Phenylboronic Acids Probing Molecular Recognition against Class A and Class C β-lactamases

 , , , ,
, , , , 

Abstract
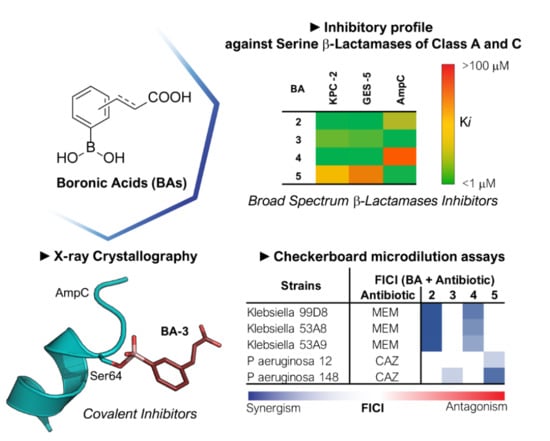
:
1. Introduction
2. Results
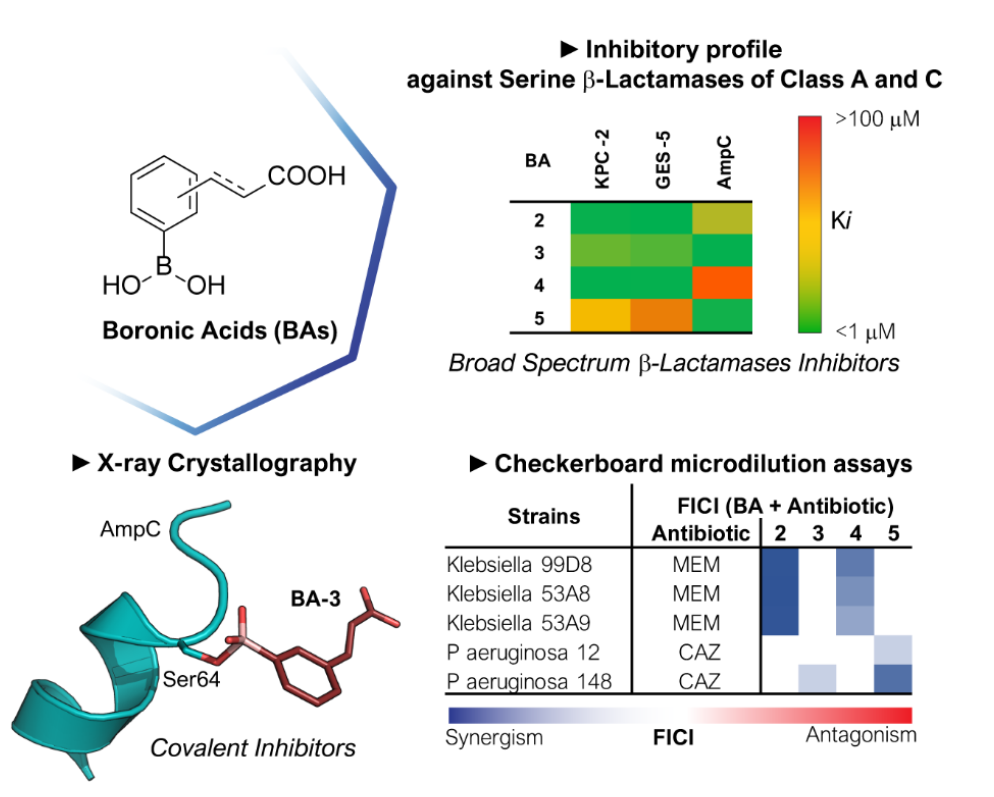
2.1. Phenyl Boronic Acid Derivatives as Cross-classes SBLs Inhibitors
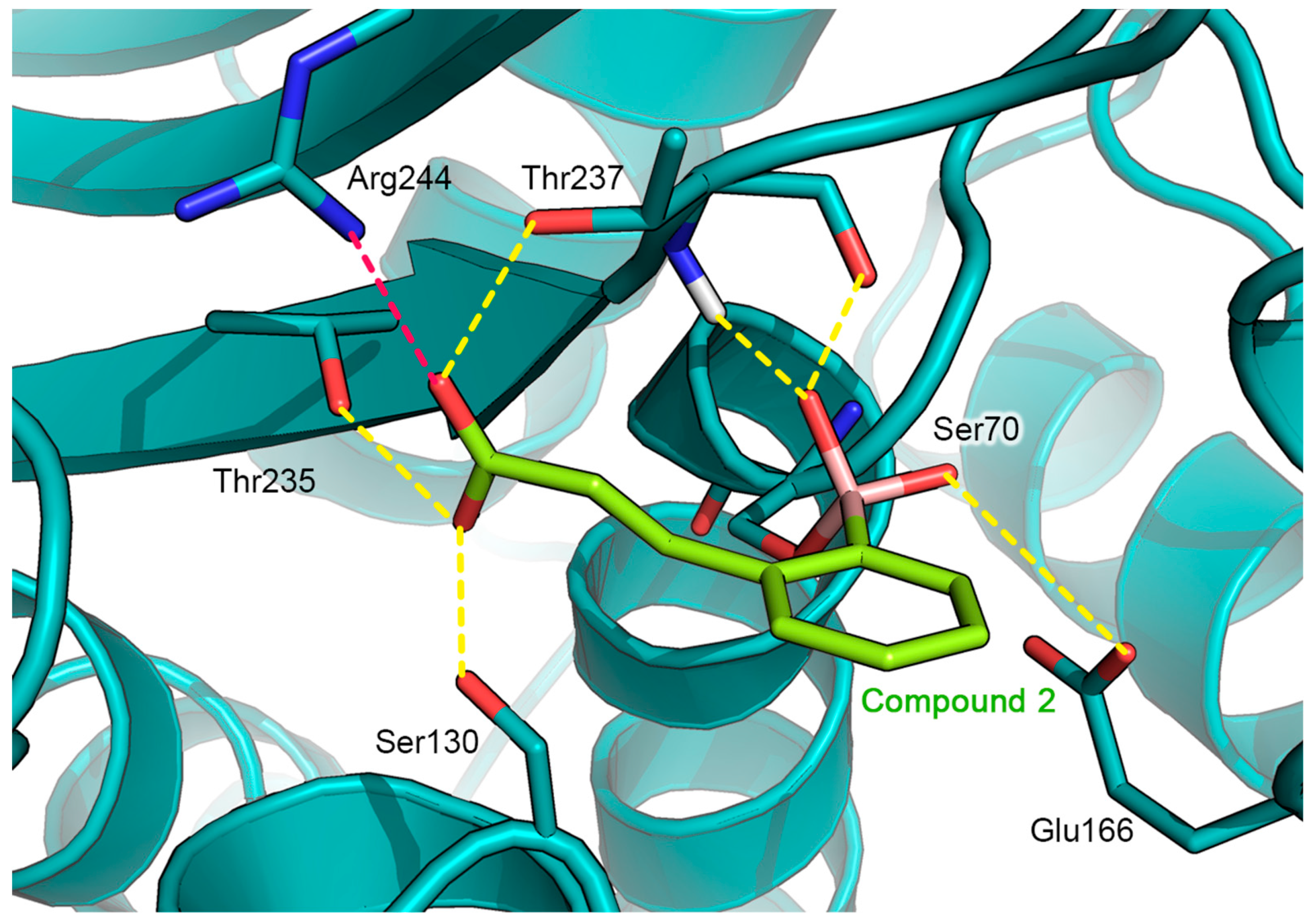
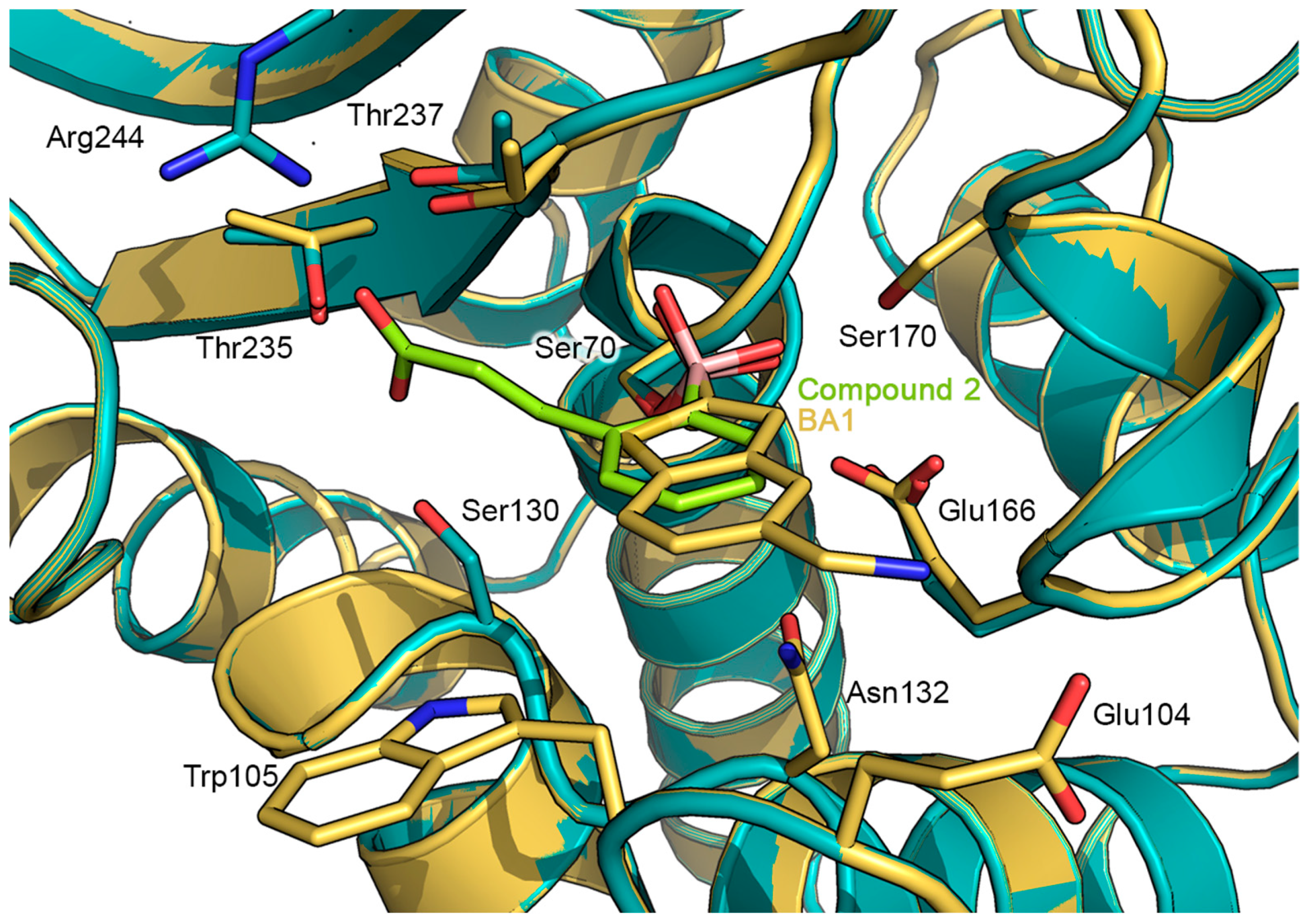
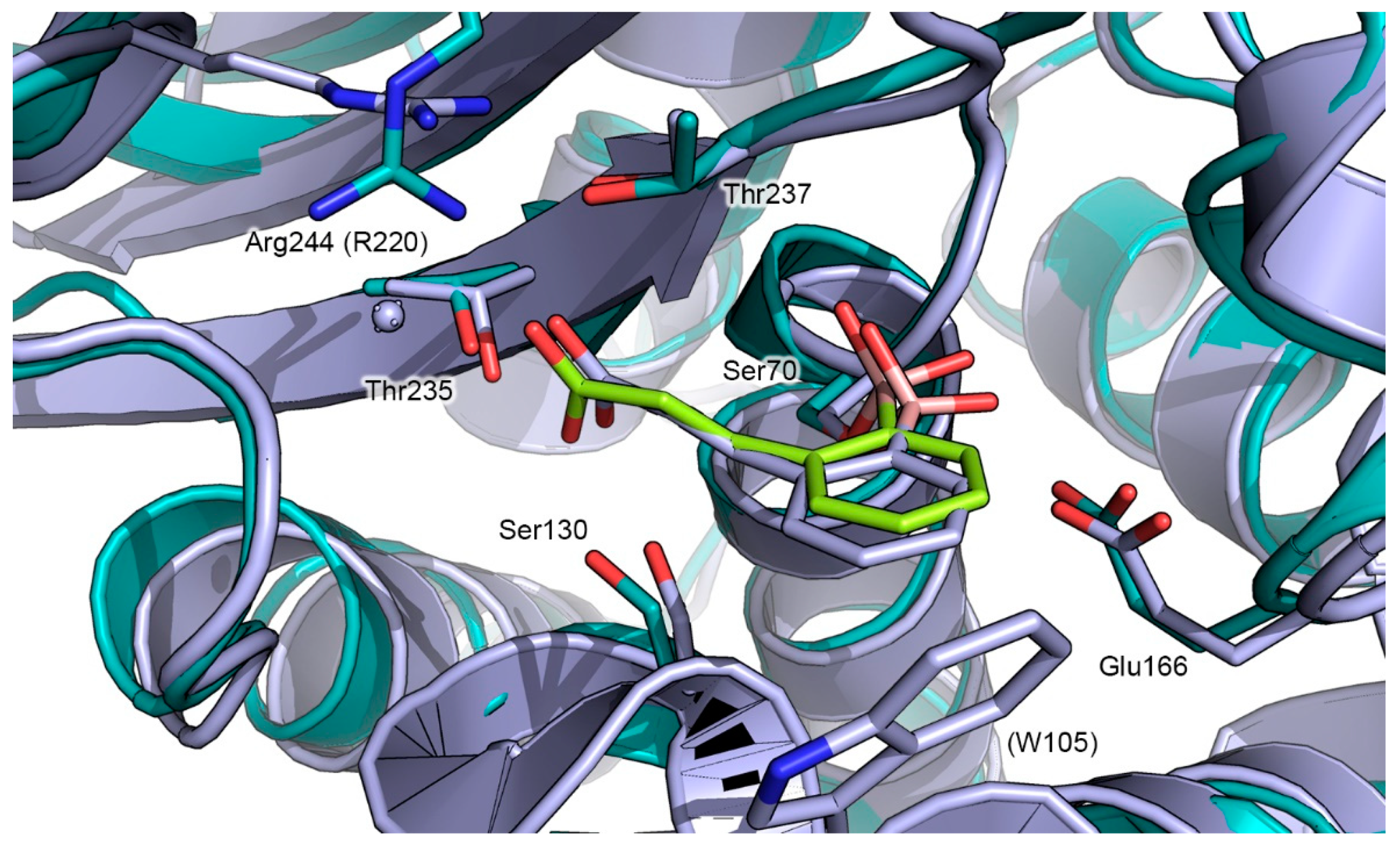
2.2. Prediction of the Binding Mode of Compound 2 in GES-5 by Docking Calculation
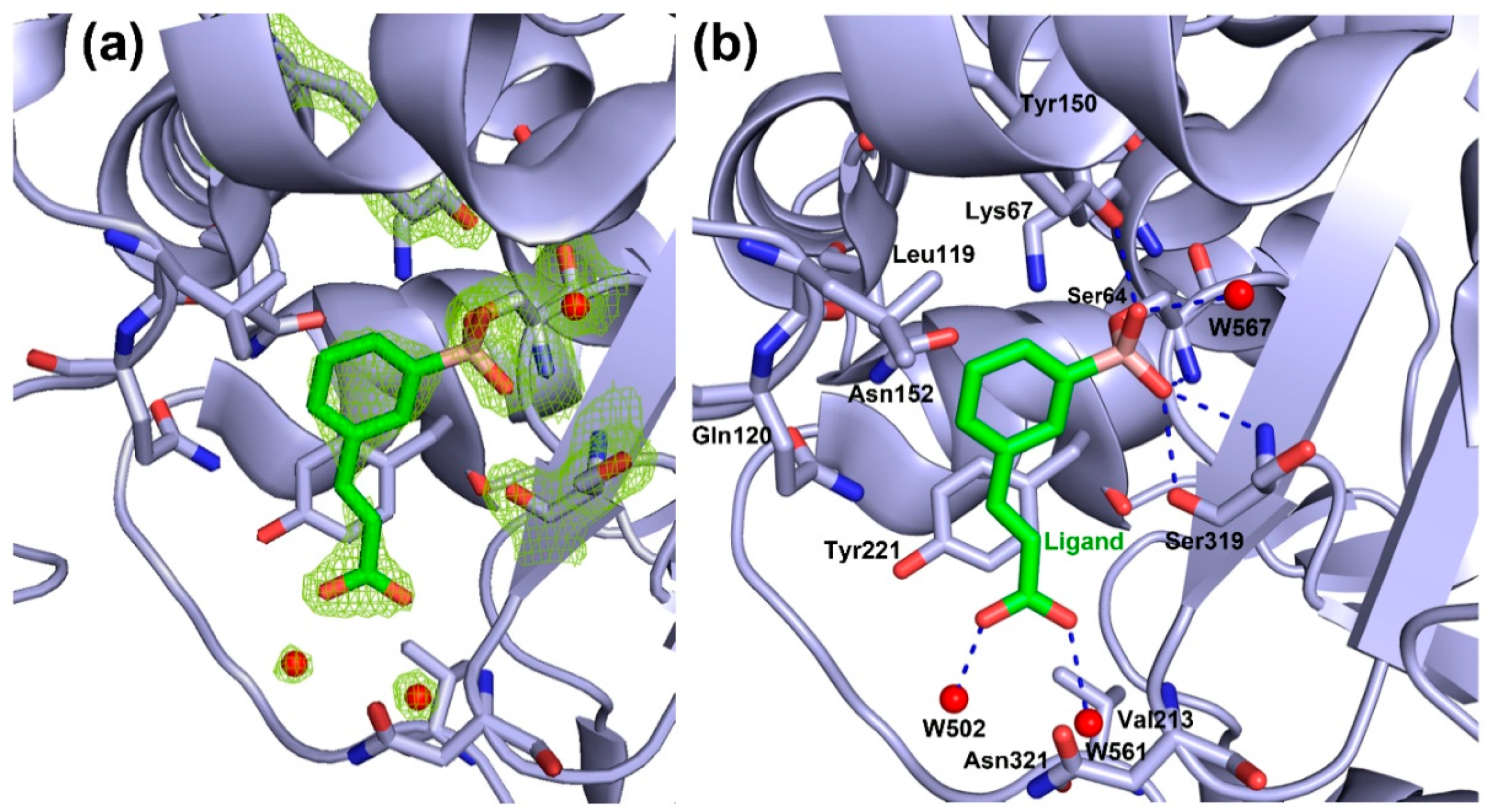
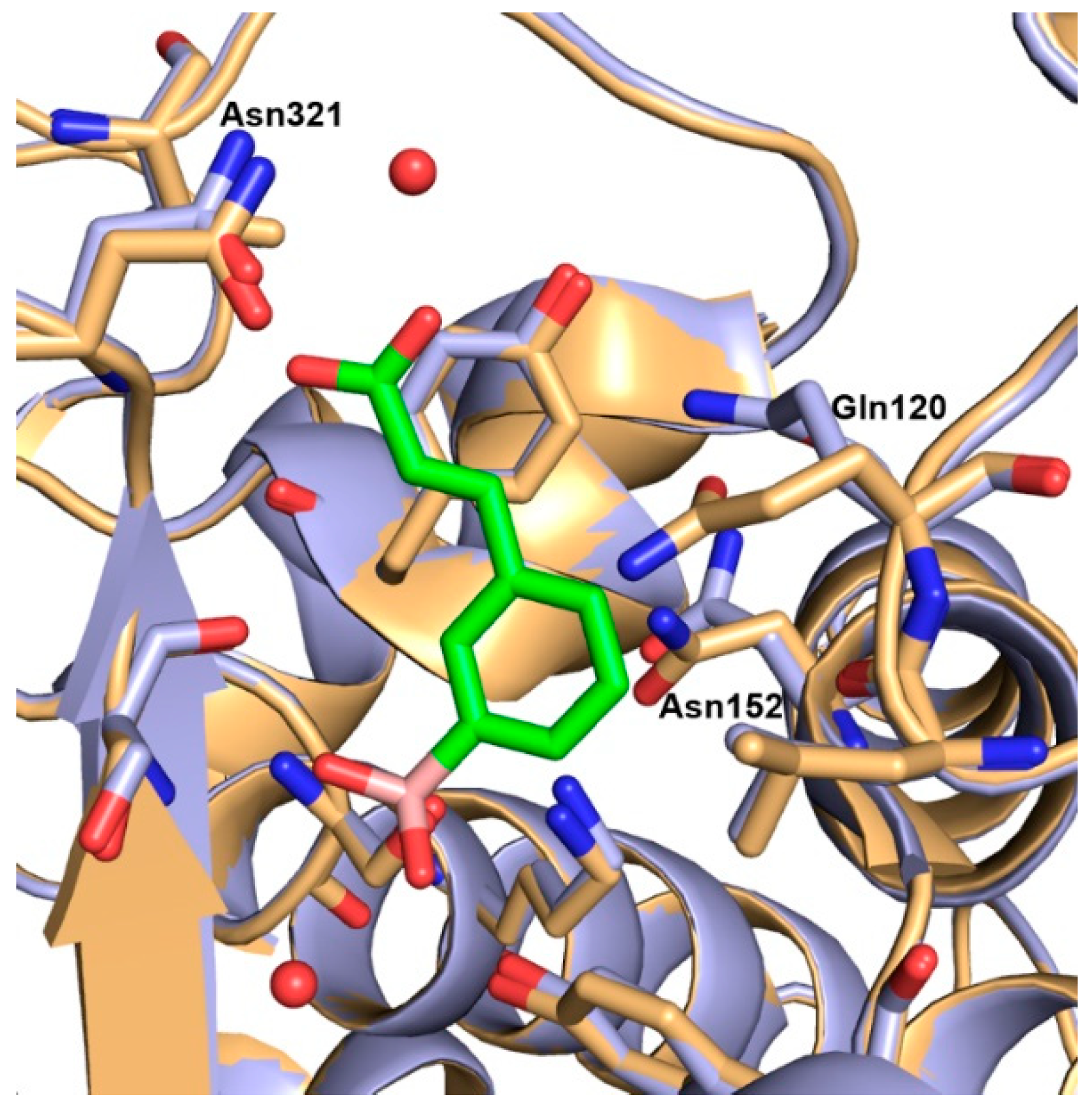
2.3. AmpC-Compound 3 Binary Complex
2.4. Biological Evaluation Against Clinical Strains
3. Discussion and Conclusions
4. Materials and Methods
4.1. Chemistry
4.2. Proteins Production and Purification
4.3. In Vitro Enzyme Inhibition Assays Against KPC-2, GES-5, and AmpC
4.4. Molecular Modeling
4.4.1. Proteins and Ligands Preparations
4.4.2. Docking Calculation
4.5. Co-Crystallization and Structure Determination
4.6. Drug Interaction Models Via Checkerboard Microdilution Assay
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Accession Code
