collecting_en.pdf (accessed on 30 March 2021).
- European Medicines Agency. Questions and Answers on Veterinary Medicinal Products Containing a Combination of Lincomycin and Spectinomycin to Be Administered Orally to Pigs and/or Poultry. Lincomycin and Spectinomycin Article 35 Referral—Annex I, II, III. Available online: https://www.ema.europa.eu/en/documents/referral/lincomycin-spectinomycin-article-35-referral-annex-i-ii-iii_en.pdf (accessed on 15 June 2021).
- Duarte, A.S.R.; Röder, T.; Van Gompel, L.; Petersen, T.N.; Hansen, R.B.; Hansen, I.M.; Bossers, A.; Aarestrup, F.M.; Wagenaar, J.A.; Hald, T. Metagenomics-Based Approach to Source-Attribution of Antimicrobial Resistance Determinants—Identification of Reservoir Resistome Signatures. Front. Microbiol. 2021, 11, 601407. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, B.E.; Bergmark, L.; Munk, P.; Lukjancenko, O.; Priemé, A.; Aarestrup, F.M.; Pamp, S.J. Impact of Sample Type and DNA Isolation Procedure on Genomic Inference of Microbiome Composition. mSystems 2016, 1, e00095-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osakunor, D.N.M.; Munk, P.; Mduluza, T.; Petersen, T.N.; Brinch, C.; Ivens, A.; Chimponda, T.; Amanfo, S.A.; Murray, J.; Woolhouse, M.E.J.; et al. The Gut Microbiome but Not the Resistome Is Associated with Urogenital Schistosomiasis in Preschool-Aged Children. Commun. Biol. 2020, 3, 155. [Google Scholar] [CrossRef] [Green Version]
- Macedo, G.; van Veelen, H.P.J.; Hernandez-Leal, L.; van der Maas, P.; Heederik, D.; Mevius, D.; Bossers, A.; Schmitt, H. Science of the Total Environment Targeted Metagenomics Reveals Inferior Resilience of Farm Soil Resistome Compared to Soil Microbiome after Manure Application. Sci. Total Environ. 2021, 770, 145399. [Google Scholar] [CrossRef]
- Van Gompel, L.; Dohmen, W.; Luiken, R.E.C.; Bouwknegt, M.; Heres, L.; van Heijnsbergen, E.; Jongerius-Gortemaker, B.G.M.; Scherpenisse, P.; Greve, G.D.; Tersteeg-Zijderveld, M.H.G.; et al. Occupational Exposure and Carriage of Antimicrobial Resistance Genes (tetW, ermB) in Pig Slaughterhouse Workers. Ann. Work Expo. Health 2020, 64, 125–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rödiger, S.; Burdukiewicz, M.; Schierack, P. ChipPCR: An R Package to Pre-Process Raw Data of Amplification Curves. Bioinformatics 2015, 31, 2900–2902. [Google Scholar] [CrossRef] [Green Version]
- Hille, K.; Felski, M.; Ruddat, I.; Woydt, J.; Schmid, A.; Friese, A.; Fischer, J.; Sharp, H.; Valentin, L.; Michael, G.B.; et al. Association of Farm-Related Factors with Characteristics Profiles of Extended-Spectrum β-Lactamase- / Plasmid-Mediated AmpC β-Lactamase-Producing Echerichia coli Isolates from German Livestock Farms. Vet. Microbiol. 2018, 223, 93–99. [Google Scholar] [CrossRef]
- Birkegård, A.C.; Halasa, T.; Græsbøll, K.; Clasen, J.; Folkesson, A.; Toft, N. Association between Selected Antimicrobial Resistance Genes and Antimicrobial Exposure in Danish Pig Farms. Sci. Rep. 2017, 7, 9683. [Google Scholar] [CrossRef] [Green Version]
- Viechtbauer, W. Conducting Meta-Analyses in {R} with the {metafor} Package. J. Stat. Softw. 2010, 36, 1–48. [Google Scholar] [CrossRef] [Green Version]
- Yoav Benjamini, Y.H. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kuznetsova, A.; Brockhoff, P.B.; Christensen, R.H.B. {lmerTest} Package: Tests in Linear Mixed Effects Models. J. Stat. Softw. 2017, 82, 1–26. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Characteristics | Country | Overall | ||
