
Identification of Therapeutic Targets in an Emerging Gastrointestinal Pathogen Campylobacter ureolyticus and Possible Intervention through Natural Products


 , , , ,
, , , ,
Abstract
:1. Introduction
2. Results
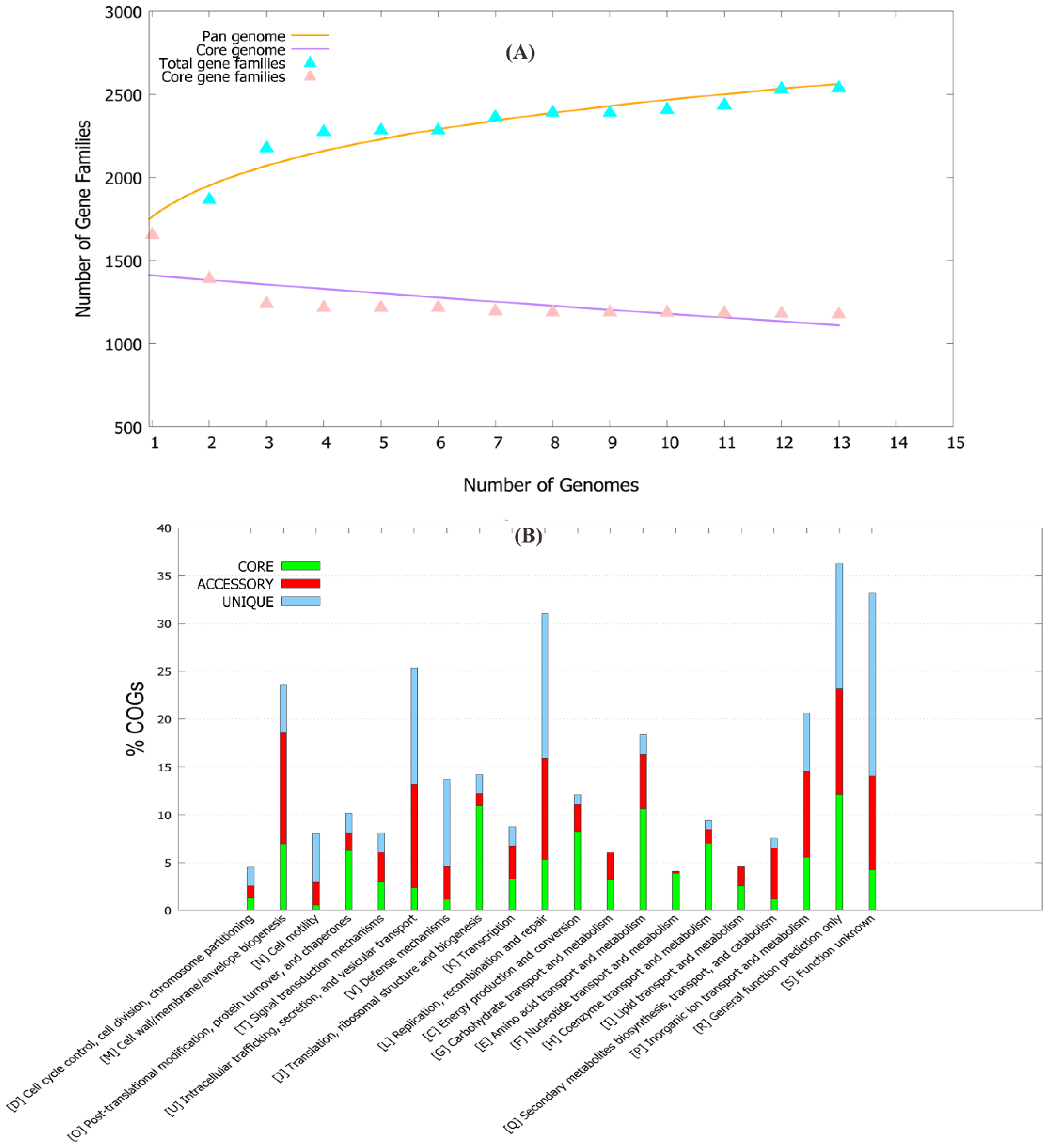
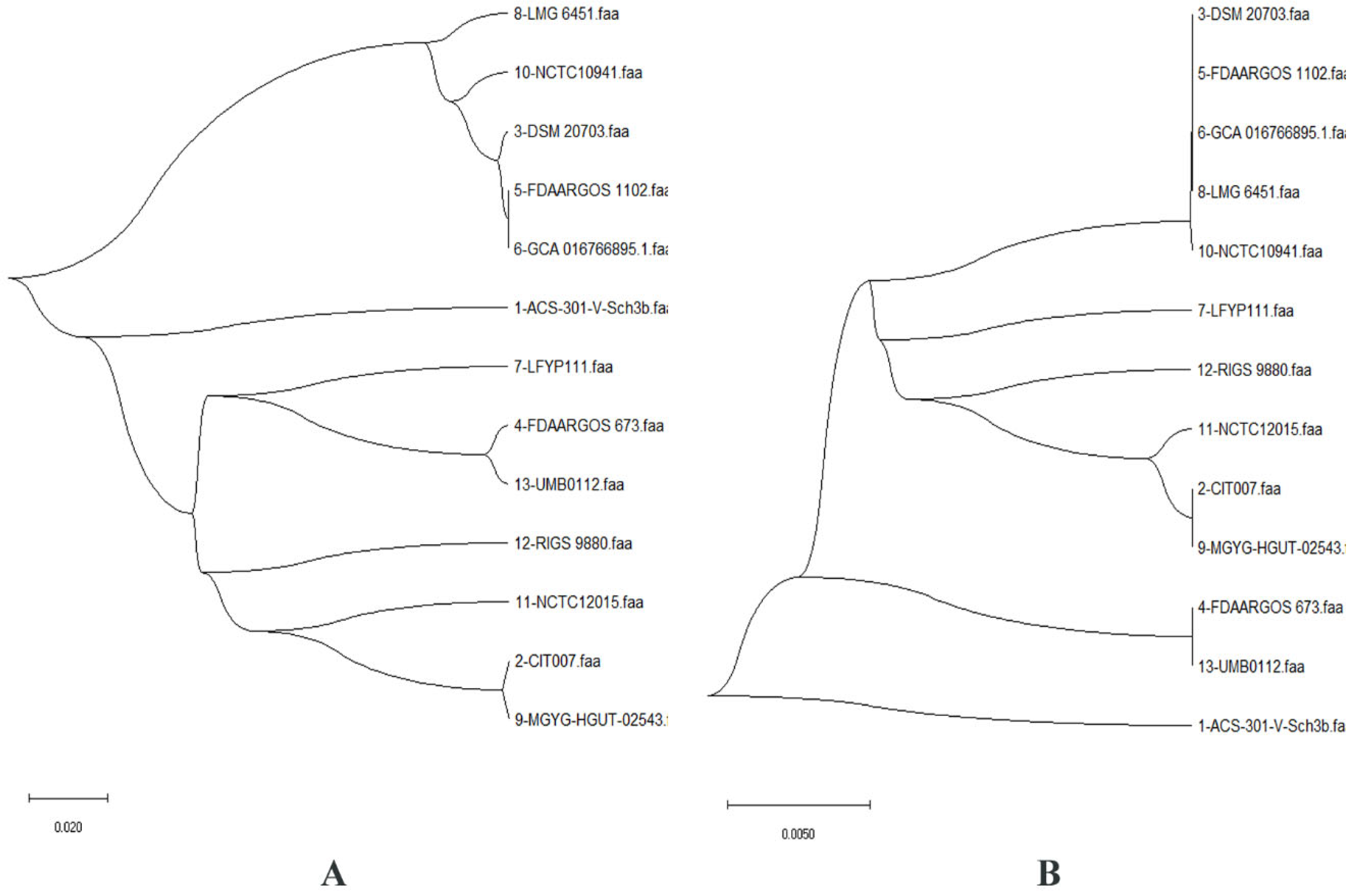
2.1. Pan-Genome and Resistome Analysis
2.2. Differential Sequence Mining and Therapeutic Target Identification
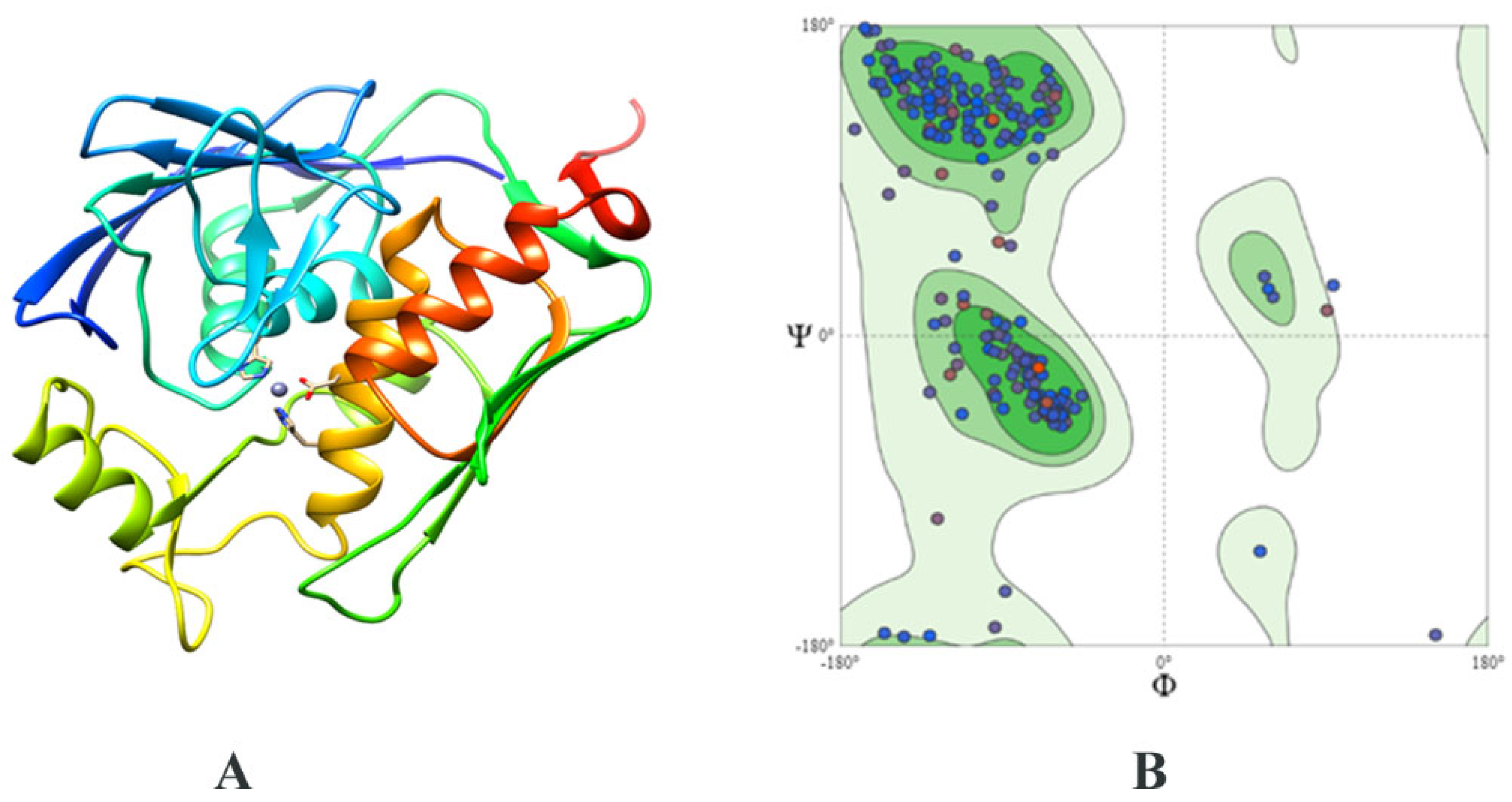
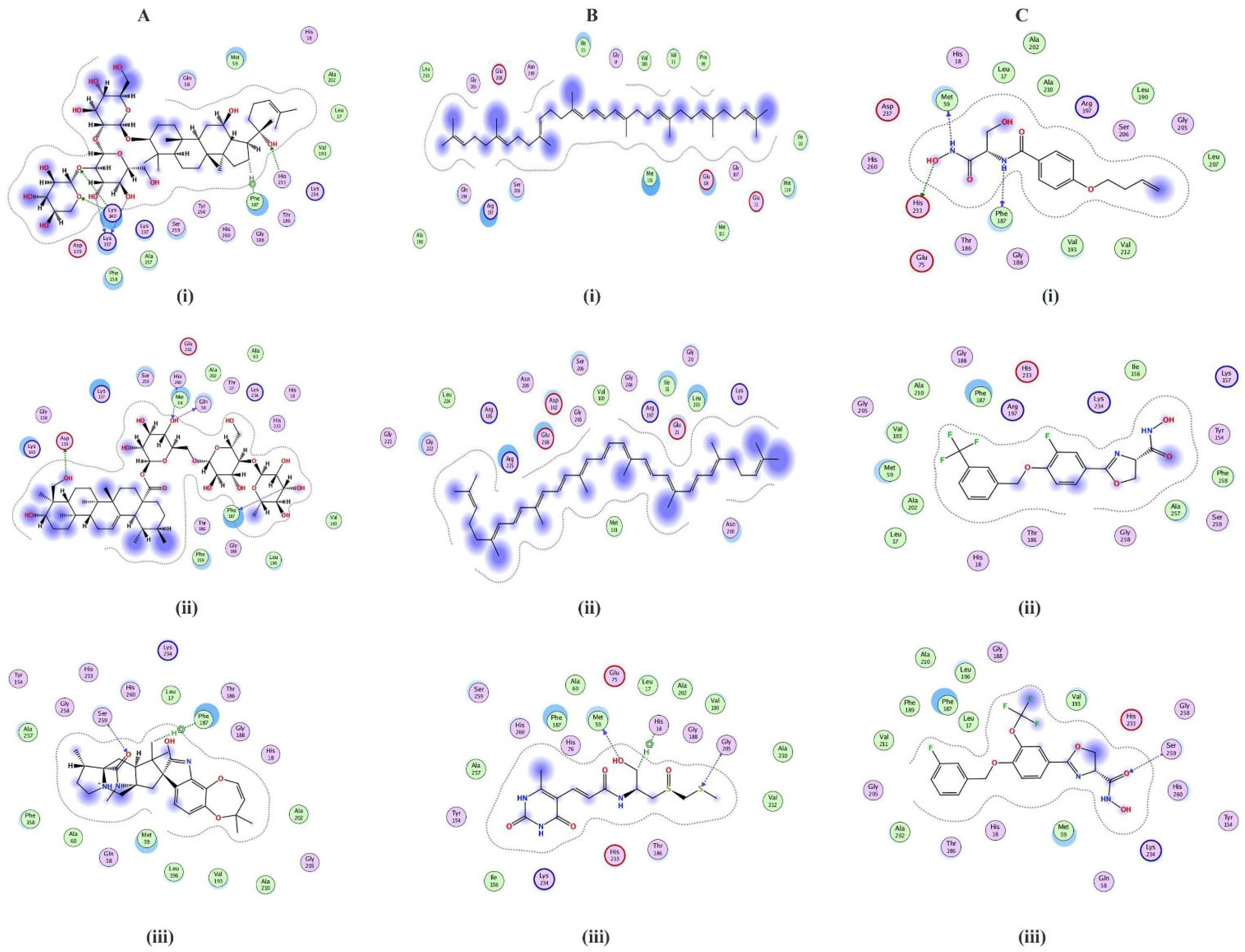
2.3. Structure Prediction and Inhibitor Screening
2.4. ADMET Profiling of Shortlisted Compounds
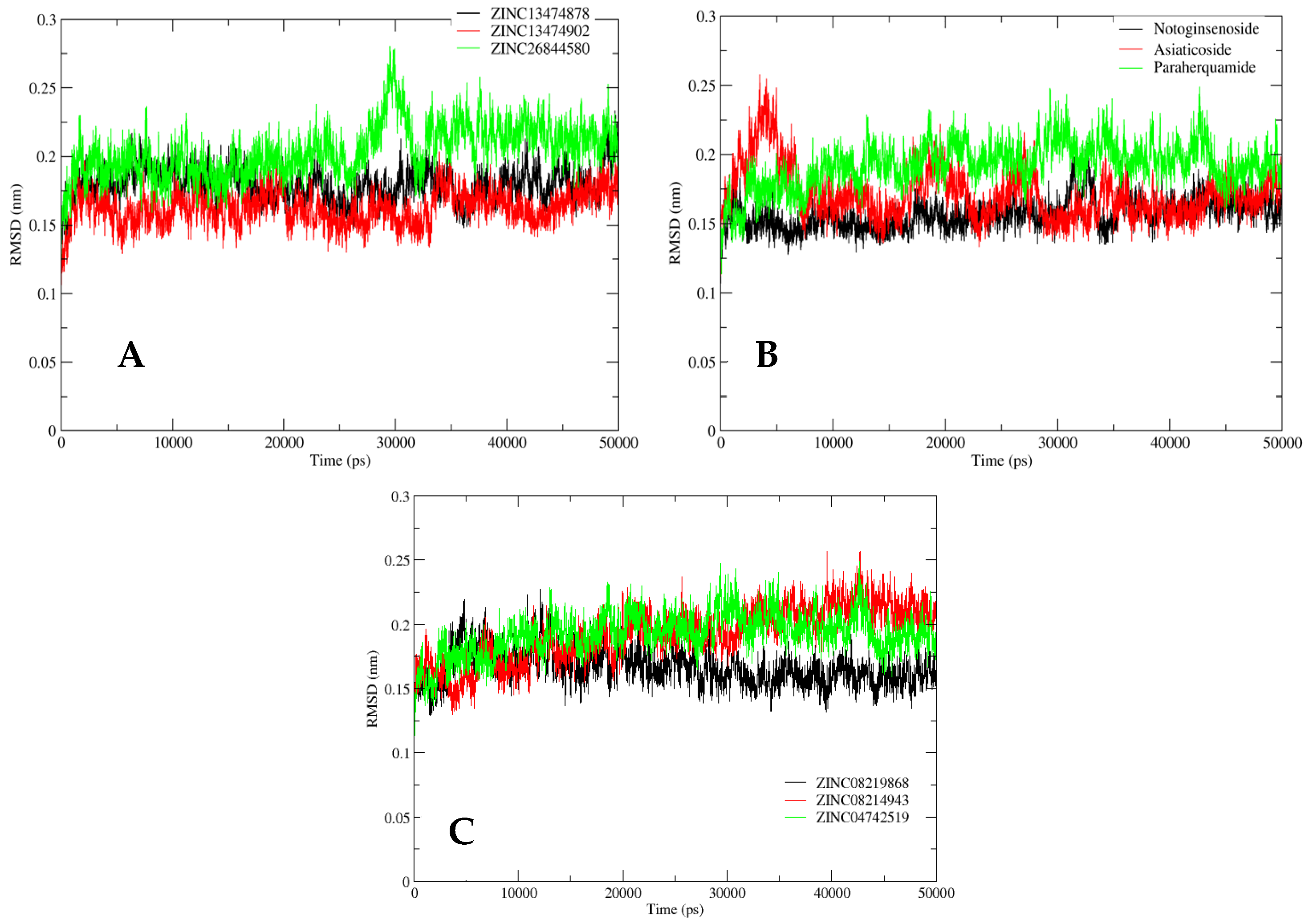
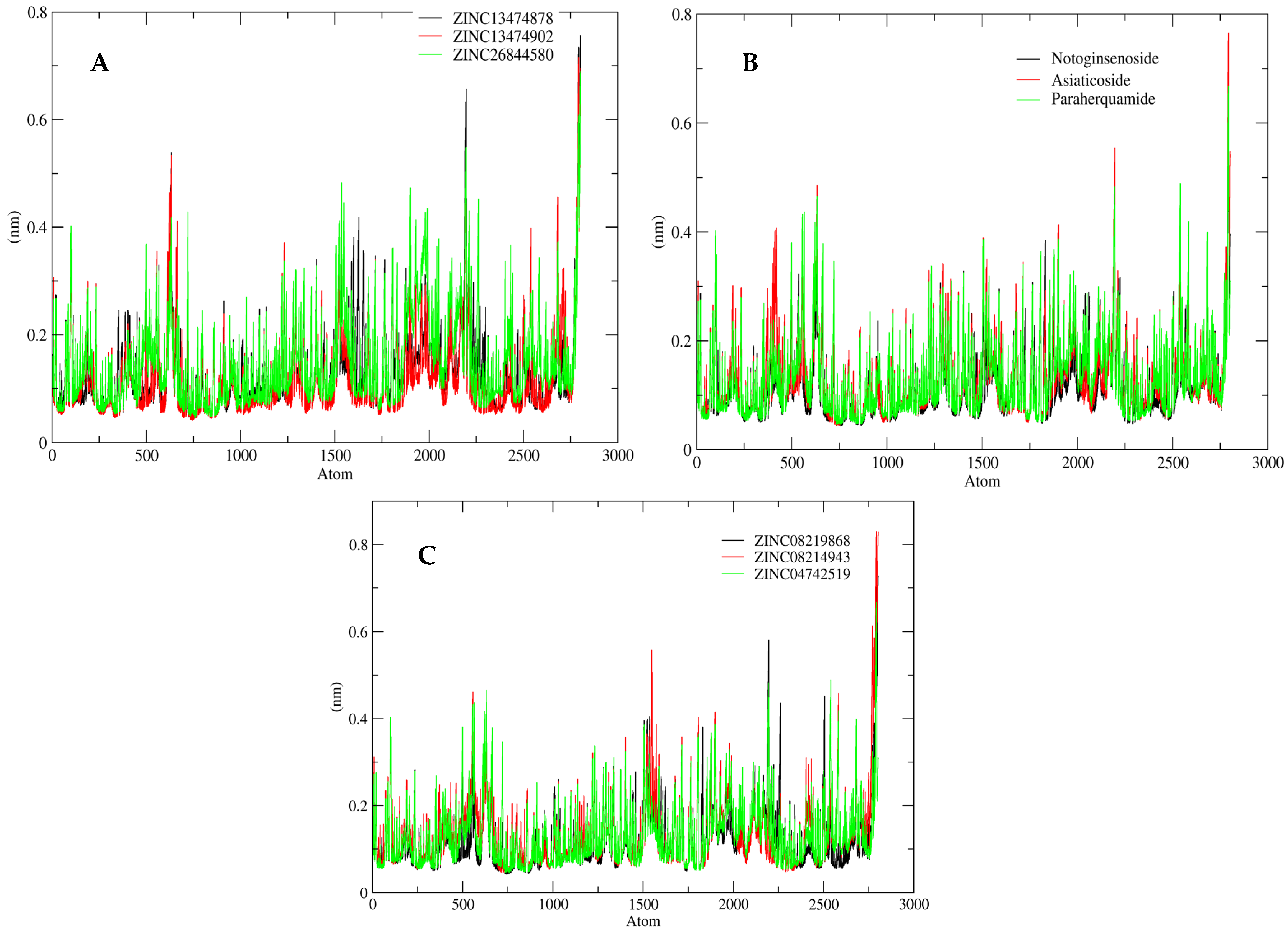
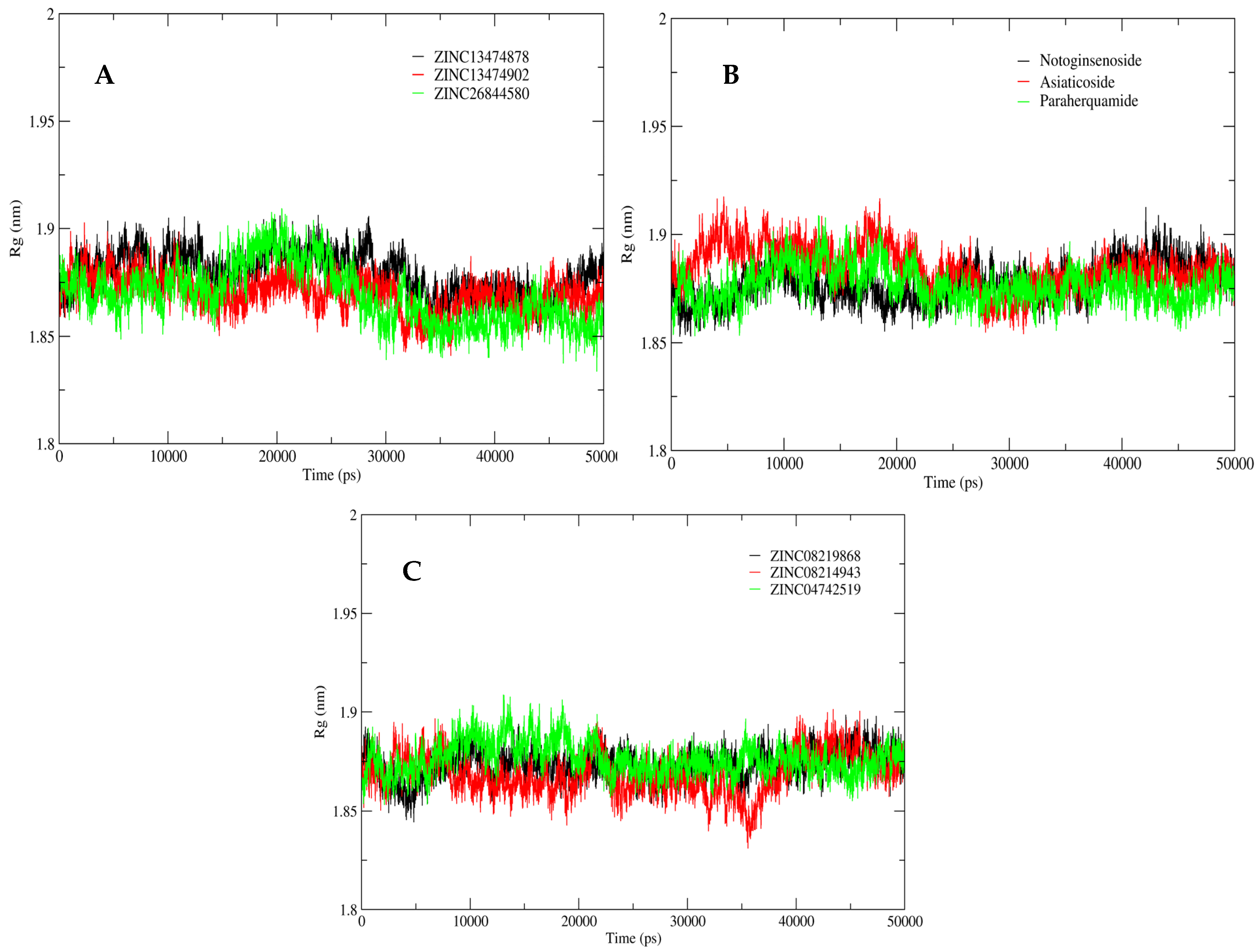
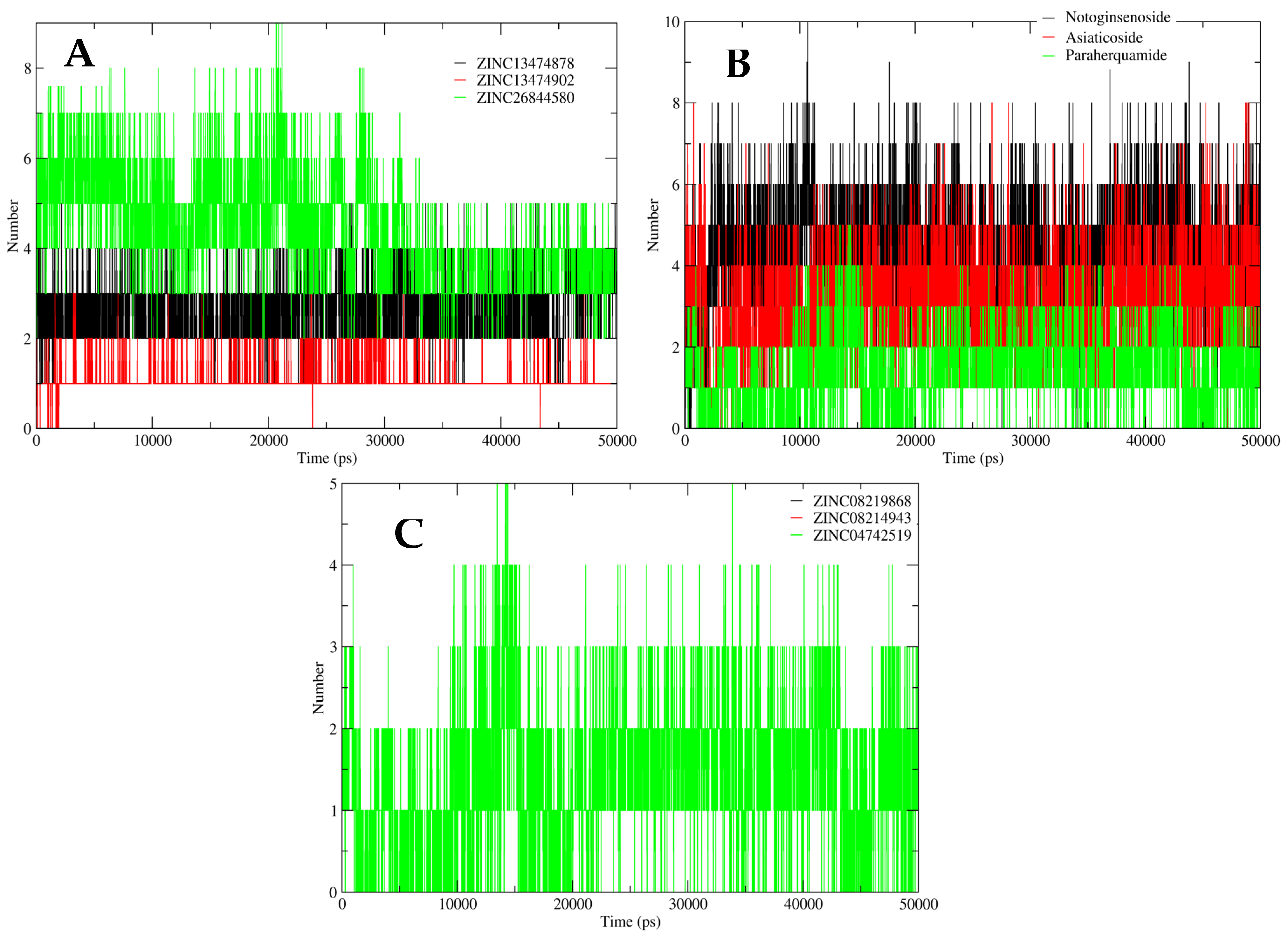
2.5. MD Simulation Analysis
3. Discussion
4. Materials and Methods
4.1. Data Retrieval
4.2. Pan-Genomic Analysis
4.3. Drug Target Prioritization
4.4. Structural Modeling
4.5. Virtual Screening
4.6. ADMET Profiling of the Shortlisted Drug Candidates
4.7. Molecular Dynamics (MD) Simulation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burgos-Portugal, J.A.; Kaakoush, N.O.; Raftery, M.J.; Mitchell, H.M. Pathogenic potential of Campylobacter ureolyticus. Infect Immun. 2012, 80, 883–890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bullman, S.; O’Leary, J.; Lucey, B.; Byrne, D.; Sleator, R.D. Campylobacter ureolyticus: An emerging gastrointestinal pathogen? FEMS Immunol. Med. Microbiol. 2011, 61, 228–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, H.; Vera, F.; Villanueva, M.P.; García, A. Occurrence of Campylobacter species in healthy well-nourished and malnourished children. Braz. J. Microbiol. 2008, 39, 56–58. [Google Scholar] [CrossRef]
- Daisy, P.; Singh, S.K.; Vijayalakshmi, P.; Selvaraj, C.; Rajalakshmi, M.; Suveena, S. A database for the predicted pharmacophoric features of medicinal compounds. Bioinformation 2011, 6, 167. [Google Scholar] [CrossRef] [Green Version]
- Serichantalergs, O.; Ruekit, S.; Pandey, P.; Anuras, S.; Mason, C.; Bodhidatta, L.; Swierczewski, B. Incidence of Campylobacter concisus and C. ureolyticus in traveler’s diarrhea cases and asymptomatic controls in Nepal and Thailand. Gut Pathog. 2017, 9, 47. [Google Scholar] [CrossRef] [Green Version]
- Boggess, K.A.; Trevett, T.N.; Madianos, P.N.; Rabe, L.; Hillier, S.L.; Beck, J.; Offenbacher, S. Use of DNA hybridization to detect vaginal pathogens associated with bacterial vaginosis among asymptomatic pregnant women. Am. J. Obs. Gynecol. 2005, 193, 752–756. [Google Scholar] [CrossRef]
- O’Donovan, D.; Corcoran, G.D.; Lucey, B.; Sleator, R.D. Campylobacter ureolyticus: A portrait of the pathogen. Virulence 2014, 5, 498–506. [Google Scholar] [CrossRef] [Green Version]
- Bennett, K.W.; Eley, A.; Woolley, P.D.; Duerden, B.I. Isolation of Bacteroides ureolyticus from the genital tract of men with and without non-gonococcal urethritis. Eur. J. Clin. Microbiol. Infect. Dis. 1990, 9, 825–826. [Google Scholar] [CrossRef]
- Basic, A.; Enerbäck, H.; Waldenström, S.; Östgärd, E.; Suksuart, N.; Dahlen, G. Presence of Helicobacter pylori and Campylobacter ureolyticus in the oral cavity of a Northern Thailand population that experiences stomach pain. J. Oral. Microbiol. 2018, 10, 1527655. [Google Scholar] [CrossRef] [Green Version]
