A New Look at the Structures of Old Sepsis Actors by Exploratory Data Analysis Tools

 ,
,  ,
,  ,
,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
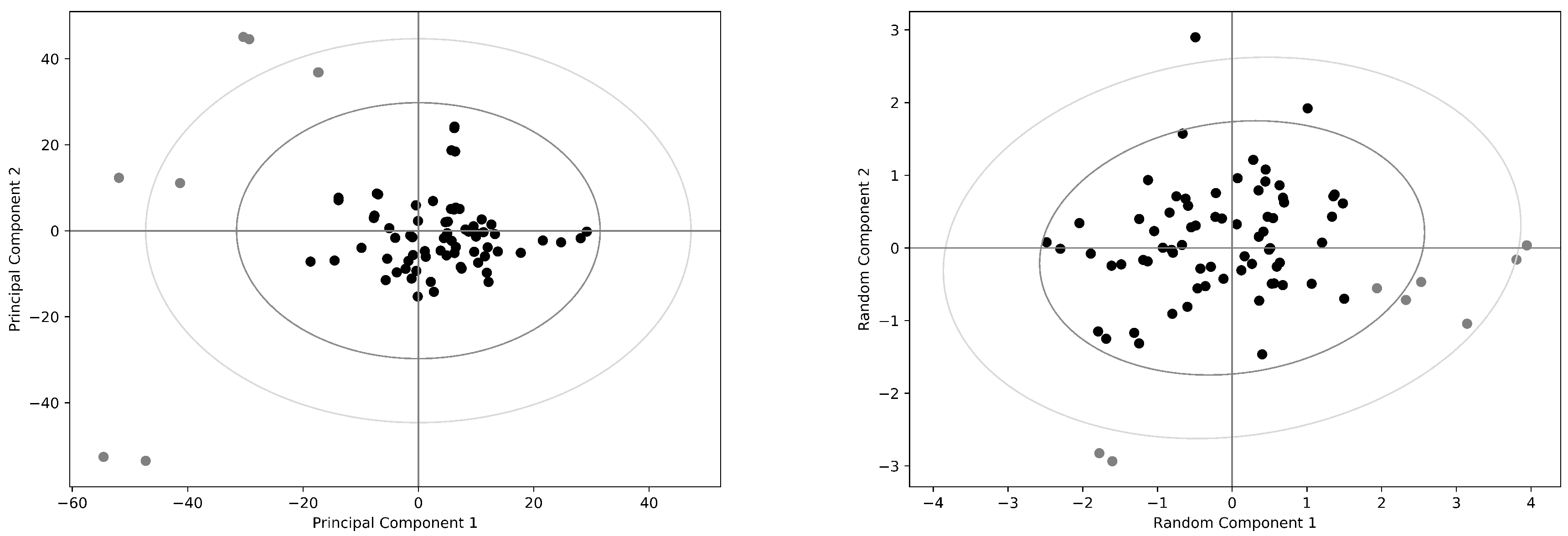
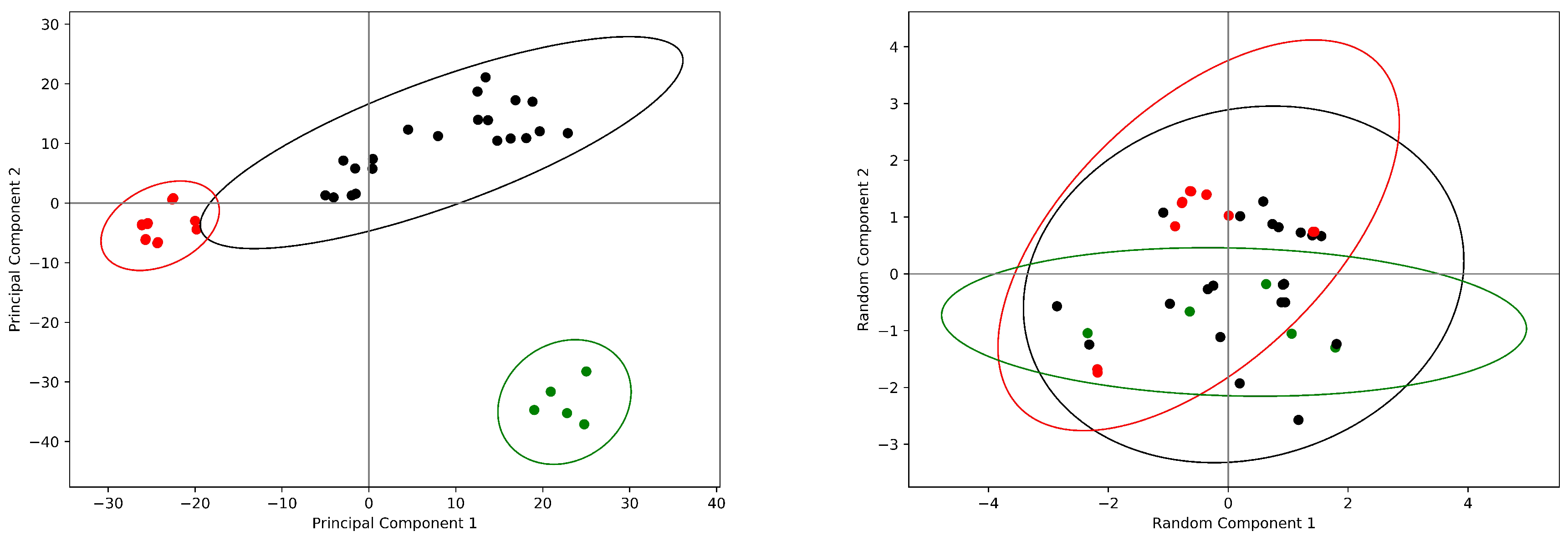
2. Results
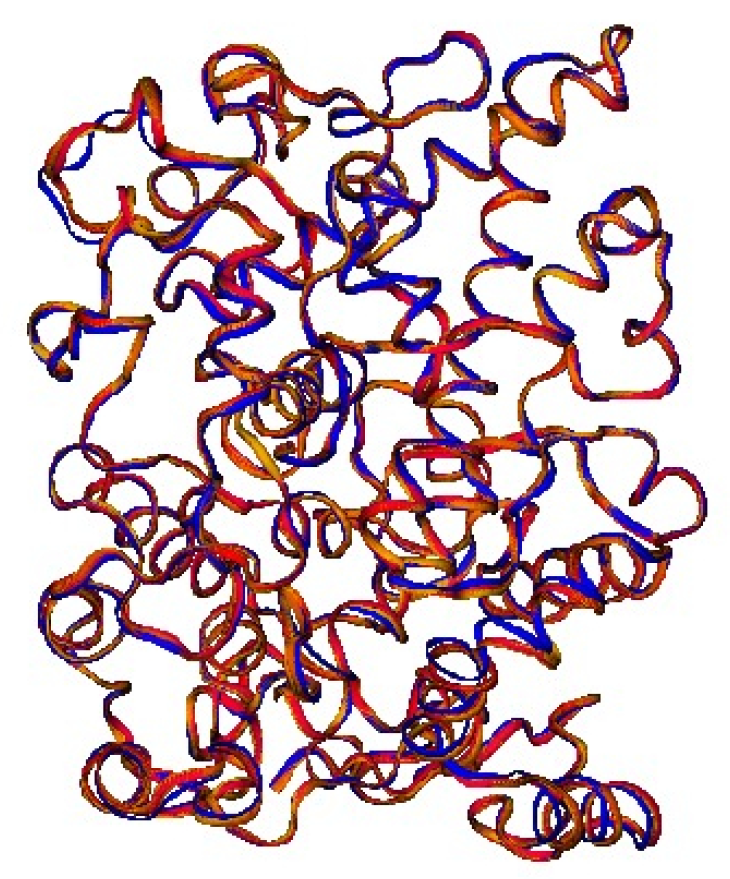
2.1. The Human Serum Albumin: Allostery in a Monomer
2.2. The Cyclooxygenase: Allostery without Conformational Change
2.3. Hemoglobin: The Quintessence of Allostery
3. Discussion
4. Methods
Author Contributions
Funding
Conflicts of Interest
References
- Berman, H.; Henrick, K.; Nakamura, H. Announcing the worldwide protein data bank. Nat. Struct. Biol. 2003, 10, 980. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlić, A.; Altunkaya, A.; Bi, C.; Bradley, A.R.; Christie, C.H.; Di Costanzo, L.; Duarte, J.M.; Dutta, S.; Feng, Z.; et al. The RCSB protein data bank: Integrative view of protein, gene and 3D structural information. Nucleic Acids Res. 2017, 45, D271–D281. [Google Scholar] [PubMed]
- Van Der Maaten, L.; Postma, E.; Van den Herik, J. Dimensionality reduction: A comparative analysis. J. Mach. Learn. Res. 2009, 10, 66–71. [Google Scholar]
- Pearson, K. LIII. On lines and planes of closest fit to systems of points in space. Philos. Mag. 1901, 2, 559–572. [Google Scholar] [CrossRef]
- Hotelling, H. Analysis of a complex of statistical variables into principal components. J. Educ. Psychol. 1933, 24, 417. [Google Scholar] [CrossRef]
- Ringnér, M. What is principal component analysis? Nat. biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef]
- Caruso, G.; Gattone, S.A.; Balzanella, A.; Di Battista, T. Cluster analysis: An application to a real mixed-type data set. In Models and Theories in Social Systems; Springer: Berlin/Heidelberg, Germany, 2019; pp. 525–533. [Google Scholar]
- Caruso, G.; Gattone, S.A.; Fortuna, F.; Di Battista, T. Cluster analysis as a decision-making tool: A methodological review. In International Symposium on Distributed Computing and Artificial Intelligence; Springer: Berlin/Heidelberg, Germany, 2017; pp. 48–55. [Google Scholar]
- Di Battista, T.; Fortuna, F. Clustering dichotomously scored items through functional k-means algorithm. Electron. J. Appl. Stat. Anal. 2016, 9, 433–450. [Google Scholar]