Abbreviations
References
- O’ Neil, J. Review on Antibiotic resisitance. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Heal. Wealth Nations 2014, 1–16. [Google Scholar]
- Bush, K. Carbapenemases: Partners in Crime. J. Glob. Antimicrob. Resist. 2013, 1, 7–16. [Google Scholar] [CrossRef]
- Papp-Wallace, K.M.; Endimiani, A.; Taracila, M.A.; Bonomo, R.A. Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother. 2011, 55, 4943–4960. [Google Scholar] [CrossRef] [Green Version]
- Meletis, G. Carbapenem resistance: Overview of the problem and future perspectives. Ther. Adv. Infect. Dis. 2016, 3, 15–21. [Google Scholar] [CrossRef]
- Watkins, R.R.; Bonomo, R.A. Overview: Global and Local Impact of Antibiotic Resistance. Infect. Dis. Clin. N. Am. 2016, 30, 313–322. [Google Scholar] [CrossRef]
- Bush, K. The ABCD of β-lactamase nomenclature. J. Inf. Chem. 2013, 19, 549–559. [Google Scholar] [CrossRef]
- Tondi, D.; Cross, S.; Venturelli, A.; Costi, M.P.; Cruciani, G.; Spyrakis, F. Decoding the Structural Basis for Carbapenem Hydrolysis by Class A β- Lactamases: Fishing for a Pharmacophore. Curr. Drug Target 2016, 17, 983–1005. [Google Scholar] [CrossRef]