- Fraczek, M.; Piasecka, M.; Gaczarzewicz, D.; Szumala-Kakol, A.; Kazienko, A.; Lenart, S.; Laszczynska, M.; Kurpisz, M. Membrane stability and mitochondrial activity of human-ejaculated spermatozoa during in vitro experimental infection with Escherichia coli, Staphylococcus haemolyticus and Bacteroides ureolyticus. Andrologia 2012, 44, 315–329. [Google Scholar] [CrossRef]
- Mukhopadhya, I.; Thomson, J.M.; Hansen, R.; Berry, S.H.; El-Omar, E.M.; Hold, G.L. Detection of Campylobacter concisus and other Campylobacter species in colonic biopsies from adults with ulcerative colitis. PLoS ONE 2011, 6, e21490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, D.; Iraola, G. Pathogenomics of emerging Campylobacter species. Clin. Microbiol. Rev. 2019, 32, e00072-18. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.P.-E.F.; Raoult, D. Genome and pan-genome analysis to classify emerging bacteria. Biol. Direct 2019, 14, 5. [Google Scholar] [CrossRef] [Green Version]
- Spreafico, R.; Soriaga, L.B.; Grosse, J.; Virgin, H.W.; Telenti, A. Advances in Genomics for Drug Development. Genes 2020, 11, 942. [Google Scholar] [CrossRef] [PubMed]
- Kareem, S.M.; Al-Kadmy, I.M.S.; Kazaal, S.S.; Ali, A.N.M.; Aziz, S.N.; Makharita, R.R.; Algammal, A.M.; Al-Rejaie, S.; Behl, T.; Batiha, G.E.; et al. Detection of gyrA and parC Mutations and Prevalence of Plasmid-Mediated Quinolone Resistance Genes in Klebsiella pneumoniae. Infect. Drug Resist. 2021, 14, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Dahl, L.G.; Joensen, K.G.; Osterlund, M.T.; Kiil, K.; Nielsen, E.M. Prediction of antimicrobial resistance in clinical Campylobacter jejuni isolates from whole-genome sequencing data. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 673–682. [Google Scholar] [CrossRef]
- Elhadidy, M.; Ali, M.M.; El-Shibiny, A.; Miller, W.G.; Elkhatib, W.F.; Botteldoorn, N.; Dierick, K. Antimicrobial resistance patterns and molecular resistance markers of Campylobacter jejuni isolates from human diarrheal cases. PLoS ONE 2020, 15, e0227833. [Google Scholar] [CrossRef] [Green Version]
- Haldenby, S.; Bronowski, C.; Nelson, C.; Kenny, J.; Martinez-Rodriguez, C.; Chaudhuri, R.; Williams, N.J.; Forbes, K.; Strachan, N.J.; Pulman, J.; et al. Increasing prevalence of a fluoroquinolone resistance mutation amongst Campylobacter jejuni isolates from four human infectious intestinal disease studies in the United Kingdom. PLoS ONE 2020, 15, e0227535. [Google Scholar] [CrossRef] [Green Version]
- Liao, Y.S.; Chen, B.H.; Teng, R.H.; Wang, Y.W.; Chang, J.H.; Liang, S.Y.; Tsao, C.S.; Hong, Y.P.; Sung, H.Y.; Chiou, C.S. Antimicrobial Resistance in Campylobacter coli and Campylobacter jejuni from Human Campylobacteriosis in Taiwan, 2016 to 2019. Antimicrob. Agents Chemother. 2022, 66, e0173621. [Google Scholar] [CrossRef]
- Espinoza, N.; Rojas, J.; Pollett, S.; Meza, R.; Patino, L.; Leiva, M.; Camina, M.; Bernal, M.; Reynolds, N.D.; Maves, R.; et al. Validation of the T86I mutation in the gyrA gene as a highly reliable real time PCR target to detect Fluoroquinolone-resistant Campylobacter jejuni. BMC Infect. Dis. 2020, 20, 518. [Google Scholar] [CrossRef]
