Impact of Strain Competition on Bacterial Resistance in Immunocompromised Populations


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
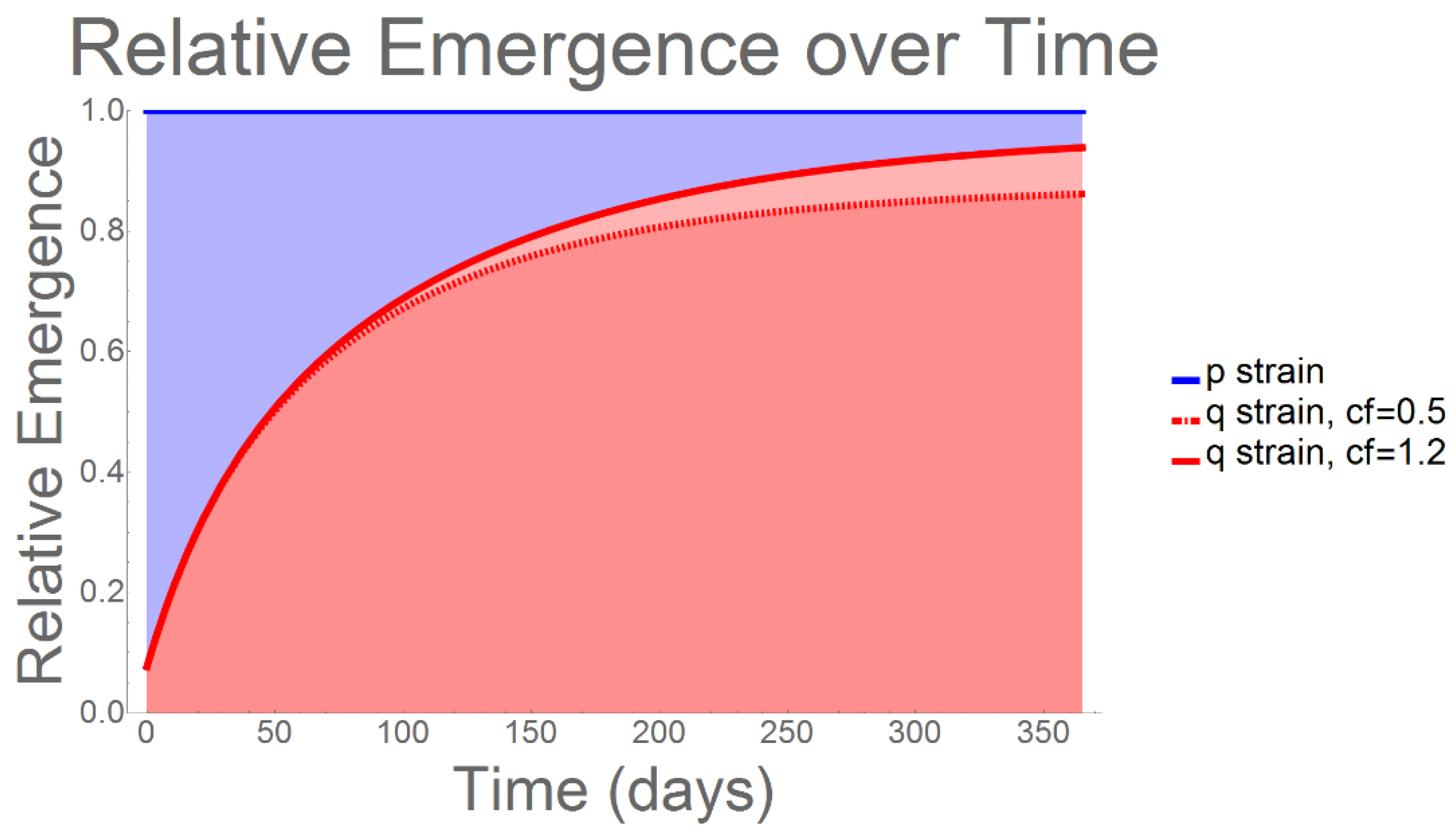
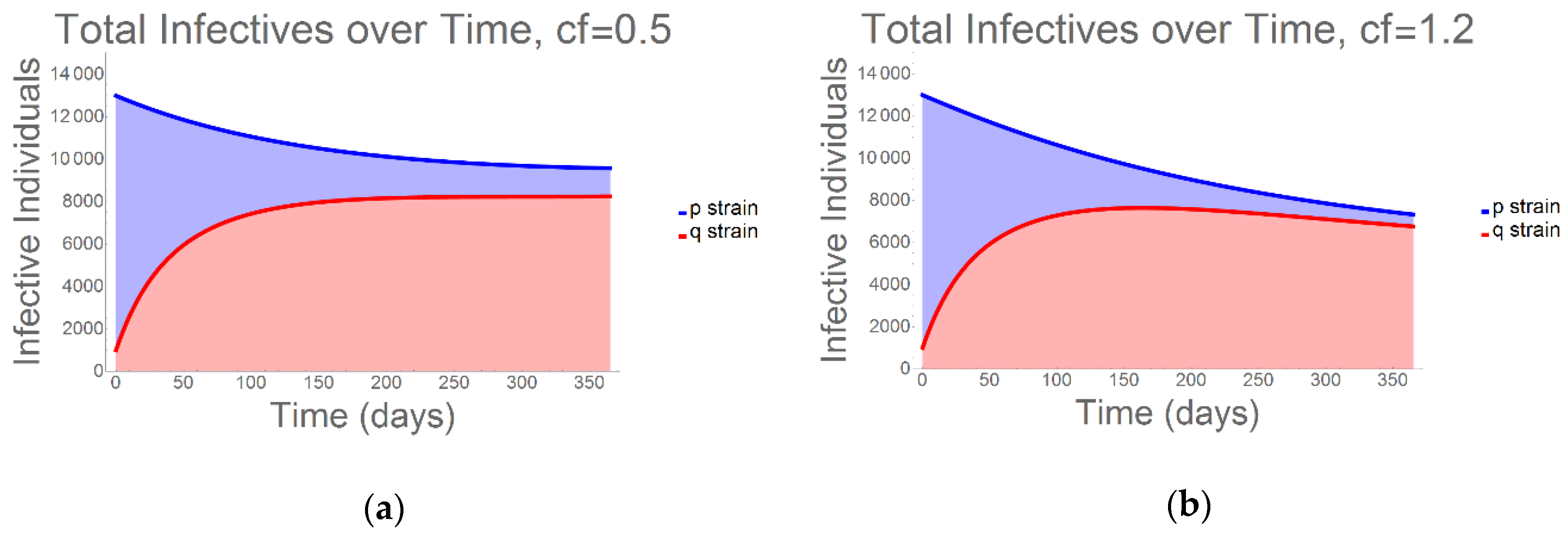
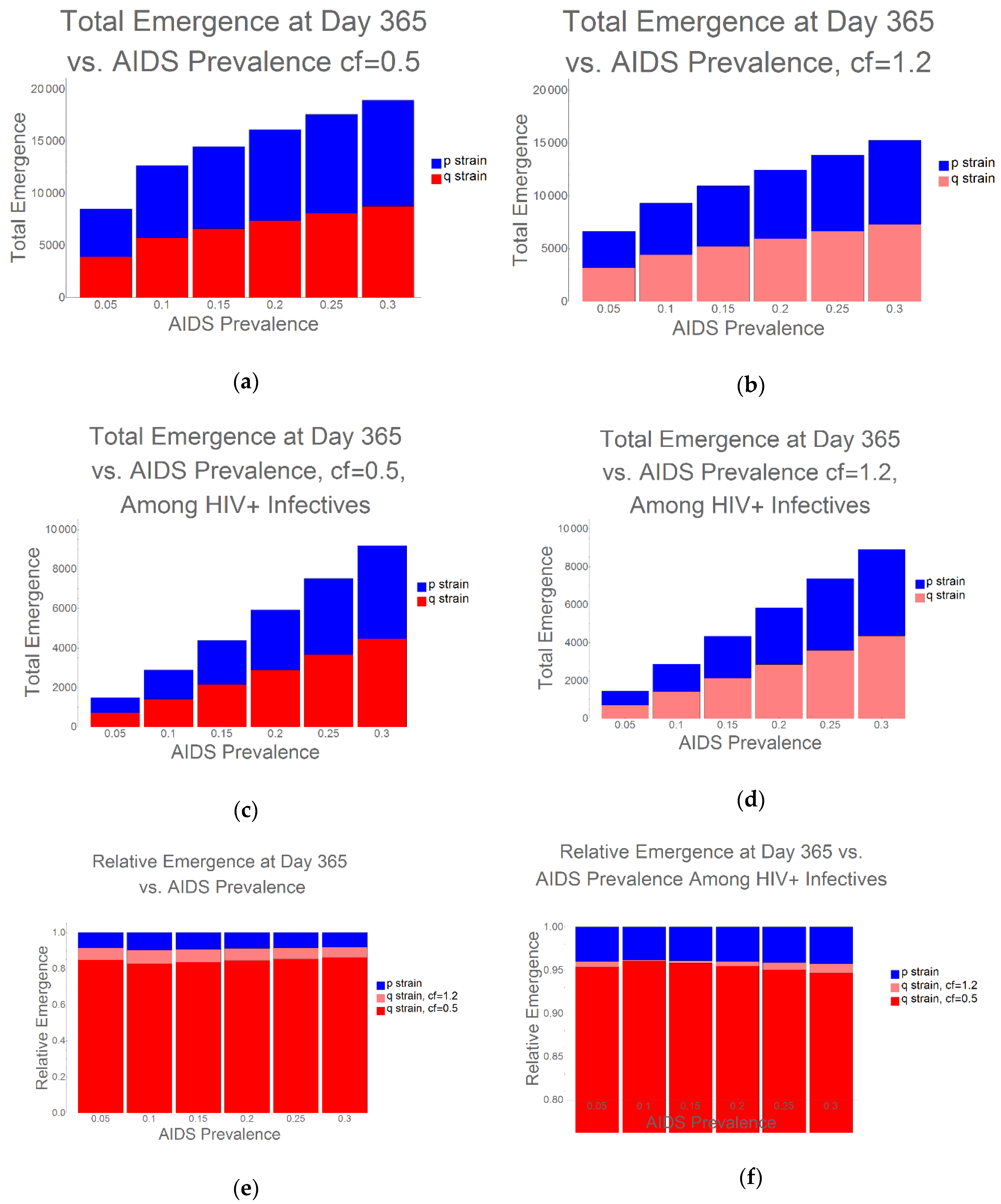
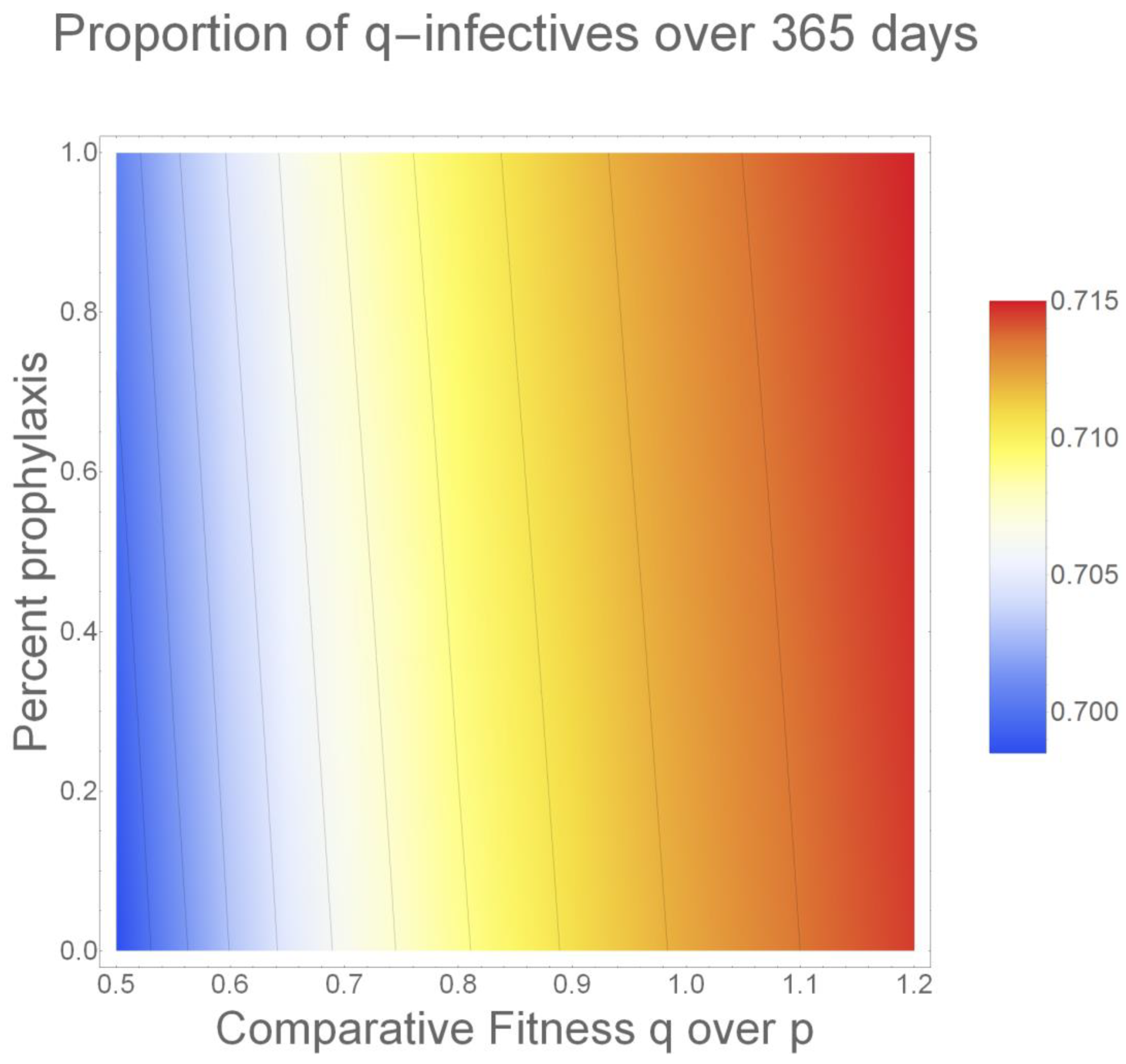
2. Results
3. Discussion
4. Materials and Methods
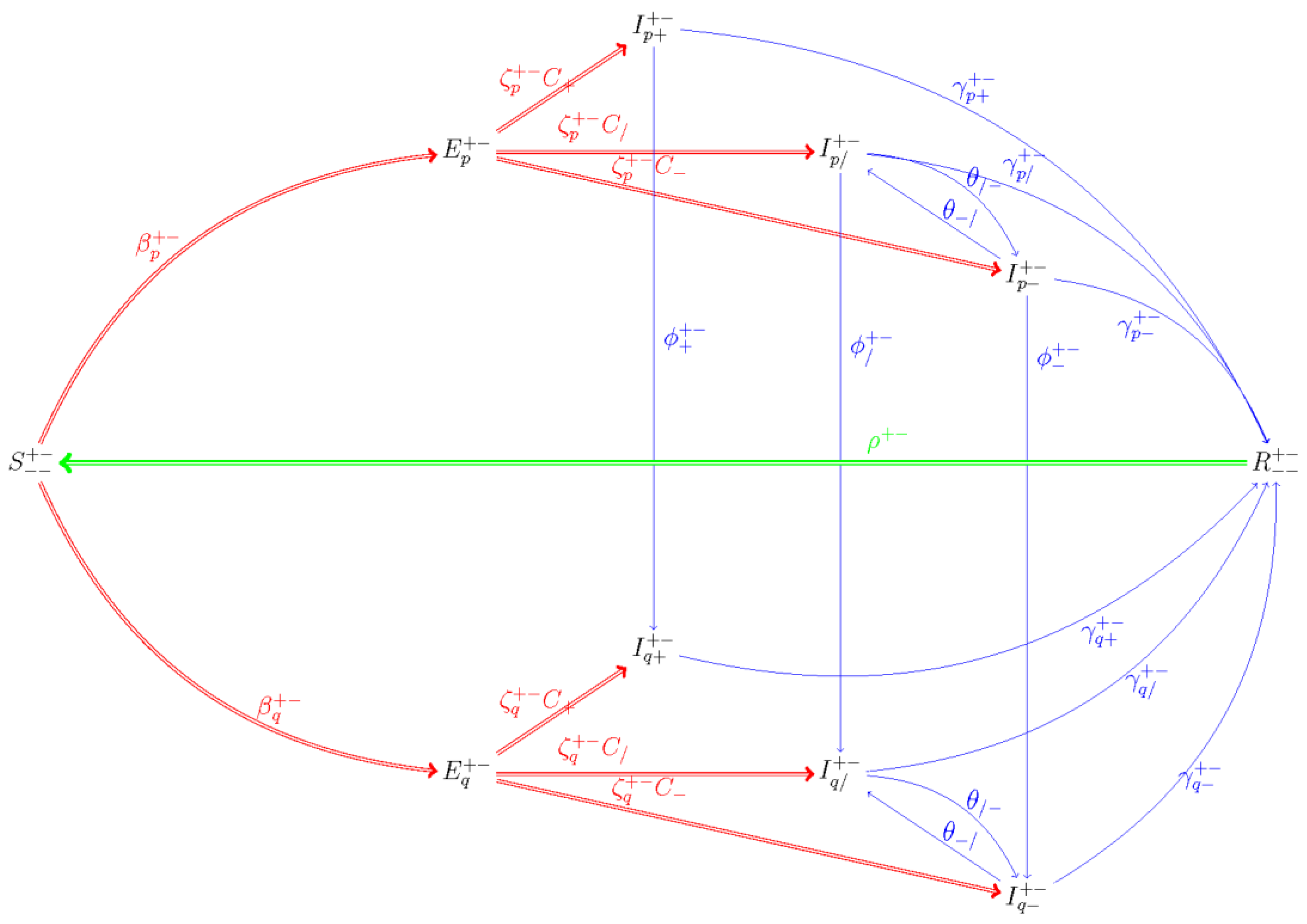
4.1. Mathematical Model
4.2. Methodological Framework
4.3. Prophylaxis-Attributable Emergence and Strain Circulation
4.4. Host-Dependent Variation in Transmission Probability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Model Description, Equations, and Parameters
Appendix A.1. Model Description and Parameters
Appendix A.2. System of Differential Equations
References
- Buchacz, K.; Baker, R.K.; Palella, F.J., Jr.; Chmiel, J.S.; Lichtenstein, K.A.; Novak, R.M.; Wood, K.C.; Brooks, J.T.; HOPS Investigators. AIDS-defining opportunistic illnesses in US patients, 1994–2007: A cohort study. Aids 2010, 24, 1549–1559. [Google Scholar]