- Farina, D.; Spyrakis, F.; Venturelli, A.; Cross, S.; Tondi, D.; Costi, M.P. The Inhibition of Extended Spectrum β-Lactamases: Hits and Leads. Curr. Med. Chem. 2014, 21, 1405–1434. [Google Scholar] [CrossRef]
- Spyrakis, F.; Bellio, P.; Quotadamo, A.; Linciano, P.; Benedetti, P.; D’Arrigo, G.; Baroni, M.; Cendron, L.; Celenza, G.; Tondi, D. First virtual screening and experimental validation of inhibitors targeting GES-5 carbapenemase. J. Comput. Aided Mol. Des. 2019, 33, 295–305. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC β-Lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef]
- Livermore, D.M.; Mushtaq, S.; Warner, M.; Zhang, J.-C.; Maharjan, S.; Doumith, M.; Woodford, N. Activity of aminoglycosides, including ACHN-490, against carbapenem-resistant Enterobacteriaceae isolates. J. Antimicrob. Chemother. 2011, 66, 48–53. [Google Scholar] [CrossRef]
- Hishinuma, T.; Tada, T.; Kuwahara-Arai, K.; Yamamoto, N.; Shimojima, M.; Kirikae, T. Spread of GES-5 carbapenemase-producing Pseudomonas aeruginosa clinical isolates in Japan due to clonal expansion of ST235. PLoS ONE. 2018, 13, e0207134. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Y. Detection of plasmid-mediated AmpC β-lactamase in Escherichia coli. Biomed. Rep. 2016, 4, 687–690. [Google Scholar] [CrossRef]
- Rodríguez-Baño, J.; Gutiérrez-Gutiérrez, B.; Machuca, I.; Pascual, A. Treatment of Infections Caused by Extended-Spectrum-Beta-Lactamase-, AmpC-, and Carbapenemase-Producing Enterobacteriaceae. Clin. Microbiol. Rev. 2018, 31, e00079-17. [Google Scholar] [CrossRef]
- Pei, W.; Cammarata, M.; Brodbelt, J.S.; Monzingo, A.F.; Pratt, R.F.; Fast, W. A Lysine-Targeted Affinity Label for Serine-β-Lactamase Also Covalently Modifies New Delhi Metallo-β-lactamase-1 (NDM-1). Biochemistry 2019, 58, 2834–2843. [Google Scholar] [CrossRef]
- Hecker, S.J.; Reddy, K.R.; Totrov, M.; Hirst, G.C.; Lomovskaya, O.; Griffith, D.C.; King, P.; Tsivkovski, R.; Sun, D.; Sabet, M.; et al. Discovery of a Cyclic Boronic Acid β-Lactamase Inhibitor (RPX7009) with Utility vs Class A Serine Carbapenemases. J. Med. Chem. 2015, 58, 3682–3692. [Google Scholar] [CrossRef]