- Voha, C.; Docquier, J.D.; Rossolini, G.M.; Fosse, T. Genetic and biochemical characterization of FUS-1 (OXA-85), a narrow-spectrum class D beta-lactamase from Fusobacterium nucleatum subsp. polymorphum. Antimicrob. Agents Chemother. 2006, 50, 2673–2679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bidell, M.R.; Lodise, T.P. Use of oral tetracyclines in the treatment of adult outpatients with skin and skin structure infections: Focus on doxycycline, minocycline, and omadacycline. Pharmacotherapy 2021, 41, 915–931. [Google Scholar] [CrossRef] [PubMed]
- Dhasmana, N.; Ram, G.; McAllister, K.N.; Chupalova, Y.; Lopez, P.; Ross, H.F.; Novick, R.P. Dynamics of Antibacterial Drone Establishment in Staphylococcus aureus: Unexpected Effects of Antibiotic Resistance Genes. mBio 2021, 12, e0208321. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Perez, R.A.; de la Torre-Luna, R.; Ahumada-Cantillano, M.; Vázquez-Garcidueñas, M.S.; MarthaPérez-Serrano, R.; ElizabethMartínez-Martínez, R.; LauraGuillén-Nepita, A. Detection of the antimicrobial resistance genes blaTEM-1, cfxA, tetQ, tetM, tetW and ermC in endodontic infections of a Mexican population. J. Glob. Antimicrob. Resist. 2018, 15, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Haubert, L.; Cunha, C.; Lopes, G.V.; Silva, W.P.D. Food isolate Listeria monocytogenes harboring tetM gene plasmid-mediated exchangeable to Enterococcus faecalis on the surface of processed cheese. Food Res. Int. 2018, 107, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Roberts, M.C.; Kenny, G.E. Dissemination of the tetM tetracycline resistance determinant to Ureaplasma urealyticum. Antimicrob. Agents Chemother. 1986, 29, 350–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaskolskiy, B.; Dementieva, E.; Leinsoo, A.; Petrova, N.; Chestkov, A.; Kubanov, A.; Deryabin, D.; Gryadunov, D. Tetracycline resistance of Neisseria gonorrhoeae in Russia, 2015-2017. Infect. Genet. Evol. 2018, 63, 236–242. [Google Scholar] [CrossRef]
- Yamada, Y.; Takashima, H.; Walmsley, D.L.; Ushiyama, F.; Matsuda, Y.; Kanazawa, H.; Yamaguchi-Sasaki, T.; Tanaka-Yamamoto, N.; Yamagishi, J.; Kurimoto-Tsuruta, R. Fragment-based discovery of novel non-hydroxamate LpxC inhibitors with antibacterial activity. J. Med. Chem. 2020, 63, 14805–14820. [Google Scholar] [CrossRef]
- Clayton, G.M.; Klein, D.J.; Rickert, K.W.; Patel, S.B.; Kornienko, M.; Zugay-Murphy, J.; Reid, J.C.; Tummala, S.; Sharma, S.; Singh, S.B.; et al. Structure of the bacterial deacetylase LpxC bound to the nucleotide reaction product reveals mechanisms of oxyanion stabilization and proton transfer. J. Biol. Chem. 2013, 288, 34073–34080. [Google Scholar] [CrossRef] [Green Version]
- Aris, S.N.A.M.; Rahman, M.Z.A.; Rahman, R.N.Z.R.A.; Ali, M.S.M.; Salleh, A.B.; Teo, C.Y.; Leow, T.C. Identification of potential riboflavin synthase inhibitors by virtual screening and molecular dynamics simulation studies. J. King Saud Univ.-Sci. 2021, 33, 101270. [Google Scholar] [CrossRef]