- Gattone, S.A.; Giordani, P.; Di Battista, T.; Fortuna, F. Adaptive cluster double sampling with post stratification with application to an epiphytic lichen community. Environ. Ecol. Stat. 2018, 25, 125–138. [Google Scholar] [CrossRef]
- Palese, L.L. Conformations of the HIV-1 protease: A crystal structure data set analysis. Biochim. Biophys. Acta 2017, 1865, 1416–1422. [Google Scholar] [CrossRef]
- Palese, L.L. Analysis of the conformations of the HIV-1 protease from a large crystallographic data set. Data Brief 2017, 15, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Halko, N.; Martinsson, P.G.; Tropp, J.A. Finding structure with randomness: Probabilistic algorithms for constructing approximate matrix decompositions. SIAM Rev. 2011, 53, 217–288. [Google Scholar] [CrossRef]
- Maida, I.; Zanna, P.; Guida, S.; Ferretta, A.; Cocco, T.; Palese, L.L.; Londei, P.; Benelli, D.; Azzariti, A.; Tommasi, S.; et al. Translational control mechanisms in cutaneous malignant melanoma: The role of eIF2α. J. Transl. Med. 2019, 17, 20. [Google Scholar] [CrossRef] [PubMed]
- Palese, L.L. Cytochrome c oxidase structures suggest a four-state stochastic pump mechanism. Phys. Chem. Chem. Phys. 2019, 21, 4822–4830. [Google Scholar] [CrossRef] [PubMed]
- Acharya, K.R.; Lloyd, M.D. The advantages and limitations of protein crystal structures. Trends Pharmacol. Sci. 2005, 26, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Palese, L.L. A random version of principal component analysis in data clustering. Comput. Biol. Chem. 2018, 73, 57–64. [Google Scholar] [CrossRef]
- Fanali, G.; di Masi, A.; Trezza, V.; Marino, M.; Fasano, M.; Ascenzi, P. Human serum albumin: From bench to bedside. Mol. Aspects Med. 2012, 33, 209–290. [Google Scholar] [CrossRef]
- Nicholson, J.; Wolmarans, M.; Park, G. The role of albumin in critical illness. Br. J. Anaesth. 2000, 85, 599–610. [Google Scholar] [CrossRef]
- Taverna, M.; Marie, A.L.; Mira, J.P.; Guidet, B. Specific antioxidant properties of human serum albumin. Ann. Intensive Care 2013, 3, 4. [Google Scholar] [CrossRef]
- Ascenzi, P.; Fasano, M. Allostery in a monomeric protein: The case of human serum albumin. Biophys. Chem. 2010, 148, 16–22. [Google Scholar] [CrossRef]
- Inchingolo, F.; Dipalma, G.; Cirulli, N.; Cantore, S.; Saini, R.; Altini, V.; Santacroce, L.; Ballini, A.; Saini, R. Microbiological results of improvement in periodontal condition by administration of oral probiotics. J. Biol. Regul. Homeost. Agents 2018, 32, 1323–1328. [Google Scholar] [PubMed]