- Zhou, M.; Yang, Q.; Lomovskaya, O.; Sun, D.; Kudinha, T.; Xu, Z.; Zhang, G.; Chen, X.; Xu, Y. In Vitro Activity of Meropenem Combined with Vaborbactam against KPC-Producing Enterobacteriaceae in China. J. Antimicrob. Chemother. 2018, 73, 2789–2796. [Google Scholar] [CrossRef]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jiménez-Castellanos, J.-C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.G.; Schofield, C.J. Structural Basis of Metallo-β-Lactamase, Serine-β-Lactamase and Penicillin-Binding Protein Inhibition by Cyclic Boronates. Nat. Commun. 2016, 7, 12406. [Google Scholar] [CrossRef]
- Genovese, F.; Lazzari, S.; Venturi, E.; Costantino, L.; Blazquez, J.; Ibacache-Quiroga, C.; Costi, M.P.; Tondi, D. Design, synthesis and biological evaluation of non-covalent AmpC β-lactamases inhibitors. Med. Chem. Res. 2017, 26, 975–986. [Google Scholar] [CrossRef]
- Tondi, D.; Venturelli, A.; Bonnet, R.; Pozzi, C.; Shoichet, B.K.; Costi, M.P. Targeting class A and C serine β-lactamases with a broad-spectrum boronic acid derivative. J. Med. Chem. 2014, 57, 5449–5458. [Google Scholar] [CrossRef]