- Cantón, R.; Morosini, M.-I. Emergence and spread of antibiotic resistance following exposure to antibiotics. FEMS Microbiol. Rev. 2011, 35, 977–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kolář, M.; Urbánek, K.; Látal, T. Antibiotic selective pressure and development of bacterial resistance. Int. J. Antimicrob. Agents 2001, 17, 357–363. [Google Scholar] [CrossRef]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, S122–S129. [Google Scholar] [CrossRef] [PubMed]
- Maciá, M.D.; Blanquer, D.; Togores, B.; Sauleda, J.; Pérez, J.L.; Oliver, A. Hypermutation Is a Key Factor in Development of Multiple-Antimicrobial Resistance in Pseudomonas aeruginosa Strains Causing Chronic Lung Infections. Antimicrob. Agents Chemother. 2005, 49, 3382–3386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, A.T.; Roque, F.; Falcão, A.; Figueiras, A.; Herdeiro, M.T. Understanding physician antibiotic prescribing behaviour: A systematic review of qualitative studies. Int. J. Antimicrob. Agents 2013, 41, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Laxminarayan, R.; Duse, A.; Wattal, C.; Zaidi, A.K.M.; Wertheim, H.F.L.; Sumpradit, N.; Vlieghe, E.; Hara, G.L.; Gould, I.M.; Goossens, H.; et al. Antibiotic resistance—The need for global solutions. Lancet Infect. Dis. 2013, 13, 1057–1098. [Google Scholar] [CrossRef] [Green Version]
- De Negre, A.A.; Ndeffo Mbah, M.L.; Myers, K.; Fefferman, N.H. Emergence of antibiotic resistance in immunocompromised host populations: A case study of emerging antibiotic resistant tuberculosis in AIDS patients. PLoS ONE 2019, 14, e0212969. [Google Scholar] [CrossRef]
- Freire-Moran, L.; Aronsson, B.; Manz, C.; Gyssens, I.C.; So, A.D.; Monnet, D.L.; Cars, O. Critical shortage of new antibiotics in development against multidrug-resistant bacteria—Time to react is now. Drug Resist. Updates 2011, 14, 118–124. [Google Scholar] [CrossRef]
