Investigation of Outbreaks of Extended-Spectrum Beta-Lactamase-Producing Klebsiella Pneumoniae in Three Neonatal Intensive Care Units Using Whole Genome Sequencing

 ,
,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
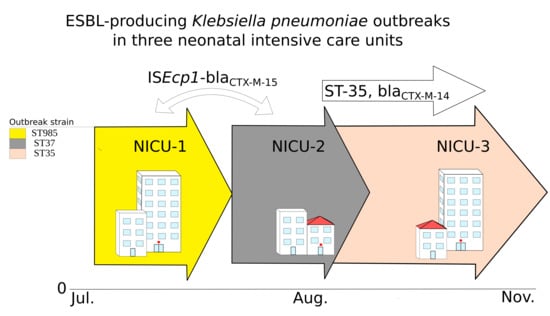
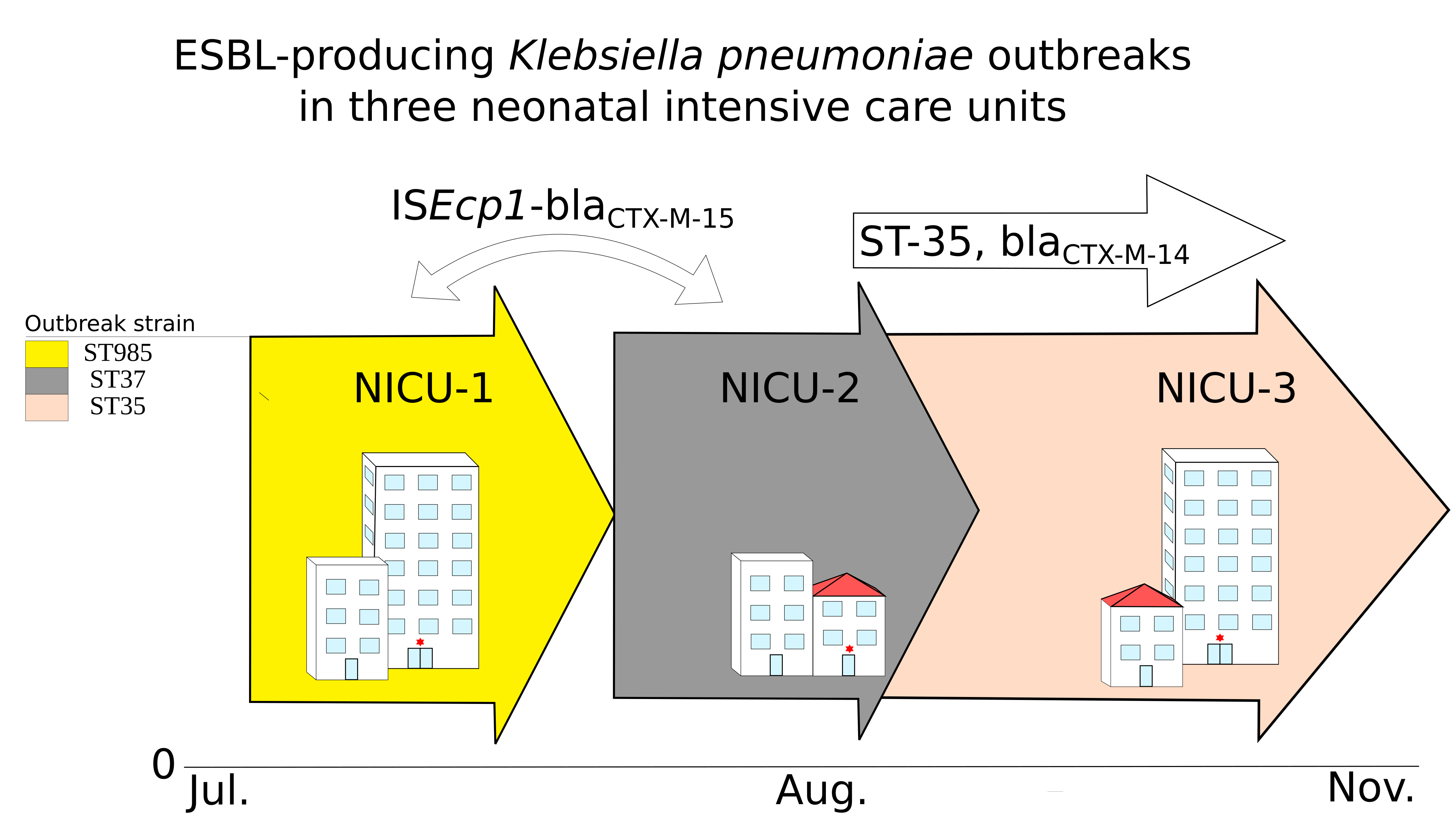