- Fujita, K.; Takata, I.; Yoshida, I.; Okumura, H.; Otake, K.; Takashima, H.; Sugiyama, H. TP0586532, a non-hydroxamate LpxC inhibitor, has in vitro and in vivo antibacterial activities against Enterobacteriaceae. J. Antibiot. 2022, 75, 98–107. [Google Scholar] [CrossRef] [PubMed]
- Krause, K.M.; Haglund, C.M.; Hebner, C.; Serio, A.W.; Lee, G.; Nieto, V.; Cohen, F.; Kane, T.R.; Machajewski, T.D.; Hildebrandt, D. Potent LpxC inhibitors with in vitro activity against multidrug-resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2019, 63, e00977-19. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Zhan, X.; Mao, X.M.; Li, Y.Q. The regulatory cascades of antibiotic production in Streptomyces. World J. Microbiol. Biotechnol. 2020, 36, 13. [Google Scholar] [CrossRef]
- Zhang, Z.; Du, C.; de Barsy, F.; Liem, M.; Liakopoulos, A.; van Wezel, G.P.; Choi, Y.H.; Claessen, D.; Rozen, D.E. Antibiotic production in Streptomyces is organized by a division of labor through terminal genomic differentiation. Sci. Adv. 2020, 6, eaay5781. [Google Scholar] [CrossRef] [Green Version]
- Zendeboodi, F.; Khorshidian, N.; Mortazavian, M.A.; Cruz, A.G. Probiotic: Conceptualization from a new approach. Curr. Opin. Food Sci. 2020, 32, 103–123. [Google Scholar] [CrossRef]
- Rajaiah, A.; Ponraj, J.G.; Swasthikka, R.P.; Abirami, G.; Ragupathi, T.; Jayakumar, R.; Ravi, A.V. Anti-QS mediated anti-infection efficacy of probiotic culture-supernatant against Vibrio campbellii infection and the identification of active compounds through in vitro and in silico analyses. Biocatal. Agric. Biotechnol. 2021, 35, 102108. [Google Scholar]
- Balmeh, N.; Mahmoudi, S.; Fard, N.A. Manipulated bio antimicrobial peptides from probiotic bacteria as proposed drugs for COVID-19 disease. Inf. Med. Unlocked 2021, 23, 100515. [Google Scholar] [CrossRef]
- Pei, Y.; Du, Q.; Liao, P.Y.; Chen, Z.P.; Wang, D.; Yang, C.R.; Kitazato, K.; Wang, Y.F.; Zhang, Y.J. Notoginsenoside ST-4 inhibits virus penetration of herpes simplex virus in vitro. J. Asian. Nat. Prod. Res. 2011, 13, 498–504. [Google Scholar] [CrossRef]
- Nhiem, N.X.; Tai, B.H.; Quang, T.H.; Kiem, P.V.; Minh, C.V.; Nam, N.H.; Kim, J.H.; Im, L.R.; Lee, Y.M.; Kim, Y.H. A new ursane-type triterpenoid glycoside from Centella asiatica leaves modulates the production of nitric oxide and secretion of TNF-alpha in activated RAW 264. 7 cells. Bioorg. Med. Chem Lett. 2011, 21, 1777–1781. [Google Scholar] [CrossRef]
- Liesch, J.M.; Wichmann, C.F. Novel antinematodal and antiparasitic agents from Penicillium charlesii. II. Structure determination of paraherquamides B, C, D, E, F, and G. J. Antibiot. 1990, 43, 1380–1386. [Google Scholar]
- Ceapa, C.D.; Vazquez-Hernandez, M.; Rodriguez-Luna, S.D.; Vazquez, A.P.C.; Suarez, V.J.; Rodriguez-Sanoja, R.; Alvarez-Buylla, E.R.; Sanchez, S. Genome mining of Streptomyces scabrisporus NF3 reveals symbiotic features including genes related to plant interactions. PLoS ONE 2018, 13, e0192618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krügel, H.; Krubasik, P.; Weber, K.; Saluz, H.P. Functional analysis of genes from Streptomyces griseus involved in the synthesis of isorenieratene, a carotenoid with aromatic end groups, revealed a novel type of carotenoid desaturase. Biochim. Et Biophys. Acta (BBA)–Mol. Cell Biol. Lipids 1999, 1439, 57–64. [Google Scholar] [CrossRef]