- De Negre, A.A.; Myers, K.; Fefferman, N.H. Impact of chemorophylaxis policy for AIDS-immunocompromised patients on emergence of bacterial resistance. PLoS ONE 2020, 15, e0225861. [Google Scholar] [CrossRef] [Green Version]
- Wong, V.K.; Baker, S.; Pickard, D.J.; Parkhill, J.; Page, A.J.; Feasey, N.A.; Kingsley, R.A.; Thomson, N.R.; Keane, J.A.; Weill, F.-X.; et al. Phylogeographical analysis of the dominant multidrug-resistant H58 clade of Salmonella Typhi identifies inter- and intracontinental transmission events. Nat. Genet. 2015. Available online: http://www.nature.com/ng/journal/vaop/ncurrent/abs/ng.3281.html#supplementary-information (accessed on 6 March 2020). [CrossRef] [PubMed] [Green Version]
- Laxminarayan, R.; Heymann, D.L. Challenges of drug resistance in the developing world. BMJ 2012, 344, e1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousaf, M.Z.; Zia, S.; Babar, M.E.; Ashfaq, U.A. The epidemic of HIV/AIDS in developing countries; the current scenario in Pakistan. Virol. J. 2011, 8, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, A.D.; Mathers, C.D.; Ezzati, M.; Jamison, D.T.; Murray, C.J.L. Global