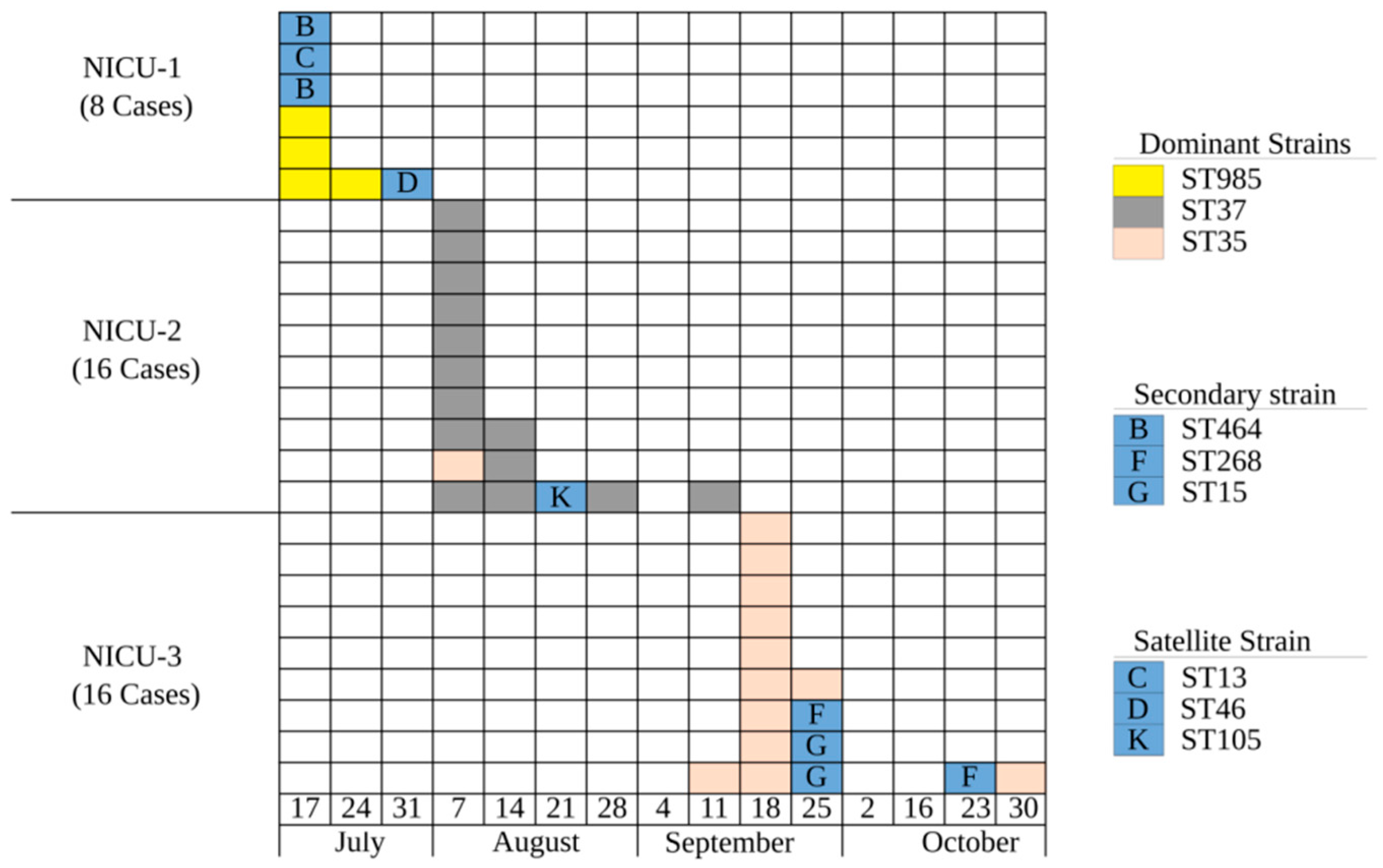
2.1. Was This a Clonal Outbreak Involving All Three Hospitals?
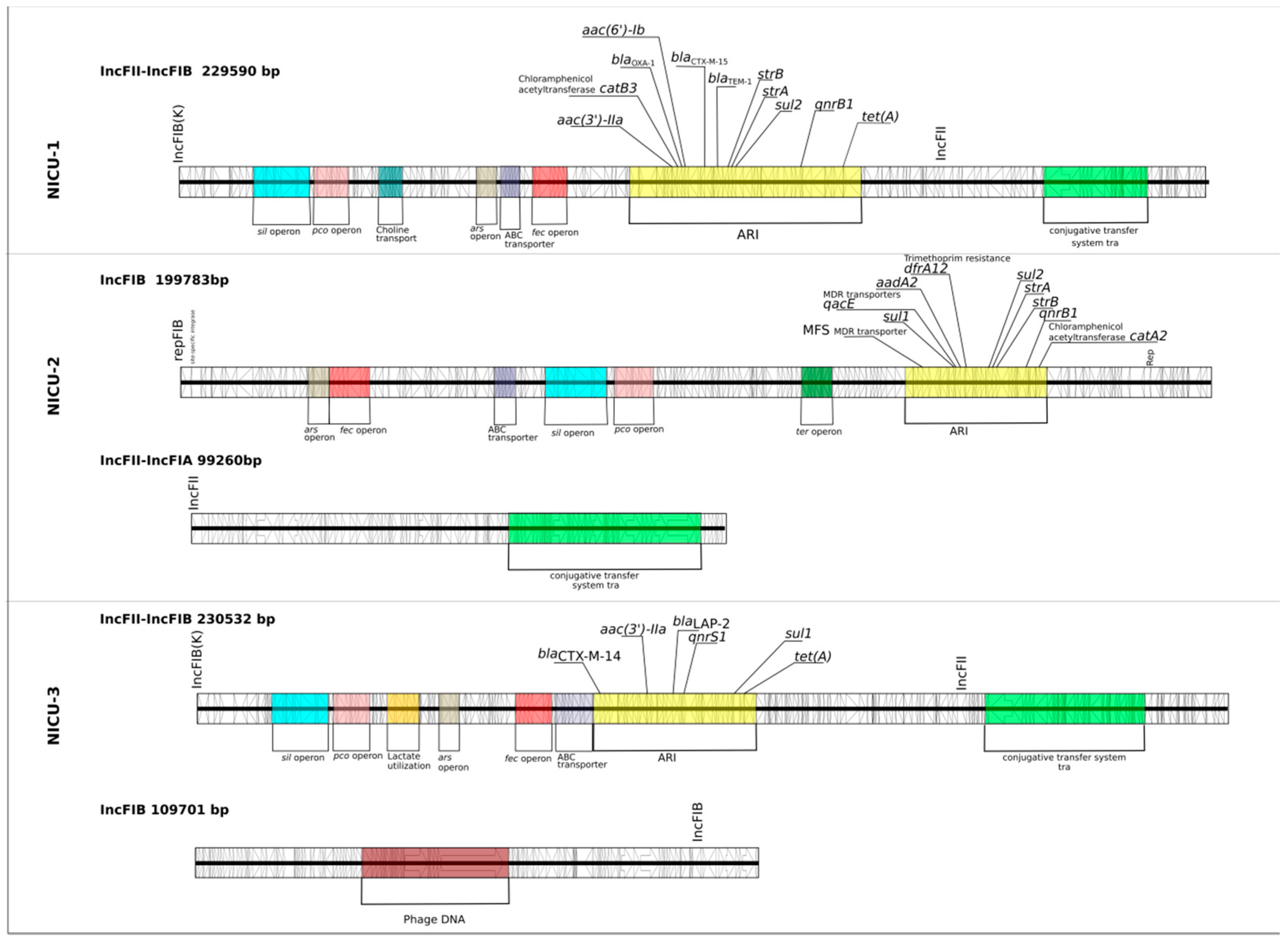
2.2. Were the Outbreaks a Result of Plasmid Dissemination Between Clones and NICUs?
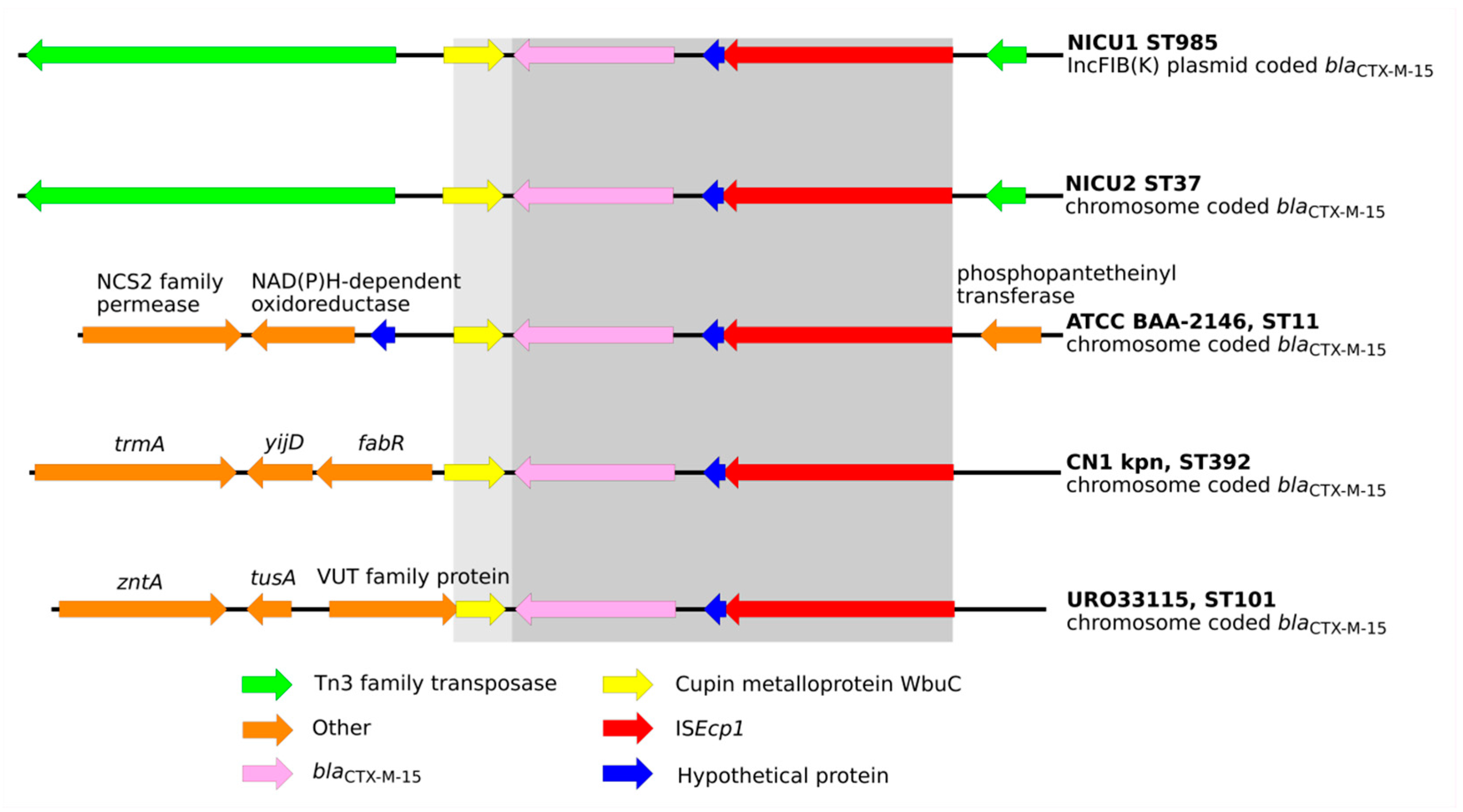
2.3. Were the Outbreaks the Result of Dissemination of a BlaCTX-M Transposable Element?
2.4. What May Have Contributed to the Success of the Dominant Clones?
3. Discussion
4. Materials and Methods
4.1. Patients and Setting
4.2. Isolates and Molecular Analysis
4.3. Phylogenomics, Resistomes, and Virulence Factors
4.4. Plasmid Content Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Centers for Disease Control and Prevention (United States). Antibiotic/Antimicrobial Resistance. Biggest Threats and Data. Available online: https://www.cdc.gov/drugresistance/biggest-threats.html?CDC_AA_refVal=https%3A%2F%2Fwww.cdc.gov%2Fdrugresistance%2Fbiggest_threats.html#groupb (accessed on 1 August 2020).