|---|---|---|---|---|
| B | E | H | ||
| Farm Information | ||||
| Included farms, n | 20 | 20 | 20 | 60 |
| No. of turkeys present on the farm, median (Min-Max) | 12,683 (5000–46,500) | 7275 (2950–38,000) | 12,609 (4404–56,850) | 10,055 (2950–56,850) |
| Farms where other livestock is present, n (%) | 4 (20) | 11 (55) | 4 (20) | 19 (32) |
| No. of people working at the farm, median (Min-Max) | 2 (1–28) | 1.5 (1–3) | 1 (1–4) | 1.5 (1–28) |
| Farms sampled in spring and summer, n (%) | 4 (20) | 5 (25) | 20 (100) | 29 (48) |
| Flock Information | ||||
| No. of turkeys at sampling, median (Min-Max) a | 4213 (2050–11,660) | 4140 (450–9155) | 6422 (302–21,356) | 4710 (450–21,356) |
| No. of turkeys at set-up in the current round in the sampled house, median (Min-Max) b | 5040 (2997–13,000) | 9180 (4240–22,000) | 7020 (3000–21,794) | 7850 (2997–22,000) |
| Weight of turkeys at set-up, kg, median (Min-Max) c | 1.5 (0.1–6.4) | 0.1 (0.1–0.5) | 1.1 (0.9–1.3) | 1.1 (0.1–6.4) |
| Age of turkeys at sampling, days, median (Min-Max) b | 134 (96–147) | 116 (74–140) | 101 (86–118) | 115 (74–147) |
| Average expected age at delivery to slaughter, days, median (Min-Max) b | 146 (106–154) | 109 (79–138) | 117 (95–127) | 118 (79–154) |
| Biosecurity at the Farm | ||||
| Visitor access more than once a month (family members, technicians, etc), n (%) | 8 (40) | 20 (100) | 16 (80) | 44 (73) |
| Outdoor access possible for turkeys, n (%) | 14 (70) | 0 (0) | 0 (0) | 14 (23) |
| Different age categories of turkeys present, n (%) | 10 (50) | 5 (25) | 0 (0) | 15 (25) |
| Bird- and vermin-proof grids placed on the air inlets, n (%) | 20 (100) | 15 (75) | 18 (90) | 53 (88) |
| Staff keeps turkeys or birds at home, n (%) | 2 (10) | 7 (35) | 1 (5) | 10 (17) |
| Disinfecting footbaths present on the farm, n (%) | 14 (70) | 10 (50) | 10 (50) | 34 (57) |
| The nearest turkey farm within 500 m, n (%) | 4 (20) | 5 (25) | 4 (20) | 13 (22) |
| Other livestock farm present within 500 m, n (%) | 12 (60) | 18 (90) | 7 (35) | 37 (62) |
| Wild birds can enter the stables, n (%) | 1 (5) | 6 (30) | 8 (40) | 15 (25) |
| AMU | AMR Gene Cluster a | Estimate | Adjusted p Value b | 95% CI | Country and Number of Farms with Reported AMU |
|---|---|---|---|---|---|
| Log10 TI beta-lactam | Beta-lactam | 1.06 | 0.033 | [0.29–1.84] | B-15, E-14, H-18 |
| Log10 TI polymixin | Colistin | 0.99 | 0.033 | [0.29–1.69] | B-4, E-11, H-5 |
| Aminoglycosides or spectinomycin used (ref:no) | Aminoglycoside | 0.92 | 0.097 | [0.08–1.76] | B-3, H-3 |
| Trimethoprim-sulphonamides used (ref:no) | Trimethoprim | 0.78 | 0.221 | [−0.15–1.71] | B-2, E-3 |
| Trimethoprim-sulphonamides used (ref:no) | Sulphonamide | 0.68 | 0.282 | [−0.26–1.61] | B-2, E-3 |
| Log10 TI quinolone | Quinolone | 0.69 | 0.338 | [−0.43–1.81] | B-5, E-4, H-12 |
| Log10 TI tetracyclines | Tetracycline | 0.09 | 0.948 | [−0.82–1.00] | B-6, E-6, H-9 |
| Log10 TI macrolides + lincomycin | Macrolide | −0.17 | 0.948 | [−1.35–1.01] | B-6, E-12, H-7 |
| Log10 TI total AMU | Total FPKM | −0.02 | 0.948 | [−0.62–0.58] | B-15, E-17, H-18 |
| Model Variables | ermB | tetW | sul2 | aph3′-III | ||||
|---|---|---|---|---|---|---|---|---|
| GMR | [95% CI] | GMR | [95% CI] | GMR | [95% CI] | GMR | [95% CI] | |
| AMU | ||||||||
| Log10 TI macrolides + lincomycin | 1.57 | [0.77, 3.23] | ||||||