- Fuke, T.; Sato, T.; Jha, S.; Tansengco, M.L.; Atomi, H. Phytoene production utilizing the isoprenoid biosynthesis capacity of Thermococcus kodakarensis. Extremophiles 2018, 22, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Sui, Y.; Mazzucchi, L.; Acharya, P.; Xu, Y.; Morgan, G.; Harvey, P.J. A Comparison of beta-Carotene, Phytoene and Amino Acids Production in Dunaliella salina DF 15 (CCAP 19/41) and Dunaliella salina CCAP 19/30 Using Different Light Wavelengths. Foods 2021, 10, 2824. [Google Scholar] [CrossRef] [PubMed]
- Pollmann, H.; Breitenbach, J.; Sandmann, G. Development of Xanthophyllomyces dendrorhous as a production system for the colorless carotene phytoene. J. Biotechnol. 2017, 247, 34–41. [Google Scholar] [CrossRef] [PubMed]
- De Lourdes Moreno, M.; Sánchez-Porro, C.; García, M.T.; Mellado, E. Carotenoids’ production from halophilic bacteria. Methods Mol. Biol. 2012, 892, 207–217. [Google Scholar] [PubMed]
- Maia, E.H.B.; Assis, L.C.; de Oliveira, T.A.; da Silva, A.M.; Taranto, A.G. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 2020, 8, 343. [Google Scholar] [CrossRef]
- Miras-Moreno, B.; Pedreño, M.A.; Romero, L.A. Bioactivity and bioavailability of phytoene and strategies to improve its production. Phytochem. Rev. Vol. 2019, 18, 356–359. [Google Scholar] [CrossRef]
- Zylicz, Z.; Hofs, H.P.; Wagener, D.J. Potentiation of cisplatin antitumor activity on L1210 leukemia s.c. by sparsomycin and three of its analogues. Cancer Lett. 1989, 46, 153–157. [Google Scholar]
- Guengerich, F.P. Cytochrome p450 and chemical toxicology. Chem. Res. Toxicol. 2008, 21, 70–83. [Google Scholar] [CrossRef]
- Lin, J.H.; Yamazaki, M. Role of P-glycoprotein in pharmacokinetics: Clinical implications. Clin Pharm. 2003, 42, 59–98. [Google Scholar] [CrossRef] [PubMed]
- Conlan, S.; Mijares, L.A.; Program, N.C.S.; Becker, J.; Blakesley, R.W.; Bouffard, G.G.; Brooks, S.; Coleman, H.; Gupta, J.; Gurson, N.; et al. Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome. Biol. 2012, 13, R64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasko, D.A.; Rosovitz, M.J.; Myers, G.S.; Mongodin, E.F.; Fricke, W.F.; Gajer, P.; Crabtree, J.; Sebaihia, M.; Thomson, N.R.; Chaudhuri, R.; et al. The pangenome structure of Escherichia coli: Comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 2008, 190, 6881–6893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hormeno, L.; Campos, M.J.; Vadillo, S.; Quesada, A. Occurrence of tet(O/M/O) Mosaic Gene in Tetracycline-Resistant Campylobacter. Microorganisms 2020, 8, 1710. [Google Scholar] [CrossRef]
- Mehla, K.; Ramana, J. Novel Drug Targets for Food-Borne Pathogen Campylobacter jejuni: An. Integrated Subtractive Genomics and Compara-tive Metabolic Pathway Study. OMICS 2015, 19, 393–406. [Google Scholar]
- Zhou, P.; Hong, J. Structure-and Ligand-Dynamics-Based Design of Novel Antibiotics Targeting Lipid A Enzymes LpxC and LpxH in Gram-Negative Bacteria. Acc. Chem. Res. 2021, 54, 1623–1634. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef] [Green Version]
- Chaudhari, N.M.; Gupta, V.K.; Dutta, C. BPGA-an ultra-fast pan-genome analysis pipeline. Sci. Rep. 2016, 6, 24373. [Google Scholar] [CrossRef] [Green Version]