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, including Tuberculosis; World Health Organization: Geneva, Switzerland, 2017.
- Jernigan, J.A.; Hatfield, K.M.; Wolford, H.; Nelson, R.E.; Olubajo, B.; Reddy, S.C.; McCarthy, N.; Paul, P.; McDonald, L.C.; Kallen, A.; et al. Multidrug-resistant bacterial infections in U.S. hospitalized patients, 2012–2017. N. Engl. J. Med. 2020, 382, 1309–1319. [Google Scholar] [CrossRef] [PubMed]
- Bush, K.; Bradford, P.A. Epidemiology of β-lactamase-producing pathogens. Clin. Microbiol. Rev. 2020, 33, 00047-19. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.; Eller, C.; Hermes, J.; Kaase, M.; Steglich, M.; Radonić, A.; Dabrowski, P.W.; Nitsche, A.; Pfeifer, Y.; Werner, G.; et al. What caused the outbreak of ESBL-producing Klebsiella pneumoniae in a neonatal intensive care unit, Germany 2009 to 2012? Reconstructing transmission with epidemiological analysis and whole-genome sequencing. BMJ Open 2015, 5, e007397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltier, F.; Choquet, M.; Decroix, V.; Adjidé, C.C.; Castelain, S.; Guiheneuf, R.; Pluquet, E. Characterization of a multidrug-resistant Klebsiella pneumoniae ST607-K25 clone responsible for a nosocomial outbreak in a neonatal intensive care unit. J. Med. Microbiol. 2019, 68, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Luo, C.; Du, P.; Hu, J.; Zhao, X.; Mo, D.; Du, X.; Xu, X.; Li, M.; Lu, H.; et al. Genomic epidemiology of an outbreak of Klebsiella pneumoniae ST471 producing extended-spectrum β-Lactamases in a neonatal intensive care unit. Infect. Drug Resist. 2020, 13, 1081–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stapleton, P.J.M.; Murphy, M.; McCallion, N.; Brennan, M.; Cunney, R.; Drew, R.J. Outbreaks of extended spectrum beta-lactamase-producing Enterobacteriaceae in neonatal intensive care units: A systematic review. Arch. Dis. Child. Fetal Neonatal Ed. 2015, 101, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Paterson, D.L.; Bonomo, R.A. Extended-spectrum β-Lactamases: A clinical update. Clin. Microbiol. Rev. 2005, 18, 657–686. [Google Scholar] [CrossRef] [Green Version]
- Hudson, C.M.; Bent, Z.W.; Meagher, R.J.; Williams, K.P. Resistance determinants and mobile genetic elements of an NDM-1-Encoding Klebsiella pneumoniae Strain. PLoS ONE 2014, 9, e99209. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.; Wang, G.; Sebra, R.; Zhuge, J.; Yin, C.; Aguero-Rosenfeld, M.E.; Schuetz, A.N.; Dimitrova, N.; Fallon, J.T. Emergence and evolution of multidrug-resistant Klebsiella pneumoniae with both blaKPC and blaCTX-M integrated in the chromosome. Antimicrob. Agents Chemother. 2017, 61, e00076-17. [Google Scholar] [CrossRef] [Green Version]
- Mshana, S.E.; Fritzenwanker, M.; Falgenhauer, L.; Domann, E.; Hain, T.; Chakraborty, T.; Imirzalioglu, C. Molecular epidemiology and characterization of an outbreak causing Klebsiella pneumoniae clone carrying chromosomally located bla CTX-M-15 at a German University-Hospital. BMC Microbiol. 2015, 15, 122. [Google Scholar] [CrossRef] [Green Version]
- Sheppard, A.E.; Stoesser, N.; Wilson, D.J.; Sebra, R.; Kasarskis, A.; Anson, L.W.; Giess, A.; Pankhurst, L.J.; Vaughan, A.; Grim, C.J.; et al. Nested Russian doll-like genetic mobility drives rapid dissemination of the Carbapenem resistance GeneblaKPC. Antimicrob. Agents Chemother. 2016, 60, 3767–3778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perdigão, J.; Modesto, A.; Pereira, A.L.; Neto, O.; Matos, V.; Godinho, A.; Phelan, J.; Charleston, J.; Spadar, A.; De Sessions, P.F.; et al. Whole-genome sequencing resolves a polyclonal outbreak by extended-spectrum beta-lactam and carbapenem-resistant Klebsiella pneumoniae in a Portuguese tertiary-care hospital. Microb. Genom. 2020. [Google Scholar] [CrossRef]