- Cendron, L.; Quotadamo, A.; Maso, L.; Bellio, P.; Montanari, M.; Celenza, G.; Venturelli, A.; Costi, M.P.; Tondi, D. X-ray Crystallography Deciphers the Activity of Broad-Spectrum Boronic Acid β-Lactamase Inhibitors. ACS Med. Chem. 2019, 10, 650–655. [Google Scholar] [CrossRef] [PubMed]
- Santucci, M.; Spyrakis, F.; Cross, S.; Quotadamo, A.; Farina, D.; Tondi, D.; De Luca, F.; Docquier, J.-D.; Prieto, A.I.; Ibacache, C.; et al. Computational and biological profile of boronic acids for the detection of bacterial serine- and metallo-β-lactamases. Sci. Rep. 2017, 7, 17716. [Google Scholar] [CrossRef] [PubMed]
- Tondi, D.; Morandi, F.; Bonnet, R.; Costi, M.P.; Shoichet, B.K. Structure-based optimization of a non-β-lactam lead results in inhibitors that do not up-regulate β-lactamase expression in cell culture. J. Am. Chem. Soc. 2005, 127, 4632–4639. [Google Scholar] [CrossRef]
- Celenza, G.; Vicario, M.; Bellio, P.; Linciano, P.; Perilli, M.; Oliver, A.; Blazquez, J.; Cendron, L.; Tondi, D. Phenylboronic Acid Derivatives as Validated Leads Active in Clinical Strains Overexpressing KPC-2: A Step against Bacterial Resistance. CheµedChem 2018, 13, 713–724. [Google Scholar] [CrossRef]