- Basharat, Z.; Jahanzaib, M.; Yasmin, A.; Khan, I.A. Pan-genomics, drug candidate mining and ADMET profiling of natural product inhibitors screened against Yersinia pseudotuberculosis. Genomics 2021, 113, 238–244. [Google Scholar] [CrossRef]
- Edgar, R. Usearch; Lawrence Berkeley National Lab. (LBNL): Berkeley, CA, USA, 2010. [Google Scholar]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic. Acids. Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic. Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome-scale analysis of protein functions and evolution. Nucleic. Acids. Res. 2000, 28, 33–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.-N.; Hua, Z.-G.; Huang, J.; Rao, N.; Guo, F.-B. CEG: A database of essential gene clusters. BMC Genom. 2013, 14, 769. [Google Scholar] [CrossRef] [Green Version]
- Luo, H.; Lin, Y.; Gao, F.; Zhang, C.-T.; Zhang, R. DEG 10, an update of the database of essential genes that includes both protein-coding genes and noncoding genomic elements. Nucleic. Acids Res. 2014, 42, D574–D580. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Pandit, S.B.; Zhang, Y.; Skolnick, J. TASSER-Lite: An automated tool for protein comparative modeling. Biophys. J. 2006, 91, 4180–4190. [Google Scholar] [CrossRef] [Green Version]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Basharat, Z.; Akhtar, U.; Khan, k.; Alotaibi, G.; Jalal, K.; Abbas, M.N.; Hayat, A.; Ahmad, D.; Hassan, S.S. Differential analysis of Orientia tsutsugamushi genomes for therapeutic target identification and possible intervention through natural product inhibitor screening. Comput. Biol. Med. 2022, 141, 105165. [Google Scholar] [CrossRef]
- Basharat, Z.; Jahanzaib, M.; Rahman, N. Therapeutic target identification via differential genome analysis of antibiotic resistant Shigella sonnei and inhibitor evaluation against a selected drug target. Infect. Genet. Evol. 2021, 94, 105004. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Ung, C. Prediction of potential toxicity and side effect protein targets of a small molecule by a ligand–protein inverse docking approach. J. Mol. Graph. Model. 2001, 20, 199–218. [Google Scholar] [CrossRef]
- Pu, L.; Liu, T.; Wu, H.-C.; Mukhopadhyay, S.; Brylinski, M. eToxPred: A machine learning-based approach to estimate the toxicity of drug candidates. BMC Pharmacol. Toxicol. 2019, 20, 2. [Google Scholar] [CrossRef] [Green Version]
- Basharat, Z.; Khan, K.; Jalal, K.; Ahmad, D.; Hayat, A.; Alotaibi, G.; al Mouslem, A.; Alkhayl, F.F.A.; Almatroudi, A. An in silico hierarchal approach for drug candidate mining and validation of natural product inhibitors against pyrimidine biosynthesis enzyme in the antibiotic-resistant Shigella flexneri. Infect Genet. Evol. 2022, 98, 105233. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No. | Protein | Functional Category | Accession | No. of Amino Acids |
|---|---|---|---|---|
| 1 | Multidrug efflux RND transporter permease subunit | Signaling and cellular processes | WP_016646469.1 | 1053 |
| 2 | Formate dehydrogenase subunit alpha | Carbohydrate metabolism | WP_081617940.1 | 974 |