- Wyres, K.L.; Lam, M.M.C.; Holt, K.E. Population genomics of Klebsiella pneumoniae. Nat. Rev. Genet. 2020, 18, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Miethke, M.; Marahiel, M.A. Siderophore-based iron acquisition and pathogen control. Microbiol. Mol. Biol. Rev. 2007, 71, 413–451. [Google Scholar] [CrossRef] [Green Version]
- Rakovitsky, N.; Frenk, S.; Kon, H.; Schwartz, D.; Temkin, E.; Solter, E.; Paikin, S.; Cohen, R.; Schwaber, M.J.; Carmeli, Y.; et al. Fourier transform infrared spectroscopy is a new option for outbreak investigation: A retrospective analysis of an extended-spectrum-beta-lactamase-producing Klebsiella pneumoniae outbreak in a neonatal intensive care unit. J. Clin. Microbiol. 2020, 58, 00098-20. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [Green Version]
- Holt, K.E.; Wertheim, H.; Zadoksm, R.N.; Baker, S.; Whitehouse, C.A.; Dance, D.; Jenney, A.; Connor, T.R.; Hsu, L.Y.; Severin, J.; et al. Genomic analysis of diversity, population structure, virulence, and antimicrobial resistance in Klebsiella pneumoniae, an urgent threat to public health. Proc. Natl. Acad. Sci. USA 2015, 112, E3574–E3581. [Google Scholar] [CrossRef] [Green Version]
- Page, A.J.; Cummins, C.A.; Hunt, M.; Wong, V.K.; Reuter, S.; Holden, M.T.; Fookes, M.; Falush, D.; Keane, J.A.; Parkhill, J. Roary: Rapid large-scale prokaryote pan genome analysis. Bioinformatics 2015, 31, 3691–3693. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2-Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In SilicoDetection and typing of plasmids using PlasmidFinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frenk, S.; Rakovitsky, N.; Temkin, E.; Schechner, V.; Cohen, R.; Kloyzner, B.S.; Schwaber, M.J.; Solter, E.; Cohen, S.; Stepansky, S.; et al. Investigation of Outbreaks of Extended-Spectrum Beta-Lactamase-Producing Klebsiella Pneumoniae in Three Neonatal Intensive Care Units Using Whole Genome Sequencing. Antibiotics 2020, 9, 705. https://doi.org/10.3390/antibiotics9100705
Frenk S, Rakovitsky N, Temkin E, Schechner V, Cohen R, Kloyzner BS, Schwaber MJ, Solter E, Cohen S, Stepansky S, et al. Investigation of Outbreaks of Extended-Spectrum Beta-Lactamase-Producing Klebsiella Pneumoniae in Three Neonatal Intensive Care Units Using Whole Genome Sequencing. Antibiotics. 2020; 9(10):705. https://doi.org/10.3390/antibiotics9100705
Chicago/Turabian StyleFrenk, Sammy, Nadya Rakovitsky, Elizabeth Temkin, Vered Schechner, Regev Cohen, Bat Sheva Kloyzner, Mitchell J. Schwaber, Ester Solter, Shoshana Cohen, Sarit Stepansky, and et al. 2020. "Investigation of Outbreaks of Extended-Spectrum Beta-Lactamase-Producing Klebsiella Pneumoniae in Three Neonatal Intensive Care Units Using Whole Genome Sequencing" Antibiotics 9, no. 10: 705. https://doi.org/10.3390/antibiotics9100705
APA StyleFrenk, S., Rakovitsky, N., Temkin, E., Schechner, V., Cohen, R., Kloyzner, B. S., Schwaber, M. J., Solter, E., Cohen, S., Stepansky, S., & Carmeli, Y. (2020). Investigation of Outbreaks of Extended-Spectrum Beta-Lactamase-Producing Klebsiella Pneumoniae in Three Neonatal Intensive Care Units Using Whole Genome Sequencing. Antibiotics, 9(10), 705. https://doi.org/10.3390/antibiotics9100705