- Vinolo, M.; Rodrigues, H.; Nachbar, R.; Curi, R. Modulation of inflammatory and immune responses by short-chain fatty acids. In Diet, Immunity and Inflammation; Elsevier: Amsterdam, The Netherlands, 2013; pp. 435–458. [Google Scholar]
- Smith, W.L.; DeWitt, D.L.; Garavito, R.M. Cyclooxygenases: Structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000, 69, 145–182. [Google Scholar] [CrossRef] [PubMed]
- Garavito, R.M.; Malkowski, M.G.; DeWitt, D.L. The structures of prostaglandin endoperoxide H synthases-1 and-2. Prostaglandins Other Lipid Mediat. 2002, 68, 129–152. [Google Scholar] [CrossRef]
- Smith, W.L.; Urade, Y.; Jakobsson, P.J. Enzymes of the cyclooxygenase pathways of prostanoid biosynthesis. Chem. Rev. 2011, 111, 5821–5865. [Google Scholar] [CrossRef] [PubMed]
- Tunctan, B.; Korkmaz, B.; Nihal Sari, A.; Kacan, M.; Unsal, D.; Sami Serin, M.; Kemal Buharalioglu, C.; Sahan-Firat, S.; Schunck, W.H.; R Falck, J.; et al. A novel treatment strategy for sepsis and septic shock based on the interactions between prostanoids, nitric oxide, and 20-hydroxyeicosatetraenoic acid. Antiinflamm. Antiallergy Agents Med. Chem. 2012, 11, 121–150. [Google Scholar]
- Dong, L.; Vecchio, A.J.; Sharma, N.P.; Jurban, B.J.; Malkowski, M.G.; Smith, W.L. Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer. J. Biol. Chem. 2011, 286, 19035–19046. [Google Scholar] [CrossRef]
- Zou, H.; Yuan, C.; Dong, L.; Sidhu, R.S.; Hong, Y.H.; Kuklev, D.V.; Smith, W.L. Human cyclooxygenase-1 activity and its responses to COX inhibitors are allosterically regulated by nonsubstrate fatty acids. J. Lipid Res. 2012, 53, 1336–1347. [Google Scholar] [CrossRef]
- Mitchener, M.M.; Hermanson, D.J.; Shockley, E.M.; Brown, H.A.; Lindsley, C.W.; Reese, J.; Rouzer, C.A.; Lopez, C.F.; Marnett, L.J. Competition and allostery govern substrate selectivity of cyclooxygenase-2. Proc. Natl. Acad. Sci. USA 2015, 112, 12366–12371. [Google Scholar] [CrossRef]
- Malkowski, M.G.; Thuresson, E.D.; Lakkides, K.M.; Rieke, C.J.; Micielli, R.; Smith, W.L.; Garavito, R.M. Structure of eicosapentaenoic and linoleic acids in the cyclooxygenase site of prostaglandin endoperoxide H synthase-1. J. Biol. Chem. 2001, 276, 37547–37555. [Google Scholar] [CrossRef]