and regional burden of disease and risk factors, 2001: Systematic analysis of population health data. Lancet 2006, 367, 1747–1757. [Google Scholar] [CrossRef]
- Central Intelligence Agency. Central Intelligence Agency World Factbook Swaziland 2013; Central Intelligence Agency: Washington, DC, USA, 2013.
- Bicego, G.T.; Nkambule, R.; Peterson, I.; Reed, J.; Donnell, D.; Ginindza, H.; Duong, Y.T.; Patel, H.; Bock, N.; Philip, N. Recent patterns in population-based HIV prevalence in Swaziland. PLoS ONE 2013, 8, e77101. [Google Scholar] [CrossRef] [PubMed]
- Earn, D.J.; Rohani, P.; Bolker, B.M.; Grenfell, B.T. A simple model for complex dynamical transitions in epidemics. Science 2000, 287, 667–670. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.; Sommers, B.; Murray, M. The effect of drug resistance on the fitness of Mycobacterium tuberculosis. Lancet Infect. Dis. 2003, 3, 13–21. [Google Scholar] [CrossRef]
- De Cock, K.M.; Jaffe, H.W.; Curran, J.W. The evolving epidemiology of HIV/AIDS. Aids 2012, 26, 1205–1213. [Google Scholar] [CrossRef]
- Kaplan, J.E.; Benson, C.; Holmes, K.K.; Brooks, J.T.; Pau, A.; Masur, H. Guidelines for prevention and treatment of opportunistic infections in HIV-infected adults and adolescents: Recommendations from CDC, the National Institutes of Health, and the HIV Medicine Association of the Infectious Diseases Society of America. MMWR Recomm. Rep. 2009, 58, 1. [Google Scholar]
- Sepkowitz, K.A. Opportunistic Infections in Patients with and Patients without Acquired Immunodeficiency Syndrome. Clin. Infect. Dis. 2002, 34, 1098. [Google Scholar] [CrossRef]
- Machado, A.; Bordalo, A.A. Prevalence of antibiotic resistance in bacteria isolated from drinking well water available in Guinea-Bissau (West Africa). Ecotoxicol. Environ. Saf. 2014, 106, 188–194. [Google Scholar] [CrossRef] [PubMed]
- U.S. Department of Health and Human Services, Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States; CDC: Atlanta, GA, USA, 2019.