| 3 | Formate dehydrogenase subunit alpha | Energy metabolism | WP_081617935.1 | 935 |
| 4 | Thiosulfate reductase PhsA | WP_024962542.1 | 761 | |
| 5 | Urease subunit alpha | Nucleotide metabolism | WP_050333258.1 | 571 |
| 6 | TolC family protein | Cellular process | WP_018713635.1 | 481 |
| 7 | Murein biosynthesis integral membrane Protein MurJ | Metabolism | WP_101637374.1 | 470 |
| 8 | 3-deoxy-7-phosphoheptulonate synthase class II | WP_018713201.1 | 447 | |
| 9 | Glutamyl-tRNA reductase | WP_101637465.1 | 436 | |
| 10 | Type II secretion system F family protein | Environmental information processing | WP_101636691.1 | 415 |
| 11 | Lipid IV(A) 3-deoxy-D-manno-octulosonic acid transferase | Metabolism | WP_018712363.1 | 382 |
| 12 | Efflux RND transporter periplasmic adaptor subunit | Genetic information processing | WP_101637141.1 | 371 |
| 13 | Lipid-A-disaccharide synthase | Metabolism | WP_018713606.1 | 347 |
| 14 | KpsF/GutQ family sugar-phosphate isomerase | WP_018713574.1 | 320 | |
| 15 | UDP-3-O-(3-hydroxymyristoyl)glucosamine N-acyltransferase | WP_101637361.1 | 317 | |
| 16 | UDP-3-O-acyl-N-acetylglucosamine deacetylase (LpxC) | WP_050333632.1 | 296 | |
| 17 | c-type cytochrome | WP_101637480.1 | 287 | |
| 18 | Pantoate--beta-alanine ligase | WP_016646546.1 | 273 | |
| 19 | 3-deoxy-manno-octulosonate cytidylyltransferase | WP_018713833.1 | 240 | |
| 20 | Carbonic anhydrase | WP_018713612.1 | 227 | |
| 21 | MotA/TolQ/ExbB proton channel family protein | Signaling and cellular processes | WP_018713282.1 | 191 |
| 22 | YceI family protein | - | WP_024962547.1 | 188 |
| 23 | Biopolymer transporter ExbD | Signaling and cellular processes | WP_018713283.1 | 130 |
| 24 | Aspartate 1-decarboxylase | Metabolism | WP_018713496.1 | 115 |
| 25 | Urease subunit beta | Nucleotide metabolism | WP_018713462.1 | 104 |
| 26 | Urease subunit gamma | Nucleotide metabolism | WP_018713461.1 | 100 |
| S. No. | Compounds | Ligand | Receptor | Interaction | Distance | E (kcal/mol) | S-Score |
|---|---|---|---|---|---|---|---|
| LpxC ZINC database inhibitors | |||||||
| 1 | ZINC26844580 | N12 | O PHE187 | H-donor | 3.43 | −0.8 | −7.42 |
| N18 | O MET59 | H-donor | 2.95 | −2.3 | |||
| O19 | NE2 HIS233 | H-donor | 2.92 | −3.1 | |||
| 2 | ZINC13474902 | Hydrophobic interactions | −7.05 | ||||
| 3 | ZINC13474878 | O1 | CA SER259 | H-acceptor | 3.44 | −0.8 | −6.90 |
| Postbiotics | |||||||
| 1 | Notoginsenoside St-4 | O63 | O LYS157 | H-donor | 2.89 | −2.6 | −8.59 |
| O64 | O LYS157 | H-donor | 3.12 | −1.0 | |||
| O35 | NE2 HIS233 | H-acceptor | 2.93 | −2.3 | |||
| O53 | NZ LYS140 | H-acceptor | 3.12 | −0.5 | |||
| O55 | NZ LYS140 | H-acceptor | 3.00 | −5.7 | |||
| C26 | 6-ring PHE187 | H-pi | 4.32 | −0.6 | |||
| 2 | Asiaticoside F | O79 | OD1 ASP139 | H-donor | 2.89 | −2.0 | −8.43 |
| O94 | O GLN58 | H-donor | 3.26 | −0.5 | |||
| C112 | O PHE187 | H-donor | 3.30 | −0.5 | |||
| O94 | N HIS260 | H-acceptor | 3.40 | −0.7 | |||
| 3 | Paraherquamide E | O43 | CA SER259 | H-acceptor | 3.43 | −0.6 | −8.02 |
| C18 | 6-ring PHE 187 | H-pi | 3.81 | −0.7 | |||
| Streptomycin compounds | |||||||
| 1 | ZINC08219868 (Phytoene) | Hydrophobic interactions | −7.20 | ||||