- Thuresson, E.D.; Malkowski, M.G.; Lakkides, K.M.; Rieke, C.J.; Mulichak, A.M.; Ginell, S.L.; Garavito, R.M.; Smith, W.L. Mutational and x-ray crystallographic analysis of the interaction of dihomo-γ-linolenic acid with prostaglandin endoperoxide H synthases. J. Biol. Chem. 2001, 276, 10358–10365. [Google Scholar] [CrossRef]
- Malkowski, M.; Ginell, S.; Smith, W.; Garavito, R. The productive conformation of arachidonic acid bound to prostaglandin synthase. Science 2000, 289, 1933–1937. [Google Scholar] [CrossRef] [PubMed]
- Harman, C.A.; Rieke, C.J.; Garavito, R.M.; Smith, W.L. Crystal structure of arachidonic acid bound to a mutant of prostaglandin endoperoxide H synthase-1 that forms predominantly 11-hydroperoxyeicosatetraenoic acid. J. Biol. Chem. 2004, 279, 42929–42935. [Google Scholar] [CrossRef] [PubMed]
- Loll, P.J.; Picot, D.; Ekabo, O.; Garavito, R.M. Synthesis and Use of Iodinated Nonsteroidal Antiinflammatory Drug Analogs as Crystallographic Probes of the Prostaglandin H2 Synthase Cyclooxygenase Active Site. Biochemistry 1996, 35, 7330–7340. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Tam, M.F.; Simplaceanu, V.; Ho, C. New look at hemoglobin allostery. Chem. Rev. 2015, 115, 1702–1724. [Google Scholar] [CrossRef] [Green Version]
- Brunori, M. Half a Century of Hemoglobin’s Allostery. Biophys. J. 2015, 109, 1077–1079. [Google Scholar] [CrossRef] [Green Version]
- Effenberger-Neidnicht, K.; Hartmann, M. Mechanisms of hemolysis during sepsis. Inflammation 2018, 41, 1569–1581. [Google Scholar] [CrossRef]
- Bateman, R.; Sharpe, M.; Singer, M.; Ellis, C. The effect of sepsis on the erythrocyte. Int. J. Mol. Sci. 2017, 18, 1932. [Google Scholar] [CrossRef] [Green Version]
- Santacroce, L.; Losacco, T. Abdominal sepsis in surgical patients. Pathophysiology and prevention. Recenti Prog. Med. 2006, 97, 411–416. [Google Scholar]
- Yoo, H.; Ku, S.K.; Kim, S.W.; Bae, J.S. Early diagnosis of sepsis using serum hemoglobin subunit Beta. Inflammation 2015, 38, 394–399. [Google Scholar] [CrossRef]
- Jiang, Y.; Jiang, F.Q.; Kong, F.; An, M.M.; Jin, B.B.; Cao, D.; Gong, P. Inflammatory anemia-associated parameters are related to 28-day mortality in patients with sepsis admitted to the ICU: A preliminary observational study. Ann. Intensive Care 2019, 9, 67. [Google Scholar] [CrossRef] [Green Version]