- Levy, S.B. Antibiotic and antiseptic resistance: Impact on public health. Pediatr. Infect. Dis. J. 2000, 19, S120–S122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neu, H.C. The Crisis in Antibiotic Resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meyer, C.N.; Skinhoj, P.; Prag, J. Bacteremia in hiv-positive and aids patients—incidence, species distribution, risk-factors, outcome, and influence of long-term prophylactic antibiotic-treatment. Scand. J. Infect. Dis. 1994, 26, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Harbarth, S.; Samore, M.H.; Lichtenberg, D.; Carmeli, Y. Prolonged Antibiotic Prophylaxis After Cardiovascular Surgery and Its Effect on Surgical Site Infections and Antimicrobial Resistance. Circulation 2000, 101, 2916–2921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Padilla, E.; Dlamini, T.; Ascorra, A.; Rüsch-Gerdes, S.; Tefera, Z.D.; Calain, P.; de la Tour, R.; Jochims, F.; Richter, E.; Bonnet, M. High prevalence of multidrug-resistant tuberculosis, Swaziland, 2009-2010. Emerg. Infect. Dis. 2012, 18, 1. [Google Scholar] [CrossRef]
- Todd, J.; Glynn, J.R.; Marston, M.; Lutalo, T.; Biraro, S.; Mwita, W.; Suriyanon, V.; Rangsin, R.; Nelson, K.E.; Sonnenberg, P. Time from HIV seroconversion to death: A collaborative analysis of eight studies in six low and middle-income countries before highly active antiretroviral therapy. AIDS 2007, 21, S55–S63. [Google Scholar] [CrossRef] [Green Version]
- Deeks, S.G.; Lewin, S.R.; Havlir, D.V. The end of AIDS: HIV infection as a chronic disease. Lancet 2013, 382, 1525–1533. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.-C.; Chang, S.-C. Impact of highly active antiretroviral therapy on incidence and management of human immunodeficiency virus-related opportunistic infections. J. Antimicrob. Chemother. 2004, 54, 849–853. [Google Scholar] [CrossRef] [Green Version]
- Giraud, A.; Matic, I.; Tenaillon, O.; Clara, A.; Radman, M.; Fons, M.; Taddei, F. Costs and benefits of high mutation rates: Adaptive evolution of bacteria in the mouse gut. Science 2001, 291, 2606–2608. [Google Scholar] [CrossRef]
- Crow, J.F. Eighty Years Ago: The Beginnings of Population Genetics. Genetics 1988, 119, 473–476. [Google Scholar] [PubMed]
- Brown, M.T.; Bussell, J.K. Medication adherence: WHO cares? Mayo Clinic Proc. 2011, 86, 304–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterberg, L.; Blaschke, T. Adherence to Medication. N. Engl. J. Med. 2005, 353, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madden, J.M.; Graves, A.J.; Zhang, F. Cost-Related Medication Nonadherence and Spending on Basic Needs Following Implementation of Medicare Part D. JAMA 2008, 299, 1922–1928. [Google Scholar] [CrossRef]
- Briesacher, B.; Gurwitz, J.; Soumerai, S. Patients At-Risk for Cost-Related Medication Nonadherence: A Review of the Literature. J. Gen. Intern. Med. 2007, 22, 864–871. [Google Scholar] [CrossRef] [Green Version]
- Di Matteo, M.; Lepper, H.S.; Croghan, T.W. Depression is a risk factor for noncompliance with medical treatment: Meta-analysis of the effects of anxiety and depression on patient adherence. Arch. Inter. Med. 2000, 160, 2101–2107. [Google Scholar] [CrossRef]
- Shepard, C.W.; Soriano-Gabarro, M.; Zell, E.R.; Hayslett, J.; Lukacs, S.; Goldstein, S.; Factor, S.; Jones, J.; Ridzon, R.; Williams, I. Antimicrobial postexposure prophylaxis for anthrax: Adverse events and adherence. Emerg. Infect. Dis. 2002, 8, 1124–1132. [Google Scholar] [CrossRef]
- Walley, J.D.; Khan, M.A.; Newell, J.N.; Khan, M.H. Effectiveness of the direct observation component of DOTS for tuberculosis: A randomised controlled trial in Pakistan. Lancet 2001, 357, 664–669. [Google Scholar] [CrossRef]