- Cheng, H.C. The power issue: Determination of KB or Ki from IC50. A closer look at the Cheng-Prusoff equation, the Schild plot and related power equations. J. Pharmacol. Toxicol. Methods 2001, 46, 61–71. [Google Scholar] [CrossRef]
- Zhu, K.; Borrelli, K.W.; Greenwood, J.R.; Day, T.; Abel, R.; Farid, R.S.; Harder, E. Docking covalent inhibitors: A parameter-free approach to pose prediction and scoring. J. Chem. Inf. Model. 2014, 54, 1932–1940. [Google Scholar] [CrossRef]
- Tondi, D.; Powers, R.A.; Caselli, E.; Negri, M.-C.; Blázquez, J.; Costi, M.P.; Shoichet, B.K. Structure-based design and in parallel synthesis of inhibitors of AmpC β-lactamase. Chem. Biol. 2001, 8, 593–610. [Google Scholar] [CrossRef]
- Venturelli, A.; Tondi, D.; Cancian, L.; Morandi, F.; Cannazza, G.; Segatore, B.; Prati, F.; Amicosante, G.; Shoichet, B.K.; Costi, M.P. Optimizing cell permeation of an antibiotic resistance inhibitor for improved efficacy. J. Med. Chem. 2007, 50, 5644–5654. [Google Scholar] [CrossRef]
- The PyMOL Molecular Graphics System, Version 1.5.0.4; Schrödinger LLC: New York, NY, USA, 2010; Available online: http://www.pymol.org (accessed on 29 September 2019).