- Perutz, M.F.; Rossmann, M.G.; Cullis, A.F.; Muirhead, H.; Will, G.; North, A. Structure of haemoglobin: A three-dimensional Fourier synthesis at 5.5-A. resolution, obtained by X-ray analysis. Nature 1960, 185, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Monod, J.; Wyman, J.; Changeux, J.P. On the nature of allosteric transitions: A plausible model. J. Mol. Biol. 1965, 12, 88–118. [Google Scholar] [CrossRef]
- Perutz, M. Stereochemistry of cooperative effects in haemoglobin: Haem–haem interaction and the problem of allostery. Nature 1970, 228, 726–734. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, J.; Chothia, C. Haemoglobin: The structural changes related to ligand binding and its allosteric mechanism. J. Mol. Biol. 1979, 129, 175–220. [Google Scholar] [CrossRef]
- Dey, S.; Chakrabarti, P.; Janin, J. A survey of hemoglobin quaternary structures. Proteins 2011, 79, 2861–2870. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, G.; Morimoto, H.; Yun, K.M.; Park, S.Y.; Nakagawa, A.; Minagawa, H.; Shibayama, N. Magnesium (II) and zinc (II)-protoporphyrin IX’s stabilize the lowest oxygen affinity state of human hemoglobin even more strongly than deoxyheme. J. Mol. Biol. 1999, 292, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- Waller, D.; Liddington, R. Refinement of a partially oxygenated T state human haemoglobin at 1.5 Å resolution. Acta Crystallogr. B 1990, 46, 409–418. [Google Scholar] [CrossRef]
- Kavanaugh, J.S.; Rogers, P.H.; Arnone, A. Crystallographic Evidence for a New Ensemble of Ligand-Induced Allosteric Transitions in Hemoglobin: The T-to-THigh Quaternary Transitions. Biochemistry 2005, 44, 6101–6121. [Google Scholar] [CrossRef]
- Sen, U.; Dasgupta, J.; Choudhury, D.; Datta, P.; Chakrabarti, A.; Chakrabarty, S.B.; Chakrabarty, A.; Dattagupta, J.K. Crystal structures of HbA2 and HbE and modeling of hemoglobin δ4: Interpretation of the thermal stability and the antisickling effect of HbA2 and identification of the ferrocyanide binding site in Hb. Biochemistry 2004, 43, 12477–12488. [Google Scholar] [CrossRef]
- Schumacher, M.A.; Dixon, M.M.; Kluger, R.; Jones, R.T.; Brennan, R.G. Allosteric transition intermediates modeled by cross-linked hemoglobins. Nature 1995, 375, 84–87. [Google Scholar] [CrossRef] [Green Version]