- Cohen, T.; Murray, M. Modeling epidemics of multidrug-resistant M. tuberculosis of heterogeneous fitness. Nat. Med. 2004, 10, 1117–1121. [Google Scholar] [CrossRef] [Green Version]
- Dormans, J.; Burger, M.; Aguilar, D.; Hernandez-Pando, R.; Kremer, K.; Roholl, P.; Arend, S.M.; Van Soolingen, D. Correlation of virulence, lung pathology, bacterial load and delayed type hypersensitivity responses after infection with different Mycobacterium tuberculosis genotypes in a BALB/c mouse model. Clin. Exp. Immunol. 2004, 137, 460–468. [Google Scholar] [CrossRef]
- Billington, O.; McHugh, T.; Gillespie, S. Physiological cost of rifampin resistance induced in vitro in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 1999, 43, 1866–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stone, K.C.; Mercer, R.R.; Gehr, P.; Stockstill, B.; Crapo, J.D. Allometric Relationships of Cell Numbers and Size in the Mammalian Lung. Am. J. Respir. Cell Mol. Biol. 1992, 6, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Gill, W.P.; Harik, N.S.; Whiddon, M.R.; Liao, R.P.; Mittler, J.E.; Sherman, D.R. A replication clock for Mycobacterium tuberculosis. Nat. Med. 2009, 15, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dye, C.; Espinal, M.A. Will Tuberculosis Become Resistant to All Antibiotics? Proc. Biol. Sci. 2001, 268, 45–52. [Google Scholar] [CrossRef] [Green Version]
- Dye, C.; Williams, B.G. Criteria for the control of drug-resistant tuberculosis. Proc. Natl. Acad. Sci. USA 2000, 97, 8180–8185. [Google Scholar] [CrossRef] [Green Version]
- Sibanda, E.L.; Weller, I.V.D.; Hakim, J.G.; Cowan, F.M. Does Trimethoprim-Sulfamethoxazole Prophylaxis for HIV Induce Bacterial Resistance to Other Antibiotic Classes? Results of a Systematic Review. Clin. Infect. Dis. 2011, 52, 1184–1194. [Google Scholar] [CrossRef]
- Homedes, N.; Ugalde, A. Patients’ compliance with medical treatments in the third world. What do we know? Health Policy Plan. 1993, 8, 291–314. [Google Scholar] [CrossRef]
- Winnick, S.; Lucas, D.O.; Hartman, A.L.; Toll, D. How Do You Improve Compliance? Pediatrics 2005, 115, e718–e724. [Google Scholar] [CrossRef] [Green Version]
- Aronson, B.S. Antibiotic-taking experiences of undergraduate college students. J. Am. Acad. Nurse Pract. 2006, 18, 591–598. [Google Scholar] [CrossRef]
- Kermack, W.O.; McKendrick, A.G. A Contribution to the Mathematical Theory of Epidemics. Proc. R. Soc. Lond. Ser. A 1927, 115, 700–721. [Google Scholar] [CrossRef] [Green Version]
- Beldomenico, P.M.; Begon, M. Disease spread, susceptibility and infection intensity: Vicious circles? Trends Ecol. Evol. 2010, 25, 21–27. [Google Scholar] [CrossRef] [PubMed]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
DeNegre, A.A.; Myers, K.; Fefferman, N.H. Impact of Strain Competition on Bacterial Resistance in Immunocompromised Populations. Antibiotics 2020, 9, 114. https://doi.org/10.3390/antibiotics9030114
DeNegre AA, Myers K, Fefferman NH. Impact of Strain Competition on Bacterial Resistance in Immunocompromised Populations. Antibiotics. 2020; 9(3):114. https://doi.org/10.3390/antibiotics9030114
Chicago/Turabian StyleDeNegre, Ashley A., Kellen Myers, and Nina H. Fefferman. 2020. "Impact of Strain Competition on Bacterial Resistance in Immunocompromised Populations" Antibiotics 9, no. 3: 114. https://doi.org/10.3390/antibiotics9030114
APA StyleDeNegre, A. A., Myers, K., & Fefferman, N. H. (2020). Impact of Strain Competition on Bacterial Resistance in Immunocompromised Populations. Antibiotics, 9(3), 114. https://doi.org/10.3390/antibiotics9030114