| 2 | ZINC08214943 (Lycopene) | Hydrophobic interactions | −7.03 | ||||
| 3 | ZINC04742519 (Sparsomycin) | O27 | O MET59 | H-donor | 2.84 | −0.7 | −7.01 |
| S37 | CA GLY205 | H-acceptor | 4.06 | −0.7 | |||
| C24 | 5-ring HIS18 | H-pi | 4.82 | −0.5 | |||
| Name | Water Solubility | CaCo2 Permeability | HIA | Skin Permeability | Max. Tolerated Dose (Human) | Minnow Toxicity | T. Pyriformis Toxicity | Oral Rat Acute Toxicity (LD50) | Hepatotoxic |
|---|---|---|---|---|---|---|---|---|---|
| ZINC26844580 | −2.348 | −0.049 | 51.882 | −2.898 | 1.177 | 2.262 | 0.269 | 2.238 | Yes |
| ZINC13474902 | −5.016 | 1.037 | 91.596 | −3.066 | 0.193 | 2.117 | 0.422 | 2.983 | Yes |
| ZINC13474878 | −4.928 | 1.103 | 90.066 | −2.993 | 0.272 | 2.587 | 0.356 | 2.96 | Yes |
| Notoginsenoside St−4 | −2.938 | −1.241 | 0 | −2.735 | −1.755 | 10.516 | 0.285 | 2.738 | No |
| Asiaticoside F | −2.922 | −1.039 | 43.096 | −2.735 | −0.991 | 11.112 | 0.285 | 2.738 | No |
| Paraherquamide E | −4.377 | 1.003 | 93.521 | −3.226 | −0.877 | 2.209 | 0.294 | 3.716 | Yes |
| ZINC08219868 (Phytoene) | −6.345 | 1.28 | 91.213 | −2.737 | −0.33 | −5.751 | 0.288 | 2.036 | No |
| ZINC08214943 (Lycopene) | −6.514 | 1.317 | 93.238 | −2.783 | −0.447 | −5.243 | 0.291 | 2.105 | No |
| ZINC04742519 (Sparsomycin) | −2.348 | −0.238 | 44.731 | −3.08 | 1.279 | 2.726 | 0.146 | 2.11 | Yes |
| Name | Ligand | Protein | Protein−Ligand Complex |
|---|---|---|---|
| ZINC1347878 | 0.04 | −19.31 | −18.95 |
| ZINC13474902 | 0.03 | −19.31 | −18.95 |
| ZINC26844580 | 0.14 | −19.31 | −19.03 |
| Notoginsenoside St-4 | −0.85 | −19.31 | −19.19 |
| Asiaticoside F | −0.82 | −19.31 | −19.20 |
| Paraherquamide E | −0.07 | −19.31 | −18.89 |
| Phytoene | −0.96 | −19.31 | −19.46 |
| Lycopene | −0.92 | −19.31 | −19.54 |
| Sparsomycin | 0.05 | −19.31 | −18.97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, K.; Basharat, Z.; Jalal, K.; Mashraqi, M.M.; Alzamami, A.; Alshamrani, S.; Uddin, R. Identification of Therapeutic Targets in an Emerging Gastrointestinal Pathogen Campylobacter ureolyticus and Possible Intervention through Natural Products. Antibiotics 2022, 11, 680. https://doi.org/10.3390/antibiotics11050680
Khan K, Basharat Z, Jalal K, Mashraqi MM, Alzamami A, Alshamrani S, Uddin R. Identification of Therapeutic Targets in an Emerging Gastrointestinal Pathogen Campylobacter ureolyticus and Possible Intervention through Natural Products. Antibiotics. 2022; 11(5):680. https://doi.org/10.3390/antibiotics11050680
Chicago/Turabian StyleKhan, Kanwal, Zarrin Basharat, Khurshid Jalal, Mutaib M. Mashraqi, Ahmad Alzamami, Saleh Alshamrani, and Reaz Uddin. 2022. "Identification of Therapeutic Targets in an Emerging Gastrointestinal Pathogen Campylobacter ureolyticus and Possible Intervention through Natural Products" Antibiotics 11, no. 5: 680. https://doi.org/10.3390/antibiotics11050680
APA StyleKhan, K., Basharat, Z., Jalal, K., Mashraqi, M. M., Alzamami, A., Alshamrani, S., & Uddin, R. (2022). Identification of Therapeutic Targets in an Emerging Gastrointestinal Pathogen Campylobacter ureolyticus and Possible Intervention through Natural Products. Antibiotics, 11(5), 680. https://doi.org/10.3390/antibiotics11050680