- Palese, L.L. Random Matrix Theory in molecular dynamics analysis. Biophys. Chem. 2015, 196, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Palese, L.L. Correlation Analysis of Trp-Cage Dynamics in Folded and Unfolded States. J. Phys. Chem. B 2015, 119, 15568–15573. [Google Scholar] [CrossRef] [PubMed]
- Palese, L.L. Protein States as Symmetry Transitions in the Correlation Matrices. J. Phys. Chem. B 2016, 120, 11428–11435. [Google Scholar] [CrossRef] [PubMed]
- Edelman, A.; Wang, Y. Random matrix theory and its innovative applications. In Advances in Applied Mathematics, Modeling, and Computational Science; Springer: Berlin/Heidelberg, Germany, 2013; pp. 91–116. [Google Scholar]
- Bossis, F.; Palese, L.L. Amyloid beta (1–42) in aqueous environments: Effects of ionic strength and E22Q (Dutch) mutation. Biochim. Biophys. Acta 2013, 1834, 2486–2493. [Google Scholar] [CrossRef] [PubMed]
- Palese, L.L. Protein dynamics: Complex by itself. Complexity 2013, 18, 48–56. [Google Scholar] [CrossRef]
- Cooper, A.; Dryden, D. Allostery without conformational change. Eur. Biophys. J. 1984, 11, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Trentadue, R.; Fiore, F.; Massaro, F.; Papa, F.; Iuso, A.; Scacco, S.; Santacroce, L.; Brienza, N. Induction of mitochondrial dysfunction and oxidative stress in human fibroblast cultures exposed to serum from septic patients. Life Sci. 2012, 91, 237. [Google Scholar]
- Bosmann, M.; Ward, P.A. The inflammatory response in sepsis. Trends Immunol. 2013, 34, 129–136. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Bro, R.; Smilde, A.K. Principal component analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef] [Green Version]
- Shlens, J. A tutorial on principal component analysis. arXiv 2014, arXiv:1404.1100. [Google Scholar]
- Raschka, S. Python Machine Learning; Packt Publishing: Birmingham, UK, 2015. [Google Scholar]
- Roweis, S. EM algorithms for PCA and SPCA. In Proceedings of the 1997 Conference on Advances in Neural Information Processing Systems, Denver, CO, USA, 29 November–4 December 1998; pp. 626–632. [Google Scholar]
- Johnson, W.B.; Lindenstrauss, J. Extensions of Lipschitz mappings into a Hilbert space. Cont. Math. 1984, 26, 1. [Google Scholar]
- Papadimitriou, C.H.; Tamaki, H.; Raghavan, P.; Vempala, S. Latent semantic indexing: A probabilistic analysis. J. Comput. Syst. Sci. 1998, 61, 159–168. [Google Scholar]