- Lahiri, S.D.; Mangani, S.; Durand-Reville, T.; Benvenuti, M.; De Luca, F.; Sanyal, G.; Docquier, J.D. Structural Insight into Potent Broad-Spectrum Inhibition with Reversible Recyclization Mechanism: Avibactam in Complex with CTX-M-15 and Pseudomonas aeruginosa AmpC β-Lactamases. Antimicrob. Agents Chemother. 2013, 57, 2496–2505. [Google Scholar] [CrossRef]
- Krishnan, N.P.; Nquyen, N.Q.; Papp-Wallace, K.M.; Bonomo, R.A.; van den Akker, F. Inhibition of Klebsiella β-Lactamases (SHV-1 and KPC-2) by Avibactam: A Structural Study. PLoS ONE 2015, 10, e0136813. [Google Scholar] [CrossRef]
- Powers, R.A.; Caselli, E.; Focia, P.J.; Prati, F.; Shoichet, B.K. Structures of ceftazidime and its transition-state analog in complex with AmpC beta-lactamase: Implications for resistance mutations and inhibitor design. Biochemistry 2001, 40, 9207–9214. [Google Scholar] [CrossRef]
- Pemberton, O.A.; Zhang, X.; Chen, Y. Molecular Basis of Substrate Recognition and Product Release by the Klebsiella pneumoniae Carbapenemase (KPC-2). J. Med. Chem. 2017, 60, 3525–3530. [Google Scholar] [CrossRef]
- Smith, C.A.; Frase, H.; Toth, M.; Kumarasiri, M.; Wiafe, K.; Munoz, J.; Mobashery, S.; Vakulenko, S.B. Structural basis for progression toward the carbapenemase activity in the GES family of beta-lactamases. J. Am. Chem. Soc. 2012, 134, 19512–19515. [Google Scholar] [CrossRef]
- Quotadamo, A.; Linciano, P.; Davoli, P.; Tondi, D.; Costi, M.P.; Venturelli, A. An Improved Synthesis of CENTA, a Chromogenic Substrate for β-Lactamases. Synlett 2016, 27, 2447–2450. [Google Scholar] [CrossRef]
- Cartwright, S.J.; Waley, S.G. Purification of beta-lactamases by affinity chromatography on phenylboronic acid-agarose. Biochem. J. 1984, 221, 505–512. [Google Scholar] [CrossRef]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef]
- Maestro, Version 11.1; Schrödinger, LLC: New York, NY, USA, 2017.
- Schrödinger Release 2017-1; Schrödinger, LLC: New York, NY, USA, 2017.
- ChemBio3D Ultra; PerkinElmer: Waltham, MA, USA, 2014.
- LigPrep, Version 3.5; Schrödinger, LLC: New York, NY, USA, 2015.
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [Green Version]
- Adams, P.D.; Afonine, P.V.; Bunkóczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.-W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Murshudov, G.N.; Skubák, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Moriarty, N.W.; Grosse-Kunstleve, R.W.; Adams, P.D. electronic Ligand Builder and Optimization Workbench (eLBOW): A tool for ligand coordinate and restraint generation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65, 1074–1080. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Code | Structure | KPC-2 [1] Ki μM | GES-5 [2] Ki μM | AmpC [3] Ki μM |
|---|---|---|---|---|
| 2 |  | 2.43 | 0.11 | 41.7 |
| 3 |  | 31.2 | 24.6 | 1.45 |
| 4 |  | 1.13 | 1.83 | 76.1 |
| 5 |  | 53.0 | 69.4 | 5.3 |
| Checkerboard Microdilution Assays and Drug Interaction Model | ||||||
|---|---|---|---|---|---|---|
| Strains | MIC [a] Antibiotic | FICI [b] Antibiotic + inhibitor | ||||
| MEM | CAZ | 2 | 3 | 4 | 5 | |
| Klebsiella 99D8 | 128 | - | 0.0156 | 2 | 0.1250 | 2 |
| Klebsiella 53A8 | 128 | - | 0.0117 | 2 | 0.1875 | 2 |
| Klebsiella 53A9 | 128 | - | 0.0175 | 1.5 | 0.2500 | 1.5 |
| P aeruginosa 12 | - | 32 | 2 | 0.5 | 2 | 0.3750 |
| P aeruginosa 148 | - | 128 | 1 | 0.375 | 1.5 | 0.0936 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linciano, P.; Vicario, M.; Kekez, I.; Bellio, P.; Celenza, G.; Martín-Blecua, I.; Blázquez, J.; Cendron, L.; Tondi, D. Phenylboronic Acids Probing Molecular Recognition against Class A and Class C β-lactamases. Antibiotics 2019, 8, 171. https://doi.org/10.3390/antibiotics8040171
Linciano P, Vicario M, Kekez I, Bellio P, Celenza G, Martín-Blecua I, Blázquez J, Cendron L, Tondi D. Phenylboronic Acids Probing Molecular Recognition against Class A and Class C β-lactamases. Antibiotics. 2019; 8(4):171. https://doi.org/10.3390/antibiotics8040171
Chicago/Turabian StyleLinciano, Pasquale, Mattia Vicario, Ivana Kekez, Pierangelo Bellio, Giuseppe Celenza, Isabel Martín-Blecua, Jesús Blázquez, Laura Cendron, and Donatella Tondi. 2019. "Phenylboronic Acids Probing Molecular Recognition against Class A and Class C β-lactamases" Antibiotics 8, no. 4: 171. https://doi.org/10.3390/antibiotics8040171
APA StyleLinciano, P., Vicario, M., Kekez, I., Bellio, P., Celenza, G., Martín-Blecua, I., Blázquez, J., Cendron, L., & Tondi, D. (2019). Phenylboronic Acids Probing Molecular Recognition against Class A and Class C β-lactamases. Antibiotics, 8(4), 171. https://doi.org/10.3390/antibiotics8040171