| Log10 TI tetracyclines | 1.54 | [0.80, 2.97] | ||||||
| Trimethoprim-sulphonamides used (ref:no) | 7.38 | [1.61, 33.8] | ||||||
| Aminoglycosides or spectinomycin used (ref:no) | 1.47 | [0.42, 5.14] | ||||||
| Technical farm characteristics | ||||||||
| Age of turkeys at sampling (standardized) | 0.73 | [0.54, 0.98] | ||||||
| Other livestock present (ref:no) | 2.89 | [1.17, 7.14] | 0.38 | [0.15, 0.95] | ||||
| Sampling season (ref: autumn, winter) | 0.21 | [0.09, 0.48] | ||||||
| Biosecurity | ||||||||
| Visitor access more than once a month (ref:no) | 0.41 | [0.22, 0.75] | 0.36 | [0.21, 0.60] | ||||
| Outdoor access possible for turkeys (ref:no) | 0.35 | [0.17, 0.75] | 0.37 | [0.19, 0.74] | ||||
| Different age categories of turkeys present (ref:no) | 0.45 | [0.25, 0.83] | ||||||
| Bird- and vermin-proof grids placed on the air inlets (ref:no) | 6.32 | [1.76, 22.73] | ||||||
| Staff keeps turkeys or birds at home (ref:no) | 0.27 | [0.09, 0.83] | ||||||
| Model Variables | AMP | TET | CIP | |||
|---|---|---|---|---|---|---|
| OR | [95% CI] | OR | [95% CI] | OR | [95% CI] | |
| AMU | ||||||
| Log10 TI aminopenicillins | 4.10 | [1.37, 12.30] | ||||
| Log10 TI tetracyclines | 3.32 | [0.75, 14.7] | ||||
| Log10 TI quinolones | 12.85 | [4.00, 41.2] | ||||
| Technical farm characteristics | ||||||
| Age of turkeys at sampling (standardized) | 0.83 | [0.53, 1.31] | 0.74 | [0.48, 1.13] | ||
| Sampling season (ref: autumn, winter) | 2.13 | [0.85, 5.31] | ||||
| Biosecurity | ||||||
| Other livestock farms present within 500 m (ref: no) | 0.48 | [0.19, 1.18] | ||||
| Wild birds can enter the stables (ref: no) | 2.67 | [0.90, 7.87] | ||||
| Different age categories of turkeys present (ref: no) | 0.48 | [0.19, 1.20] | ||||
| The nearest turkey farm within 500 m (ref: no) | 0.28 | [0.11, 0.69] | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Horie, M.; Yang, D.; Joosten, P.; Munk, P.; Wadepohl, K.; Chauvin, C.; Moyano, G.; Skarżyńska, M.; Dewulf, J.; Aarestrup, F.M.; et al. Risk Factors for Antimicrobial Resistance in Turkey Farms: A Cross-Sectional Study in Three European Countries. Antibiotics 2021, 10, 820. https://doi.org/10.3390/antibiotics10070820
Horie M, Yang D, Joosten P, Munk P, Wadepohl K, Chauvin C, Moyano G, Skarżyńska M, Dewulf J, Aarestrup FM, et al. Risk Factors for Antimicrobial Resistance in Turkey Farms: A Cross-Sectional Study in Three European Countries. Antibiotics. 2021; 10(7):820. https://doi.org/10.3390/antibiotics10070820
Chicago/Turabian StyleHorie, Mayu, Dongsheng Yang, Philip Joosten, Patrick Munk, Katharina Wadepohl, Claire Chauvin, Gabriel Moyano, Magdalena Skarżyńska, Jeroen Dewulf, Frank M. Aarestrup, and et al. 2021. "Risk Factors for Antimicrobial Resistance in Turkey Farms: A Cross-Sectional Study in Three European Countries" Antibiotics 10, no. 7: 820. https://doi.org/10.3390/antibiotics10070820
APA StyleHorie, M., Yang, D., Joosten, P., Munk, P., Wadepohl, K., Chauvin, C., Moyano, G., Skarżyńska, M., Dewulf, J., Aarestrup, F. M., Blaha, T., Sanders, P., Gonzalez-Zorn, B., Wasyl, D., Wagenaar, J. A., Heederik, D., Mevius, D., Schmitt, H., Smit, L. A. M., & Van Gompel, L., on behalf of the EFFORT-Group . (2021). Risk Factors for Antimicrobial Resistance in Turkey Farms: A Cross-Sectional Study in Three European Countries. Antibiotics, 10(7), 820. https://doi.org/10.3390/antibiotics10070820