- Kaski, S. Dimensionality reduction by random mapping: Fast similarity computation for clustering. In Proceedings of the 1998 IEEE World Congress on Computational Intelligence, Anchorage, Alaska, 4–9 May 1998; Volume 1, pp. 413–418. [Google Scholar]
- Achlioptas, D. Database-friendly random projections. In Proceedings of the Twentieth ACM SIGMOD-SIGACT-SIGART Symposium on Principles of Database Systems, Santa Barbara, CA, USA, 21–23 May 2001; pp. 274–281. [Google Scholar]
- Bingham, E.; Mannila, H. Random projection in dimensionality reduction: Applications to image and text data. In Proceedings of the Seventh ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, San Francisco, CA, USA, 26–29 August 2001; pp. 245–250. [Google Scholar]
- Pérez, F.; Granger, B.E. IPython: A system for interactive scientific computing. Comput. Sci. Eng. 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Van Der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy array: A structure for efficient numerical computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Oliphant, T.E. Python for scientific computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pedregosa, F.; Varoquaux, G.; Gramfort, A.; Michel, V.; Thirion, B.; Grisel, O.; Blondel, M.; Prettenhofer, P.; Weiss, R.; Dubourg, V.; et al. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011, 12, 2825–2830. [Google Scholar]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gnoni, A.; De Nitto, E.; Scacco, S.; Santacroce, L.; Palese, L.L. A New Look at the Structures of Old Sepsis Actors by Exploratory Data Analysis Tools. Antibiotics 2019, 8, 225. https://doi.org/10.3390/antibiotics8040225
Gnoni A, De Nitto E, Scacco S, Santacroce L, Palese LL. A New Look at the Structures of Old Sepsis Actors by Exploratory Data Analysis Tools. Antibiotics. 2019; 8(4):225. https://doi.org/10.3390/antibiotics8040225
Chicago/Turabian StyleGnoni, Antonio, Emanuele De Nitto, Salvatore Scacco, Luigi Santacroce, and Luigi Leonardo Palese. 2019. "A New Look at the Structures of Old Sepsis Actors by Exploratory Data Analysis Tools" Antibiotics 8, no. 4: 225. https://doi.org/10.3390/antibiotics8040225
APA StyleGnoni, A., De Nitto, E., Scacco, S., Santacroce, L., & Palese, L. L. (2019). A New Look at the Structures of Old Sepsis Actors by Exploratory Data Analysis Tools. Antibiotics, 8(4), 225. https://doi.org/10.3390/antibiotics8040225